


Abstract
Despite high levels of homology, transcription coactivators p300 and CREB binding protein (CBP) are both indispensable during embryogenesis. They are largely known to regulate the same genes. To identify genes preferentially regulated by p300 or CBP, we performed an extensive genome-wide survey using the ChIP-seq on cell-cycle synchronized cells. We found that 57% of the tags were within genes or proximal promoters, with an overall preference for binding to transcription start and end sites. The heterogeneous binding patterns possibly reflect the divergent roles of CBP and p300 in transcriptional regulation. Most of the 16 103 genes were bound by both CBP and p300. However, after stimulation 89 and 1944 genes were preferentially bound by CBP or p300, respectively. Target genes were found to be primarily involved in the regulation of metabolic and developmental processes, and transcription, with CBP showing a stronger preference than p300 for genes active in negative regulation of transcription. Analysis of transcription factor binding sites suggest that CBP and p300 have many partners in common, but AP-1 and Serum Response Factor (SRF) appear to be more prominent in CBP-specific sequences, whereas AP-2 and SP1 are enriched in p300-specific targets. Taken together, our findings further elucidate the distinct roles of coactivators p300 and CBP in transcriptional regulation.
INTRODUCTION
The primary mechanism to control cellular processes, such as proliferation and differentiation, is by regulation of gene expression [reviewed in (1–3)]. Gene expression is a highly coordinated process that results in the synthesis of messenger RNA after recruitment of the pre-initiation complex, histone modifying factors and transcription factors (TFs) to regulatory regions of the chromatin. The histone modifications that take place during this process, including methylation and acetylation, play a critical role in gene regulation, and defects have been implicated in many pathological conditions from cancer to autoimmune diseases (4–6). Recently, chromatin immunoprecipitation (ChIP) has been extensively applied in combination with high-throughput sequencing to map genome-wide chromatin modification profiles in human T cells (7,8) and in mouse ES cells (9). Binding sites of the insulator binding protein CTCF (7), RNA pol II (7,10) and several TFs (11–14) have also been mapped. The acetylation profile in primary human T cells was further investigated by determining the binding of several histone deacetylases (15) and histone acetyltransferases (HATs) including p300. Binding of p300 was found both at genes and at intergenic DNase hypersensitive sites, consistent with binding to enhancers, found in other p300 ChIP-seq experiments (16,17).
The HAT p300 and its family member CREB-binding protein (CBP) are transcription coactivators for a broad range of genes involved in multiple cellular processes such as proliferation, differentiation, apoptosis and DNA repair [reviewed in (18,19)]. In addition, a number of studies suggested the involvement of p300 and CBP in pathological disorders such as the Rubinstein–Taybi Syndrome [reviewed in (20)] and the development of cancer [reviewed in (21)]. Originally, CBP was identified through its association with the phosphorylated TF CREB (22), but CBP and p300 also interact with many other TFs, such as cJun (23), p53 (24) and MyoD (25) via conserved domains (CH1, CH3, KIX and SID). Apart from the transcriptional regulation through acetylation of histones and other factors, p300 and CBP can also act as a bridge or as a scaffold between upstream TFs and the basal transcription machinery.
A crucial role for both p300 and CBP in development was shown in mice with a homozygous deletion of either gene (Ep300 and Crebbp for the proteins p300 and CBP) resulting in embryonic lethality at a very early stage (26,27). Interestingly, the double heterozygous Ep300+/–/Crebbp+/– mice also die in utero (26), indicating that a fine-tuned balance in the expression of both proteins is needed to ensure the normal development. From phenotypic changes in the knock-out mice it is indicated that p300 and CBP have different functions, which has been further illustrated in additional in vivo studies (28–30). A comparison between the acetyltransferase domains of p300 and CBP showed that they differ structurally (31). In part, this might contribute to their functional differences. However, the current detailed mechanism of action of p300 and CBP and the differences between these transcription coactivators is not clear.
In contrast to the in vivo situation, most studies with tissue culture cells show similar functions for p300 and CBP, and only limited differential roles for p300 and CBP have been described [reviewed in (18)]. To obtain a better insight into genes regulated by the general transcription coactivators p300 or CBP next-generation sequencing of ChIP genomic fragments (ChIP-seq) (14) was performed. ChIP-seq and ChIP-on-microarray (ChIP-chip) have high correspondence in results, but ChIP-seq offers the advantages of requiring less input material, potential to identify binding sites with low affinity, not being limited to target regions (i.e. probes on a microarray), not having hybridization errors and it is less costly for whole genome analysis (14). In this study, we used the glioblastoma cell line T98G. This cell line can easily be synchronized by serum-deprivation and reintroduced into the cell cycle upon stimulation with serum and TPA. Previously, RNA pol II ChIP was performed in growth factor stimulated T98G cells (32), and this showed that 30 min upon growth factor stimulation pol II occupancy at the promoter of immediate early genes was maximal. We observed that maximal occupancy of p300 and CBP at the promoter of immediate early genes was also around 30 min (Y.F.M.R., unpublished results).
We show p300 and CBP binding to the chromatin in quiescent and stimulated cells, and alterations in their binding to a large number of genes after stimulation. In most cases there is overlap between regions bound by p300 and CBP, but we also identified distinct regions of binding, indicating specific targets for each of these acetyltransferases. Bound regions were analyzed genome-wide for their position relative to genes and were found to have a preference for transcription start sites (TSSs) and transcript ends. Interestingly, functional classification of target genes suggests that CBP is more involved in the regulation of transcription inhibition than p300. A list of TFs that might be involved in the transcription regulation of the identified genes together with p300 and/or CBP was obtained by searching for enriched TF binding sites (TFBSs) in the bound regions. Results show previously established binding partners, and suggest differences for p300 and CBP in their preferences for TFs.
MATERIALS AND METHODS
Cell culture, ChIP, qPCR and sequencing
Human glioblastoma T98G cells were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS), penicillin (100 μg/ml) and streptomycin (100 μg/ml). Prior to stimulation with serum (20%) and tetradecanoyl phorbol acetate (TPA 100 ng/ml; Sigma), cells were serum starved for 2–3 days (DMEM supplemented with 0.1% FBS).
For sequencing, chromatin was isolated from serum-starved cells (T0) and from cells stimulated for 30 min with serum and TPA (T30). Chromatin from T30 samples were prepared in duplicate, each being used for individual ChIPs, sequencing and downstream analysis. In addition, for more time-point specific data (analyzed only by ChIP and quantitative PCR) we isolated chromatin at 0, 2, 5, 15, 30, 60, 90, 120 and 360 min following stimulation. Chromatin was prepared and ChIPs were carried out as previously described, including fragmentation by sonification (33) (fragment size ∼500 bp). Immunoprecipitations were performed for p300 using the p300-(2) antibody produced in our lab (23), and for CBP with a commercially available antibody (A22 from Santa Cruz).
For Reverse Transcriptase-Polymerase Chain Reaction (RT-PCR) analysis, RNA was isolated using the SV Total RNA isolation System (Promega Corporation Benelux), according to the manufacturers' protocol, and first-strand cDNA synthesis was performed using 1 μg of RNA and ImProm II reverse transcriptase (Promega Corporation Benelux).
