


Abstract
Long non-coding RNA (lncRNA), highly up-regulated in liver cancer (HULC) plays an important role in tumorigenesis. Depletion of HULC resulted in a significant deregulation of several genes involved in liver cancer. Although up-regulation of HULC expression in hepatocellular carcinoma has been reported, the molecular mechanisms remain unknown. In this study, we used in vivo and in vitro approaches to characterize cancer-dependent alterations in the chromatin organization and find a CREB binding site (encompassing from −67 to −53 nt) in the core promoter. Besides, we also provided evidence that PKA pathway may involved in up-regulation of HULC. Furthermore, we demonstrated HULC may act as an endogenous ‘sponge’, which down-regulates a series of microRNAs (miRNAs) activities, including miR-372. Inhibition of miR-372 leads to reducing translational repression of its target gene, PRKACB, which in turn induces phosphorylation of CREB. Over-expression of miR-372 decreases the association of CREB with the proximal promoter, followed by the dissociation of P300, resulting in a change of the histone ‘code’, such as in deacetylation and methylation. The study elucidates that fine tuning of HULC expression is part of an auto-regulatory loop in which it’s inhibitory to expression and activity of miR-372 allows lncRNA up-regulated expression in liver cancer.
INTRODUCTION
Eukaryotic genomes are not the simple, well-order substrates of gene transcription that was once believed. We now know them to transcribe a broad spectrum of RNA molecules, ranging from long protein-coding mRNAs to short non-coding transcripts, which frequently overlap or are interleaved on either strand. The large proportion of a eukaryotic genome that is transcribed thus produces a huge array of RNA molecules differing in size, abundance and protein-coding capability (1).
In stark contrast to this diversity of RNA species, only a small number of non-protein-coding transcripts currently have experimentally-derived functions. Moreover, only rarely have disease-associated mutations been identified outside of protein-coding genes. Might, therefore, this colorful pageant of genomic transcription be a mirage? Might much of a genome’s repertoire of non-protein-coding transcripts be inconsequential transcriptional ‘noise’? Wang et al. (2) reported that non-protein-coding transcripts identified in mouse cDNA collections are poorly conserved and therefore argued that they are likely to be non-function. By contrast, many members of known functional classes of non-coding RNAs (ncRNAs), including microRNAs (miRNAs) are well-conserved across a diverse range of species (3–5).
NcRNAs are grouped into three subclasses according to their number of nucleotides (6,7). The growing class of miRNAs (19–25 nt) has been related to cell differentiation and cancer in recent publications (8–11). Small ncRNAs with a length of 100–200 nt are commonly found as translational repressors, and long ncRNAs (>200 nt) are reported involved in gene silencing (7). According to their structural features, these three subclasses of heterogeneous transcriptional units can be further subcategorized.
Hepatocellular carcinoma (HCC) is one of the deadliest cancers, causing about half a million deaths each year (12). Using a HCC-specific cDNA microarray platform, Panzitt et al. (13) identified a novel mRNA-like long ncRNA, highly up-regulated in liver cancer (HULC) as one of the most up-regulated genes in HCC. Up-regulation of HULC in colorectal carcinomas that metastasize to the livers was independently described by Matouk et al., in a later study, indicating that HULC is not restricted to HCC, however, to some extent, dependent on the liver micro-environment (14). However, the mechanism involved in up-regulating expression of HULC in HCC remains unknown.
Depletion of HULC in Hep3B cell line resulted in a significant deregulation of several genes, some of which have already been reported in the context of liver cancer (13). This indicates that there may not only be one single HULC target gene, but rather suggests a more general regulatory role for HULC. Disappointedly, preliminary studies revealed no significant homologies between HULC and its putative target sequences, thus indicating that the effect of HULC on its targets may not base on direct RNA–RNA interactions. Whether the mechanism by which HULC affects target gene expression constitutes a common regulatory principle remains to be resolved.
The aim of the present study was to define the mechanism responsible for the up-regulation of HULC in HCC and thereby provide a basic model to aid future studies of Hulc promoter regulation and the lncRNA function. We find that one cAMP response element binding protein (CREB) binding site within the Hulc proximal promoter region can specifically bind phospho-CREB transcription factors through the PKA pathway and that mutation or deletion of this site diminishes Hulc promoter activity in liver cancer cells. Moreover, phospho-CREB is able to ‘open’ and maintain the local chromatin structure across the Hulc promoter. We also demonstrated that HULC RNA has the ability of inhibiting a series of miRNAs activities, including miR-372. Inhibition of miR-372 leads to reducing translational repression of its target gene, Prkacb, which can induce phosphorylation of CREB (15). The regulatory circuitry described in this study provides another example of how gene reprogramming during tumorigenesis relies on fine modulation of gene expression. It is also interesting to note that fine tuning of lncRNA, HULC expression is part of an autoregulatory loop in which it’s inhibitory to expression and activity of miR-372 allows the establishment of the lncRNA up-regulated expression in liver cancer.
MATERIALS AND METHODS
Patients and materials
Tumorous liver tissues and corresponding adjacent non-tumoral liver tissue were collected from 14 patients who underwent curative surgery for HCC at Ruijin Hospital, Shanghai, China. Informed consent was obtained from each patient recruited, and the study protocol was approved by the Clinical Research Ethics Committee of the Shanghai Jiaotong University school of medicine. QSG-7701, Chang liver, HL-7702, Huh-1, Huh-4, Hep3B, HepG2, Huh-7, SNU-449, SNU-475 cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) with 10% fetal bovine serum (FBS), 100 U/ml penicillin and 100 mg/ml streptomycin. Antibodies were purchased from the following companies: P300 (SC-584), Brg1 (SC-10768), Prkacb (SC-904), CREB (SC-186), p-CREB (SC-7978), CBP (SC-7300) and RNA polymerase II (SC-9001) from Santa Cruz Biotechnology (Santa Cruz, CA, USA); tetra-acetyl-H4 (06-866) and diacetyl-H3 (07-593) from Upstate (Charlottesville, VA, USA); dimethyl-H3-K9 (ab1220) from Abcam (Cambridge, MA, USA) and trimethyl-H3-K4 (9751) from Cell Signaling Technology (Boston, MA, USA). PKA inhibitor H89, and inhibitor of transcription, α-amanitin were purchased from Sigma (St. Louis, MO, USA).
Cell culture
To examine the effects of H89 on HULC expression, cells were incubated for 1 day with indicated amount