Quantitative PCR for ChIP and for cDNA samples was carried out on Applied Biosystems 7900HT Fast Real-Time PCR System with SYBR Green PCR Master Mix (Applied Biosystems Europe). Primers were designed using the Primer Express program from Applied Biosystems (for sequences of primers see Supplementary Table S1). Efficiency of the ChIP is presented as percentage of the input. Expression levels of the genes as determined by quantitative RT-PCR were normalized to GAPDH, and fold induction was calculated with reference to the untreated samples (t = 0 minutes).
For ChIP-seq all samples were prepared with Illumina's DNA sampleprep Kit (FC-102-1001) according to the manufacturer's protocol. Single ends of each sample were then sequenced on a single lane of the Illumina Genome Analyzer (GAI for samples CBP T0 and T30-1 and p300 T0 and T30-1, GAII for samples CBP T30-2 and p300 T30-2) for 36 cycles.
Illumina genome analyzer sequencing analysis
Sequencing results were run through the standard Illumina GAPipeline (v1.0 for GAI runs and v1.3 for GAII runs) to convert images to reads (unaligned sequences produced by the Illumina Genome Analyzer) and edit for quality (FIRECREST, Bustard and GERALD). A general overview of the entire ChIP-seq analysis is provided in Supplementary Figure S1A. The reads were then trimmed to the first 32 bp to remove lower quality base calls at the 3′-end of the read. These were then run through the developing GAPSS_R (www.lgtc.nl/GAPSS) pipeline. This pipeline took the reads, removed the first base pair (often low quality compared to other 5′ nucleotides), converted to FASTA format, aligned to the human reference genome (NCBI build 36) with Rmap v0.41 (34), permitting up to two mismatches, and exported tags (the term for aligned reads) into region files (merging adjacent nucleotides with at least one aligned read into one region, followed by compressing those regions within 100 bp into one (based on a range of compression sizes, see below and Supplementary Figures S1B and S2). The pipeline also created wiggle files [viewable in the UCSC genome browser (35)]. These tracks had positions with only a single read removed, in order to create more manageable files.
All unedited wiggle files were concatenated to one with custom Perl scripts and converted to a region file [a range of compression windows (20, 50, 100, 150 and 200 bp) were used] with GAPSS_R scripts. The compression windows account for small gaps in the genomic sequences covered, such as the result of non-unique genomic sequences (Rmap does not map to these). An appropriate compression size is hard to determine, considering a bigger window results in less regions (Supplementary Figure S2) and therefore specificity, but covers larger genomic repeats. We settled on a window of 100 bp to retain a large number of regions, while at the same time accounting for small repetitive elements. This consensus region file had the number of tags from the individual region files mapped to it with a custom Perl script. To make data more manageable and reduce background or very low affinity binding we removed regions with <6 tags (total over all samples). To further reduce the noise only regions with at least 1 tag/million reads aligned (18.1 tags across all total samples) were evaluated. Without applying this threshold performance was poorer, as addressed in the results.
To annotate regions we downloaded from Ensembl 54 Biomart (36,37) for all genes (with an HGNC ID) the chromosomal location, strand, gene start, gene end, transcript start, transcript end and gene ID. These were loaded into a custom mysql database that was queried to annotate regions for overlap with genes (including flanking 1 kb). We also annotated for distance to the nearest TSSs and transcript ends. Histograms were plotted in the statistical language R to visualize the distance to TSSs and transcript ends.
Statistical analysis
The statistical language R was used to evaluate reproducibility and overlap across samples and to determine genes with differential TF binding across different conditions. To be able to compare data across samples, samples were scaled to the average total number of tags per condition/coactivator. A square root transformation was applied before calculating the reproducibility and comparability across samples. This was to stabilize the variance, inherent to the count process, over the entire intensity range (38), and to spread the data points better over the intensity range (Supplementary Figure S3). After this, to give a better estimation of the comparability of the data from the different samples Pearson’s correlations were calculated in R. This was done on all regions with abundance >1 tag/million tags and a square root transformation applied before calculating the correlations. The Pearson’s correlations on the linear scale were slightly lower.
Subsequently, data were summarized at the gene level by adding all tags within a gene or its 1 kb flanking regions. To determine the genes different between conditions/coactivators Fisher’s exact P-values were calculated in R. For each individual gene, a two-by-two table was created containing the number of tags for this gene in condition 1 and condition 2 and the total number of tags in condition 1 and condition 2. We then applied the method of Benjamini and Hochberg to correct for multiple testing.
Functional classification
A list of 250 genes, identified as most significantly different between the time points for each coactivator (T30/T0 with adj. P-value <0.001), was uploaded in DAVID 2008 (39,40) for functional enrichment analysis. To obtain a general impression of the types of processes in which CBP and p300 are involved, functional annotation charts were generated for the Gene Ontology (GO) term ‘GOTERM_BP_ALL’ (41,42) using a human background.
In addition, significantly different genes at T30 were divided into two groups where either CBP or p300 binding was higher. From these groups, the 250 genes most significantly different were uploaded in DAVID 2008 for functional enrichment analysis. Individual GO-terms with a P-value <0.001 are shown for genes with higher CBP or p300 binding.
CORE_TF analysis for TF partners of p300/CBP
We took the same significant gene sets as from the functional analysis and retrieved the most substantially sequenced region (most number of tags in this particular region) for these genes. These regions were extended at both sides to a final length of 2 kb and sequences retrieved with Ensembl Perl API. As a background set, we retrieved 3000 random genes' TSSs from Ensembl Biomart that were located on chromosomes 1–22, X and Y and retrieved the sequences ±1 kb from these TSSs. The regions based on significantly different genes were entered into CORE_TF (43) as experimental sequences and the random TSS sequences were entered as background regions. We evaluated enrichment of TFBSs [defined as TRANSFAC (44) position weight matrices] in the experimental sequences using the most stringent match setting (44,45) to minimize false positives. P-values representing the significance of over-representation were calculated with a binomial test.
RESULTS
Initial sequencing analysis
Stringent regulation of gene expression is fundamental to control cellular processes such as proliferation and differentiation. The general coactivators p300 and CBP play an important role in the regulation of gene transcription by virtue of their acetyltransferase activity. We set out to determine and compare genes regulated by p300 and CBP. Chromatin was isolated from serum-starved (T0) and from stimulated (T30, done in duplo) human glioblastoma cells and ChIP-seq performed using CBP-and p300-specific antibodies.
Sequence files generated by the Illumina GAPipeline were submitted to the NCBI Sequence Read Archive (SRA; http://www.ncbi.nlm.nih.gov/Traces/sra: SRS009476, SRS009457, SRS009477, SRS009478, SRS009479 and SRS009480) (46). The reads passing quality control were mapped to the human reference genome and adjacent tags were joined into regions (Table 1). We also have made sequencing data available as UCSC hg18 viewable wiggle tracks (excluding positions with only one tag aligned, Supplementary materials 1–6).
Table 1.
Sequencing results
| CBP |
p300 |
|||||||
|---|---|---|---|---|---|---|---|---|
| t (min) | No. of reads | No. aligned | Percentage of aligned | No. of regionsa | No. of reads | No. aligned | Percentage of aligned | No. of regionsa |
| T0 | 5 498 759 | 4 018 590 | 73 | 713 141 | 6 327 413 | 5 086 340 | 80 | 841 029 |
| T301 | 6 389 605 | 4 849 826 | 76 | 889 781 | 6 446 269 | 5 156 450 | 80 | 802 627 |
| T302 | 6 047 530 | 5 001 204 | 83 | 851 988 | 6 065 594 | 5 124 836 | 84 | 684 222 |
The total number of reads, reads aligned, percentage aligned and number of regions created (aafter compressing regions within 100 bp into one and excluding regions composed of only a single tag) for each condition (T0: quiescent cells and T30: 30 min after growth factor stimulation) and for each transcription coactivator (CBP or p300). For T30-independent biological replicates were sequenced as indicated by 1 and 2.
Preferential binding in genes and promoters
Without applying a threshold of 1 tag/million tags, we found low overlap of identified regions in the replicated samples indicating that regions with low abundance represent noise (data not shown). With the threshold of 1 tag/million tags, we found a high consistency in the identified regions between all samples (47.96 and 47.43% overlap between CBP and p300 at T0 and T30, respectively; Table 2). Concordantly, the reproducibility between biological replicates was high (Pearson’s correlation: 0.77 and 0.87 for CBP and p300, respectively). A similarly high correlation was found across the different samples (Pearson’s correlation 0.81 on average between all time points and coactivators; Supplementary Table S2), indicating relatively minor differences in the distribution of p300 and CBP binding sites across the genome. In subsequent analyses, datasets of the T30 biological replicates were summed and treated as one sample, which provided us with high-quality results.
Table 2.
Region overlap
| CBP T0 | CBP T302 | CBP T301 | p300 T0 | p300 T302 | p300 T301 | |
|---|---|---|---|---|---|---|
| CBP T0 | 267 562 | 129 089 | 133 493 | 140 245 | 120 092 | 134 799 |
| CBP T302 | 315 020 | 143 160 | 152 520 | 136 405 | 148 669 | |
| CBP T301 | 322 556 | 151 519 | 136 685 | 150 180 | ||
| p300 T0 | 322 354 | 138 933 | 158 708 | |||
| p300 T302 | 267 804 | 139 592 | ||||
| p300 T301 | 304 880 |
The number of regions, after applying thresholds (>1 tag/million tags), overlapping between conditions (T0 and T30) and coactivators (CBP and p300). For T30-independent biological replicates were sequenced as indicated by 1 and 2.
To study the biological implications of our data, we annotated the regions obtained from sequencing with Ensembl and found that the sequenced regions covered 16 103 annotated genes in total. When looking at conditions and coactivators independently there were 16 045, 16 075, 15 684, and 15 996 genes identified as bound by CBP at T0 and T30, and by p300 at T0 and T30, respectively. We observed similar percentages of tag in genes and their 1 kb flanking regions in all samples (57.08, 57.10, 57.30 and 59.93% for CBP-T0, CBP-T30, p300-T0 and p300-T30, respectively). Therefore, both CBP and p300 appear to be needed to maintain basal levels of expression in quiescent cells as well as to activate or repress transcription after serum stimulation.
Previous studies have focused on the binding of p300 to enhancers (15,16). First, we evaluated the distance for all regions bound by CBP or p300 to the nearest TSS and transcript end (polyadenylation site). We found that genome-wide, 57% of all tags could be annotated to genes (±1 kb) and a clear preference for TSSs and transcript ends was observed (Figure 1A and B). There were no apparent differences between the profiles of CBP and p300 (data not shown). Also, different from what has been shown before for most TFs or histone modification maps, p300 and CBP show three distinct patterns of binding, including a distinguished peak (binding to a specific site like the TSS, e.g. ZNF688; Figure 1C), binding across the gene with no clear preference for a specific region (e.g. EGR1; Figure 1D), and so-called ‘U-shaped binding’ (binding across the gene with a bias toward the TSS and transcript end, e.g. DUSP1; Figure 1E).
Figure 1.

Histogram for the compilation of ChIP-seq regions showing the frequency of the distance from the localization of a sequenced region to the nearest transcription start site (blue) and transcript end (red) [full plot in (A), zoomed in (B)], which indicates a preference for binding to TSSs and transcript ends. Representative examples of the different types of binding are shown as custom tracks on the UCSC genome browser: binding to a specific site resulting in a ‘peak’ (C), binding across the gene (D), and ‘U-shaped binding’, with binding across the gene with preference for both TSS and transcript end (E). The y-axis indicates the number of tags aligned at each position in the genome. The black line in Figure 1C–E indicates a value of 5 tags in the custom tracks.
Differential binding by CBP and p300
With most data corresponding to a genic region, we focused our following analyses to genes, and on those regions within 1 kb upstream of TSS and 1 kb downstream of the transcript end, (16 103 genes across all four samples). Since we were especially interested in genes that were preferentially regulated by p300 or CBP during entry in the cell cycle, a Fisher’s exact test was performed to determine statistically significant differences in the total number of tags localized to a certain gene in different conditions (between time points or between coactivators) studied.
Despite high overlap in regions bound by CBP and p300 in quiescent and in stimulated cells (Table 2), there was also a considerable number of quantitative changes in CBP and p300 binding upon stimulation. Significant differences between p300 and CBP binding was found for 120 and for 1611 genes at a false discovery rate of 0.1% at T0 and T30, respectively (Figure 2A). At a false discovery rate of 1% this was 256 and 2502 at T0 and T30, respectively (Supplementary Table S3). From the genes differentially bound by p300 and CBP in quiescent cells (T0), only 25 did not have significantly different binding upon growth factor stimulation (Figure 2A). These results indicate very high overlap in genes bound by p300 as well as by CBP in the quiescent state and a divergence of the roles of CBP and p300 mainly during periods of activated transcription. Analysis of the 250 genes that were most significantly different in our data, showed that for the majority p300 binding was higher than CBP binding (191 and 227 of 250 genes, for T0 and T30, respectively).
Figure 2.

Genes differentially bound by CBP and p300 (A) and between time points (B). P-values (Fisher’s exact test) for the indicated comparisons were sorted in rising order and plotted (Upper panels). Under the null hypothesis of no significant differences, this would give a straight line on the diagonal. However, as becomes evident by the curve shape there is a bias towards low P-values. The number of genes with significant differences between conditions are indicated in the graphs (false discovery rate of 0.1%). Venn diagrams (Lower panels) demonstrate the number of significantly different bound genes, as shown in the plots above, that overlap between time points (A) or coactivators (B).
When comparing between time points, we found 765 genes differentially bound by CBP and 2620 genes differentially bound by p300 (Figure 2B). Of the 250 genes, which were most significantly different between time points, the majority (209 and 155 for CBP and p300, respectively) demonstrated higher binding for both, p300 and CBP, at T30 when compared to T0. In addition, the majority of genes with changed binding of CBP after stimulation also demonstrated difference in binding by p300 (676 out of 765 and 2620 genes, respectively; Figure 2B). The apparently higher number of genes with significant changes in p300 binding is likely due to the higher efficiency of the p300 antibody causing better signal-to-noise ratios and higher sensitivity in the detection of quantitative changes in binding profiles (see below). The high level of overlap between coactivators can explain the restricted number of differences found thus far in functions of p300 and CBP.
We present a full list of genes bound by CBP and p300 at T0 and T30 in Supplementary material 7. Table 3 lists the 10 genes for which levels of binding differ most significantly between the four samples. Among the genes with strongest CBP and p300 binding and most significantly different between T30 and T0 are many immediate-early genes that are bound by both p300 and CBP (Table 3; e.g. ATF3, FOSB and DUSP1).
Table 3.
Gene Top 10
| CBP T30 Versus T0 | p300 T30 Versus T0 | ||||
|---|---|---|---|---|---|
| Gene | P-value | Ratio | Gene | P-value | Ratio |
| CATSPER3 | 0 | 2.58 | THBS1 | 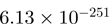 |
6.07 |
| ATF3 | 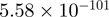 |
4.51 | ATF3 | 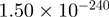 |
5.83 |
| TRIP13 | 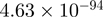 |
11.07 | FOSB | 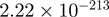 |
14.03 |
| CYR61 | 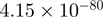 |
6.43 | CYR61 | 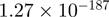 |
6.33 |
| FOSB | 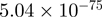 |
10.03 | EGR1 | 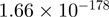 |
14.11 |
| SMAD3 | 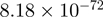 |
2.09 | TPM1 | 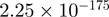 |
4.81 |
| TMEM49 | 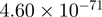 |
2.32 | DUSP1 | 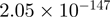 |
6.81 |
| MYH9 | 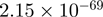 |
2.39 | MYH9 | 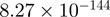 |
2.84 |
| CRISPLD2 | 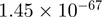 |
2.89 | NR4A1 | 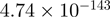 |
13.16 |
| THBS1 | 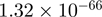 |
4.15 | CRISPLD2 | 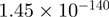 |
4.04 |
| T0 p300 Versus CBP | T30 p300 Versus CBP | ||||
|---|---|---|---|---|---|
| Gene | P-value | Ratio | Gene | P-value | Ratio |
| CXXC1 | 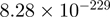 |
22.71 | CXXC1 | 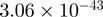 |
20.32 |
| AKT1S1 | 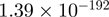 |
6.03 | MKKS | 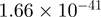 |
3.87 |
| FBXL19 | 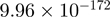 |
5.56 | CATSPER3 | 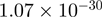 |
1.44 |
| MKKS | 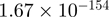 |
4.73 | AKT1S1 | 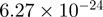 |
4.46 |
| C3orf19 | 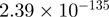 |
7.93 | FAM40A | 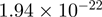 |
4.21 |
| BSCL2 | 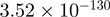 |
8.25 | FBXL19 | 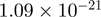 |
3.27 |
| THBS1 | 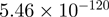 |
2.1 | ZNF350 | 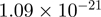 |
9.14 |
| MADCAM1 | 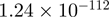 |
7.85 | METTL3 | 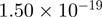 |
13.47 |
| ZNF175 | 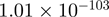 |
17.86 | MADCAM1 | 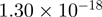 |
7.2 |
| C1orf174 | 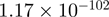 |
9.84 | C1orf174 | 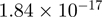 |
7.25 |
Top 10 genes that are most significantly different between time points and coactivators according to the P-values of the Fisher’s exact test. The ratio shows the quantitative difference in binding as expressed by the number of tags between the two samples that are compared: from T30 and T0 (upper half of the table) and from p300 and CBP (lower half of the table).
Validation
To validate our results and to refine the temporal resolution of the experiment, genes were selected to further characterize with ChIP and quantitative PCR in a time-course from 0 to 360 min following stimulation with serum and TPA. The genes included genes bound by both CBP and p300 and genes unique to one of the coactivators, and spanned a wide range of significance values (Figure 3E). In general, the recovery obtained (as a percentage of the input) for CBP is lower than for p300 (Figure 3A–D), consistent with the generally lower number of tags for CBP in each region of the ChIP-seq experiment (significant quantitative correlation between results of the qPCR and ChIP-seq experiment for the genes presented here are shown in Supplementary Figure S4). The qPCR results also confirm the differential binding across time points established with ChIP-seq analysis for all genes analyzed (Figure 3A–D and Supplementary Figure S5), and demonstrate that for most genes the temporal binding pattern is comparable between CBP (black bars) and p300 (white bars). This is true for the increased binding to the promoter of CTGF, as well as for the decreased binding to the promoter of ZNF608 in stimulated cells compared to unstimulated cells (Figure 3A and B). Binding to the promoter of CDK5 differs for p300 and CBP (Figure 3C). Binding of p300 is increased in time with a maximum at 60 min post-stimulation, while there is hardly any change in the binding of CBP. These results correlate with the statistical analysis, that demonstrated significant changes between P300 and CBP at T30, and a significant increase in P300 but not CBP binding between T30 and T0 (Figure 3E). The binding of P300 and CBP to the SERPINE gene increased significantly over time (P-value of 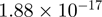 for CBP T30 versus T0 and
for CBP T30 versus T0 and 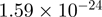 for p300 T30 versus T0). Inspection of the wiggle track (Figure 3D) revealed that p300 and CBP bound mainly to the 3′-UTR and to a lesser extent to the region around the TSS of the SERPINE1 gene. Also, the small increase observed around the TSS could be confirmed by qPCR. The wiggle file for SERPINE1 also shows a stronger binding >2 kb upstream of the TSS (Figure 3D). The interaction to this putative enhancer region and the change upon stimulation was also confirmed by qPCR of ChIP samples (Supplementary Figure S5J).
for p300 T30 versus T0). Inspection of the wiggle track (Figure 3D) revealed that p300 and CBP bound mainly to the 3′-UTR and to a lesser extent to the region around the TSS of the SERPINE1 gene. Also, the small increase observed around the TSS could be confirmed by qPCR. The wiggle file for SERPINE1 also shows a stronger binding >2 kb upstream of the TSS (Figure 3D). The interaction to this putative enhancer region and the change upon stimulation was also confirmed by qPCR of ChIP samples (Supplementary Figure S5J).
Figure 3.


ChIP-analysis for time-course experiment (0, 2, 5, 15, 30, 60, 90, 120 and 360 min after stimulation of serum-starved T98G cells with serum and TPA). Shown are graphs for qPCR results [x-axis: time in minutes; left y-axis: ChIP recovery in percentages of the input; right y-axis: fold induction for the RT-qPCR with reference to the untreated samples (t = 0 min)] and screen-shots from custom tracks of the UCSC genome browser for the ChIP-seq data (T0 and T30 only) for CTGF (A), ZNF608 (B), CDK5 (C) and SERPINE1 (D). White bars: p300 ChIP; black bars: CBP ChIP; RT-qPCR data are indicated as dots, interconnected; arrows in the screen-shots indicate the position of the PCR-amplicon. Also indicated for these genes is the adj. P-value and the ratio difference between time-points (T30 versus T0) and coactivators (p300 versus CBP) of the total number of reads along the whole gene, plus 1 kb up- or downstream from all ChIP-seq data (E).
To evaluate whether changes in p300 and CBP binding also affected gene expression, we performed quantitative RT-PCR for the genes CTGF, ZNF608, CDK5 and SERPINE1 (Figure 3A–D: the line in the graphs shows fold induction in the time course). For the three genes with increased binding, two (CTGF and SERPINE1) show increased expression. The gene ZNF608 shows a decrease in expression over time, consistent with decreased binding of p300/CBP. CDK5 did not show any differences in expression. This is consistent with the uniform levels of CBP binding over time, but not with the increased binding of p300. Most likely, for CDK5 and possibly also for other genes binding of p300/CBP is not sufficient to induce the expression but other factors that play a critical role are also required. Obviously, gene expression is a complex process and highly variable between genes, so only detailed studies can unravel the role of specific factors.
Biological processes coordinated by p300 and CBP
To get an impression of the biological implications of p300 and CBP binding, we clustered genes regulated by CBP and p300 into functional pathways. We used DAVID 2008 (39,40) to classify the 250 genes most significantly differing between time points (for both CBP and p300). The analysis (P-value < 0.001) shows that CBP as well as p300 are mainly involved in transcription regulation of genes controlling developmental processes, metabolic processes (such as NR4A, CRISPLD2, CRIM1, CYCLIN-L1 and PER1) and of genes coding for proteins that control gene expression (such as ATF3, FOSB, SP3 and HES1; see Supplementary material 8). Next, using DAVID 2008 we wanted to specify in more detail whether certain groups of genes were preferentially bound by CBP or by p300. Remarkably, in the cluster of genes regulating transcription, those with significantly higher CBP than p300 binding are involved in negative regulation of transcription (Table 4, and Supplementary Table S4C and D). Another interesting observation for genes preferentially bound by CBP is the presence of clusters related to signal transcription/cell communication. In the list obtained for higher levels of p300, mainly clusters relate to transcription and metabolic processes are found.
Table 4.
Functional classification for genes bound by CBP or p300
| T30 p300 higher than CBP | |||||
|---|---|---|---|---|---|
| ID (GO:#) | Term | Count | % | P-value | Enrichment |
| 0010467 | Gene expression | 81 | 38.76 | 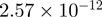 |
2.06 |
| 0044237 | Cellular metabolic process | 127 | 60.77 | 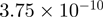 |
1.44 |
| 0008152 | Metabolic process | 135 | 64.59 | 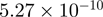 |
1.38 |
| 0044238 | Primary metabolic process | 126 | 60.29 | 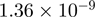 |
1.42 |
| 0043170 | Macromolecule metabolic process | 111 | 53.11 |  |
1.44 |
| 0006350 | Transcription | 56 | 26.79 | 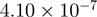 |
1.95 |
| 0006139 | Nucleobase, nucleoside, nucleotide and nucleic acid metabolic process | 72 | 34.45 |  |
1.66 |
| 0045449 | regulation of transcription | 53 | 25.36 | 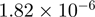 |
1.91 |
| 0016070 | RNA metabolic process | 58 | 27.75 |  |
1.8 |
| 0019219 | Regulation of nucleobase, nucleoside, nucleotide and nucleic acid metabolic process | 53 | 25.36 |  |
1.87 |
| 0010468 | Regulation of gene expression | 54 | 25.84 |  |
1.83 |
| 0031323 | Regulation of cellular metabolic process | 54 | 25.84 |  |
1.76 |
| 0043283 | Biopolymer metabolic process | 84 | 40.19 |  |
1.47 |
| 0019222 | Regulation of metabolic process | 55 | 26.32 |  |
1.73 |
| 0006351 | Transcription, DNA-dependent | 48 | 22.97 |  |
1.81 |
| 0032774 | RNA biosynthetic process | 48 | 22.97 |  |
1.81 |
| 0006355 | regulation of transcription, DNA-dependent | 46 | 22.01 |  |
1.77 |
| 0006979 | Response to oxidative stress | 7 | 3.35 |  |
6.83 |
| 0050794 | Regulation of cellular process | 67 | 32.06 |  |
1.42 |
| T30 CBP higher than p300 | |||||
|---|---|---|---|---|---|
| ID (GO:#) | Term | Count | % | P-value | Enrichment |
| 0051056 | Regulation of small GTPase mediated signal transductio | 18 | 7.06 |  |
5.97 |
| 0046578 | Regulation of Ras protein signal transduction | 13 | 5.10 |  |
5.36 |
| 0007242 | Intracellular signaling cascade | 38 | 14.90 |  |
2.06 |
| 0009966 | Regulation of signal transduction | 21 | 8.24 |  |
2.98 |
| 0007154 | Cell communication | 77 | 30.20 |  |
1.52 |
| 0007165 | Signal transduction | 71 | 27.84 |  |
1.54 |
| 0007265 | Ras protein signal transduction | 13 | 5.10 |  |
4.03 |
| 0007264 | Sall GTPase mediated signal transduction | 18 | 7.06 | 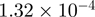 |
2.94 |
| 0007399 | Nervous system development | 23 | 9.02 |  |
2.4 |
| 0016481 | Negative regulation of transcription | 13 | 5.10 |  |
3.52 |
| 0045934 | Negative regulation of nucleobase, nucleoside, nucleotide and nucleic acid metabolic process | 13 | 5.10 |  |
3.22 |
Significantly enriched GO categories for genes that show higher binding of CBP or p300 at 30 min after stimulation with TPA and serum (ID: GO-category-number, term: description of the GO category; count: number of significant genes in this GO category; %: percentage of signifiacnt genes in this GO category; P-value: statistical significance of the GO category (P-value from hypergeometric test for over-representation); enrichment: fold enrichment of significant genes compared to the background.
Analysis of ChIP-seq regions for consensus TFBSs
p300 and CBP do not bind DNA directly, but regulate by binding to many different protein partners. Therefore, to identify (DNA-binding) partners of p300/CBP, we looked for enrichment of TFBSs in and around the regions bound by CBP and/or p300 in the 250 genes that differ most significantly between time points (the same genes that were used for DAVID analysis). We found a significant over-representation of AP-1, CREB, NFKB and SRF binding sites in the gene regions bound by both CBP and p300 (Table 5 and Supplementary material 9), which are known to be regulated by CBP and/or p300 (47). As mentioned before, there is more binding of p300 and CBP to the chromatin at T30 after stimulation. Therefore, enrichment of the TFBSs in our sequences likely reflects increased binding of these factors upon growth factor stimulation.
Table 5.
Enrichment for TFBSs in CBP/p300 bound sequences
| TFBS | T30 Versus T0 |
TFBS | T30 |
||
|---|---|---|---|---|---|
| CBP | p300 | CBP > p300 | p300 > CBP | ||
| AP-1 | 0 | 0 | AP-1 | 0 |  |
| CREB |  |
 |
AP-2 |  |
 |
| NFKB | 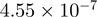 |
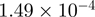 |
CREB |  |
 |
| SRF |  |
 |
E2F |  |
0 |
| SP1 |  |
 |
|||
| SRF |  |
 |
|||
| YY1 |  |
0 | |||
TFBSs with the most significant P-values for enrichment in regions bound by CBP and p300 at 0 and 30 min after stimulation with serum and TPA.
We also compared genes significantly different between coactivators at T30 using the same lists of 250 genes as used for the functional classification. CREB and YY1 are significantly enriched in both gene sets (Table 5; all results are presented in Supplementary material 9). However, CBP binding was found to correlate more with AP-1 and SRF binding partners than p300, whereas p300 binding was more correlated to AP-2, E2F and SP1-binding. These results indicate that CBP and p300 share some, but not all, regulatory partners.
DISCUSSION
Transcription coactivators CBP and p300 share high levels of homology and, in many cases, the same regulatory regions are targeted for transcription regulation. This is in contrast with the fact that both proteins are indispensable during embryogenesis. To investigate which genes are regulated, and whether there is a difference in those regulated by p300 and by CBP upon growth factor stimulation a genome-wide screen was performed in T98G cells. Although there is a high concordance between binding targets of p300 and CBP, and both seem to regulate the same biological pathways, we have identified significant differences in the levels and targets of binding. These differences include the diversity in the regulation of genes involved in transcription, and in cell death and cell adhesion. In addition, regulatory regions of these genes showed significant differences in binding sites of other TFs and TF families such as AP-1, AP-2, SP1, E2F and SRF.
It is well established that p300/CBP associate to both enhancers and TSSs. Previous studies have focused on the enhancer-binding of p300 (15–17). Although we also found examples of enhancer-binding, over 57% of all tags are within genes or proximal promoters (±1 kb), and genome-wide we find that binding is primarily located around TSSs and to some extend also to transcript ends (Figure 1A and B). Therefore, we chose to focus our analysis of CBP/p300 in relation to genic regions. In all, we found 16 103 genes bound by CBP or p300 at T0 or T30, with over 97.4% of genes bound by both coactivators at both time points.
When analyzing the binding of CBP/p300 to genes, we did not only observe distinct regions of binding. There was a high variety in binding patterns for both coactivators. This includes binding to a clear and distinct region (e.g. to the TSS; referred to as ‘peak’), binding across the gene, or a combination of more prominent binding around the TSS and the transcript end, as well as binding across the gene (in the text referred to as ‘U-shaped binding’; Figure 1C–E).
At present, the mechanisms that determine the diverse binding patterns remain to be established. Possibly, it is dependent on the way p300/CBP regulate transcription of a particular gene. Both, p300 and CBP can bind to specific TFs, and this might result in a distinguished peak around the TSS and transcript end. In addition, p300 and CBP regulate chromatin structure via the acetylation of histones, thereby making the chromatin more prone to be targeted for transcription. This might account for binding (to the histones) across the gene. Binding across a gene was previously described to occur also by protein kinases (48). Chow et al. (48) propose that in this way the kinases may contribute to transcription initiation and elongation, or processes such as 5′ capping, and splicing. Binding to both the TSS and transcript end has previously been observed for RNA polymerase II (49). Interaction between CBP/p300 and RNA polymerase II (50) may explain the presence of similar ChIP-seq patterns for these acetyltransferases. Enrichment at the TSS might correlate to the longer time needed for the transcription initiation compared to transcript elongation. The peak at the transcript end might correlate to widespread transcription of antisense transcripts (51), a phenomena that is particularly prominent in the 3′-end of genes (52).
Our ChIP-seq data are from arrested cells and from cells 30 min after stimulation. Therefore, over-representation of genes required early in the cell cycle was expected at T30. The number of reads correlates roughly to the binding affinity of proteins for that region and immediate-early genes are among the genes with the highest number of tags. Analysis of a number of these genes with quantitative RT-PCR (Figure 3A, Supplementary Figure S5 and data not shown), also showed increase in gene expression for immediate-early genes. Our data suggest that at T30 CBP and p300 are more intimately involved in the regulation of transcriptional activation of immediate-early genes compared to other groups of genes. Consistently, Tullai et al. (32) previously published microarray data on gene expression of serum-starved T98G cells upon growth stimulation. We found that from 49 immediate-early genes that were identified, 36 demonstrated significantly increased binding by CBP and p300 at T30 compared to T0 in our analysis (3 out of 49 could not be identified in Ensembl).
With the time-course experiment, most genes that were analyzed show maximal binding between 30 and 60 min after stimulation. The time-course experiment confirms high accuracy of our data since all genes tested, although different levels of significance (from 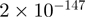 to
to 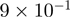 ) and variable ratios of difference (from 1 to 9) were chosen, confirm binding of the coactivators and changes in time.
) and variable ratios of difference (from 1 to 9) were chosen, confirm binding of the coactivators and changes in time.
Binding of p300 and CBP to the chromatin occurs through the interaction with TFs. To obtain more insight in transcription regulatory complexes bound by p300 and/or CBP, we set out to identify possible partners of CBP and p300 for the genes identified in our experiment. Therefore, we analyzed for the enrichment of TFBSs. When looking at genes with significant binding at T30 for each coactivator, we found some examples of TFBSs that were found to be specific only for CBP or for p300. For example, AP-1 and SRF binding sites were significantly enriched in CBP bound regions, while AP-2, E2F and SP1 binding sites were more abundant in p300 bound regions. This may represent TFs that are regulated during the cell cycle, in most cases, solely by CBP or p300 and contribute to their unique functions.
We observed overlap in enrichment of TFBSs for proteins such as YY1 and CREB. Interestingly, YY1 is known to contribute to cell-cycle regulation and can serve both, as a transcriptional repressor and an activator (53). Also, YY1 is known to interact with p300/CBP, as well as with other TFs identified in this study (AP-1, AP-2, NFKB, E2, SP1 and CREB) (53,54,55). Our functional classification suggested that CBP is more associated with transcriptional repression, whereas p300 is more associated with transcriptional activation. It could be speculated that YY1 is a putative partner involved in this functional difference between p300 and CBP, while the p300-YY1 complex might activate transcription in vivo, the CBP-YY1 complex might account for transcriptional repression.
In the future, it would be valuable to perform ChIP-seq in the same cell line and conditions with antibodies for the coactivator specific TFs in this study (AP-1, AP-2, SP1, E2F and SRF, and YY1). This will confirm whether genome-wide CBP/p300 and their specific regulatory partners cooperate, and it will help to further elucidate their role in cell-cycle control. In addition, ChIP-seq with antibodies specific to open chromatin states will be helpful to unravel the mechanisms leading to the diverse binding patterns.
CONCLUSION
Transcription coactivators CBP and p300 share high levels of homology and, in many cases, the same regulatory regions are targeted for transcription regulation. This is in contrast with the fact that both proteins are indispensable during embryogenesis. To investigate which genes are regulated, and whether there is a difference in those regulated by p300 and by CBP upon growth factor stimulation a genome-wide screen was performed in T98G cells. Although there is a high concordance between binding targets of p300 and CBP and both seem to regulate the same biological pathways, we have identified significant differences in the levels and targets of binding. In addition, regulatory regions of these genes also showed significant differences in binding sites of other TFs such as AP-1, AP-2, SP1, E2F and SRF.
Besides the differences in targets of p300 and CBP, we identified various binding-patterns that potentially correlate with different types of transcription regulation by p300 and CBP. Most interestingly, we observed a so-called ‘U-shaped binding’ with high levels of p300/CBP at both, TSS and transcript end. Possibly, the acetyltransferases contribute to other processes such as transcription elongation and reverse transcription. Taken together, our data contribute to the improvement of our knowledge of processes that regulate gene expression by the transcription coactivators p300 and CBP, and confirm that regulation by these coactivators is not identical.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Centre for Medical Systems Biology within the framework of the Netherlands Genomics Initiative (NGI)/Netherlands Organisation for Scientific Research (NWO); Center for Biomedical Genetics (in the Netherlands).
Conflict of interest statement. None declared.
ACKNOWLEDGEMENTS
We wish to thank Michel P. Villerius for his computational support and Dorien JM Peters for her review and comments about the manuscript.
REFERENCES
- 1.Farnham PJ. Insights from genomic profiling of transcription factors. Nat. Rev. Genet. 2009;10:605–616. doi: 10.1038/nrg2636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Moore MJ, Proudfoot NJ. Pre-mRNA processing reaches back to transcription and ahead to translation. Cell. 2009;136:688–700. doi: 10.1016/j.cell.2009.02.001. [DOI] [PubMed] [Google Scholar]
- 3.Sonenberg N, Hinnebusch AG. Regulation of translation initiation in eukaryotes: mechanisms and biological targets. Cell. 2009;136:731–745. doi: 10.1016/j.cell.2009.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lu TY, Kao CF, Lin CT, Huang DY, Chiu CY, Huang YS, Wu HC. DNA methylation and histone modification regulate silencing of OPG during tumor progression. J. Cell. Biochem. 2009;108:315–325. doi: 10.1002/jcb.22256. [DOI] [PubMed] [Google Scholar]
- 5.Selaru FM, David S, Meltzer SJ, Hamilton JP. Epigenetic events in gastrointestinal cancer. Am. J. Gastroenterol. 2009;104:1910–1912. doi: 10.1038/ajg.2008.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Szyf M. Epigenetic therapeutics in autoimmune disease. Clin. Rev. Allergy Immunol. 2009;37 doi: 10.1007/s12016-009-8172-8. [31 July 2009, Epub ahead of Print] [DOI] [PubMed] [Google Scholar]
- 7.Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, Wei G, Chepelev I, Zhao K. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129:823–837. doi: 10.1016/j.cell.2007.05.009. [DOI] [PubMed] [Google Scholar]
- 8.Wang Z, Zang C, Rosenfeld JA, Schones DE, Barski A, Cuddapah S, Cui K, Roh TY, Peng W, et al. Combinatorial patterns of histone acetylations and methylations in the human genome. Nat. Genet. 2008;40:897–903. doi: 10.1038/ng.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mikkelsen TS, Ku M, Jaffe DB, Issac B, Lieberman E, Giannoukos G, Alvarez P, Brockman W, Kim TK, Koche RP, et al. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature. 2007;448:553–560. doi: 10.1038/nature06008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Welboren WJ, van Driel MA, Janssen-Megens EM, van Heeringen SJ, Sweep FC, Span PN, Stunnenberg HG. ChIP-Seq of ERalpha and RNA polymerase II defines genes differentially responding to ligands. EMBO J. 2009;28:1418–1428. doi: 10.1038/emboj.2009.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Smeenk L, van Heeringen SJ, Koeppel M, van Driel MA, Bartels SJJ, Akkers RC, Denissov S, Stunnenberg HG, Lohrum M. Characterization of genome-wide p53-binding sites upon stress response. Nucleic Acids Res. 2008;36:3639–3654. doi: 10.1093/nar/gkn232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wederell ED, Bilenky M, Cullum R, Thiessen N, Dagpinar M, Delaney A, Varhol R, Zhao YJ, Zeng T, Bernier B, et al. Global analysis of in vivo Foxa2-binding sites in mouse adult liver using massively parallel sequencing. Nucleic Acids Res. 2008;36:4549–4564. doi: 10.1093/nar/gkn382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Johnson DS, Mortazavi A, Myers RM, Wold B. Genome-wide mapping of in vivo protein-DNA interactions. Science. 2007;316:1497–1502. doi: 10.1126/science.1141319. [DOI] [PubMed] [Google Scholar]
- 14.Robertson G, Hirst M, Bainbridge, Bilenky M, Zhao Y, Zeng T, Euskirchen G, Bernier B, Varhol R, Delaney A, et al. Genome-wide profiles of STAT1 DNA association using chromatin immunoprecipitation and massively parallel sequencing. Nat. Methods. 2007;4:651–657. doi: 10.1038/nmeth1068. [DOI] [PubMed] [Google Scholar]
- 15.Wang Z, Zang C, Cui K, Schones DE, Barski A, Peng W, Zhao K. Genome-wide mapping of HATs and HDACs reveals distinct functions in active and inactive genes. Cell. 2009;138:1019–1031. doi: 10.1016/j.cell.2009.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Visel A, Blow MJ, Li Z, Zhang T, Akiyama JA, Holt A, Plajzer Frick I, Shoukry M, Wright C, Chen F, et al. ChIP-seq accurately predicts tissue-specific activity of enhancers. Nature. 2009;457:854–858. doi: 10.1038/nature07730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Heintzman ND, Stuart RK, Hon G, Fu Y, Ching CW, Hawkins RD, Barrera LO, Calcar SV, Qu C, Ching KA, et al. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat. Genet. 2007;39:311–318. doi: 10.1038/ng1966. [DOI] [PubMed] [Google Scholar]
- 18.Kalkhoven E. CBP and p300: HATs for different occasions. Biochem. Pharmacol. 2004;68:1145–1155. doi: 10.1016/j.bcp.2004.03.045. [DOI] [PubMed] [Google Scholar]
- 19.Goodman RH, Smolik S. CBP/p300 in cell growth, transformation, and development. Genes. Dev. 2000;14:1553–1577. [PubMed] [Google Scholar]
- 20.Roelfsema JH, Peters DJM. Rubinstein-Taybi syndrome: clinical and molecular overview. Expert Rev. Mol. Med. 2007;9:1–16. doi: 10.1017/S1462399407000415. [DOI] [PubMed] [Google Scholar]
- 21.Iyer NG, Ozdag H, Caldas C. p300/CBP and cancer. Oncogene. 2004;23:4225–4231. doi: 10.1038/sj.onc.1207118. [DOI] [PubMed] [Google Scholar]
- 22.Chrivia JC, Kwok RP, Lamb N, Hagiwara M, Montminy MR, Goodman RH. Phosphorylated CREB binds specifically to the nuclear protein CBP. Nature. 1993;365:855–859. doi: 10.1038/365855a0. [DOI] [PubMed] [Google Scholar]
- 23.Duyndam MC, van Dam H, Smits PH, Verlaan M, van der Eb AJ, Zantema A. The N-terminal transactivation domain of ATF2 is a target for the co-operative activation of the c-jun promoter by p300 and 12S E1A. Oncogene. 1999;18:2311–2321. doi: 10.1038/sj.onc.1202584. [DOI] [PubMed] [Google Scholar]
- 24.Grossman SR. p300/CBP/p53 interaction and regulation of the p53 response. Eur. J. Biochem. 2001;268:2773–2778. doi: 10.1046/j.1432-1327.2001.02226.x. [DOI] [PubMed] [Google Scholar]
- 25.Puri PL, Avantaggiati ML, Balsano C, Sang N, Graessmann A, Giordano A, Levrero M. p300 is required for MyoD-dependent cell cycle arrest and muscle-specific gene transcription. EMBO J. 1997;16:369–383. doi: 10.1093/emboj/16.2.369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yao TP, Oh SP, Fuchs M, Zhou ND, Ch'ng LE, Newsome D, Bronson RT, Li E, Livingston DM, Eckner R. Gene dosage-dependent embryonic development and proliferation defects in mice lacking the transcriptional integrator p300. Cell. 1998;93:361–372. doi: 10.1016/s0092-8674(00)81165-4. [DOI] [PubMed] [Google Scholar]
- 27.Tanaka Y, Naruse I, Hongo T, Xu M, Nakahata T, Maekawa T, Ishii S. Extensive brain hemorrhage and embryonic lethality in a mouse null mutant of CREB-binding protein. Mech. Dev. 2000;95:133–145. doi: 10.1016/s0925-4773(00)00360-9. [DOI] [PubMed] [Google Scholar]
- 28.Partanen A, Motoyama J, Hui CC. Developmentally regulated expression of the transcriptional cofactors/histone acetyltransferases CBP and p300 during mouse embryogenesis. Int. J. Dev. Biol. 1999;43:487–494. [PubMed] [Google Scholar]
- 29.Kung AL, Rebel VI, Bronson RT, Ch'ng LE, Sieff CA, Livingston DM, Yao TP. Gene dose-dependent control of hematopoiesis and hematologic tumor suppression by CBP. Genes Dev. 2000;14:272–277. [PMC free article] [PubMed] [Google Scholar]
- 30.Shikama N, Lutz W, Kretzschmar R, Sauter N, Roth J-F, Marino S, Wittwer J, Scheidweiler A, Eckner R. Essential function of p300 acetyltransferase activity in heart, lung and small intestine formation. EMBO J. 2003;22:5175–5185. doi: 10.1093/emboj/cdg502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bordoli L, Husser S, Luthi U, Netsch M, Osmani H, Eckner R. Functional analysis of the p300 acetyltransferase domain: the PHD finger of p300 but not of CBP is dispensable for enzymatic activity. Nucleic Acids Res. 2001;29:4462–4471. doi: 10.1093/nar/29.21.4462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tullai JW, Schaffer ME, Mullenbrock S, Sholder G, Kasif S, Cooper GM. Immediate-early and delayed primary response genes are distinct in function and genomic architecture. J. Biol. Chem. 2007;282:23981–23995. doi: 10.1074/jbc.M702044200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Denissov S, van Driel M, Voit R, Hekkelman M, Hulsen T, Hernandez N, Grummt I, Wehrens R, Stunnenberg H. Identification of novel functional TBP-binding sites and general factor repertoires. EMBO J. 2007;26:944–954. doi: 10.1038/sj.emboj.7601550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Smith AD, Xuan Z, Zhang MQ. Using quality scores and longer reads improves accuracy of Solexa read mapping. BMC Bioinformatics. 2008;9:128. doi: 10.1186/1471-2105-9-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, Haussler D. The human genome browser at UCSC. Genome Res. 2002;12:996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hubbard TJP, Aken BL, Ayling S, Ballester B, Beal K, Bragin E, Brent S, Chen Y, Clapham P, Clarke L, et al. Ensembl 2009. Nucleic Acids Res. 2009;37:D690–D697. doi: 10.1093/nar/gkn828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kasprzyk A, Keefe D, Smedley D, London D, Spooner W, Melsopp C, Hammond M, Rocca-Serra P, Cox T, Birney E. EnsMart: a generic system for fast and flexible access to biological data. Genome Res. 2004;14:160–169. doi: 10.1101/gr.1645104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Freeman MF, Tukey JW. Transformations related to the angular and the square root. Ann. Math. Statist. 1950;21:607–611. [Google Scholar]
- 39.Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 40.Dennis G, Jr, Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, Lempicki RA. DAVID: database for annotation, visualization, and integrated discovery. Genome Biol. 2003;4:P3. [PubMed] [Google Scholar]
- 41.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Camon E, Barrell D, Lee V, Dimmer E, Apweiler R. The Gene Ontology Annotation (GOA) Database-an integrated resource of GO annotations to the UniProt Knowledgebase. In Silico Biol. 2004;4:5–6. [PubMed] [Google Scholar]
- 43.Hestand MS, van Galen M, Villerius MP, van Ommen G-JB, den Dunnen JT, Hoen PAC't. CORE TF: a user-friendly interface to identify evolutionary conserved transcription factor binding sites in sets of co-regulated genes. BMC Bioinformatics. 2008;9:495. doi: 10.1186/1471-2105-9-495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Matys V, Fricke E, Geffers R, Gossling E, Haubrock M, Hehl R, Hornischer K, Karas D, Kel AE, Kel-Margoulis OV, et al. TRANSFAC: transcriptional regulation, from patterns to profiles. Nucleic Acids Res. 2003;31:374–378. doi: 10.1093/nar/gkg108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kel AE, Gossling E, Reuter I, Cheremushkin E, Kel-Margoulis OV, Wingender E. MATCH: a tool for searching transcription factor binding sites in DNA sequences. Nucleic Acids Res. 2003;31:3576–3579. doi: 10.1093/nar/gkg585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wheeler DL, Barrett T, Benson DA, Bryant SH, Canese K, Chetvernin V, Church DM, Dicuccio M, Edgar R, Federhen S, et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2008;36:D13–D21. doi: 10.1093/nar/gkm1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Deng L, de la Fuente C, Fu P, Wang L, Donnelly R, Wade JD, Lambert P, Li H, Lee CG, Kashanchi F. Acetylation of HIV-1 Tat by CBP/P300 increases transcription of integrated HIV-1 genome and enhances binding to core histones. Virology. 2000;277:278–295. doi: 10.1006/viro.2000.0593. [DOI] [PubMed] [Google Scholar]
- 48.Chow CW, Davis RJ. Protein kinases: chromatin-associated enzymes? Cell. 2006;127:887–890. doi: 10.1016/j.cell.2006.11.015. [DOI] [PubMed] [Google Scholar]
- 49.Gilchrist DA, Fargo DC, Adelman K. Using ChIP-chip and ChIP-seq to study the regulation of gene expression: genome-wide localization studies reveal widespread regulation of transcription elongation. Methods. 2009;48:398–408. doi: 10.1016/j.ymeth.2009.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cho H, Orphanides G, Sun X, Yang XJ, Ogryzko V, Lees E, Nakatani Y, Reinberg DA. A human RNA polymerase II complex containing factors that modify chromatin structure. Mol. Cell. Biol. 1998;18:5355–5363. doi: 10.1128/mcb.18.9.5355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Katayama S, Tomaru Y, Kasukawa T, Waki K, Nakanishi M, Nakamura M, Nishida H, Yap CC, Suzuki M, Kawai J, et al. Antisense transcription in the mammalian transcriptome. Science. 2005;309:1564–1566. doi: 10.1126/science.1112009. [DOI] [PubMed] [Google Scholar]
- 52.Hoen PAC't, Ariyurek Y, Thygesen HH, Vreugdenhil E, Vossen RHAM, de Menezes RX, Boer JM, van Ommen G-JB, den Dunnen JT. Deep sequencing-based expression analysis shows major advances in robustness, resolution and inter-lab portability over five microarray platforms. Nucleic Acids Res. 2008;36:e141. doi: 10.1093/nar/gkn705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gordon S, Akopyan G, Garban H, Bonavida B. Transcription factor YY1: structure, function, and therapeutic implications in cancer biology. Oncogene. 2006;25:1125–1142. doi: 10.1038/sj.onc.1209080. [DOI] [PubMed] [Google Scholar]
- 54.Seto E, Lewis B, Shenk T. Interaction between transcription factors Sp1 and YY1. Nature. 1993;365:462–464. doi: 10.1038/365462a0. [DOI] [PubMed] [Google Scholar]
- 55.Zhou Q, Gedrich RW, Engel DA. Transcriptional repression of the c-fos gene by YY1 is mediated by a direct interaction with ATF/CREB. J. Virol. 1995;69:4323–4330. doi: 10.1128/jvi.69.7.4323-4330.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]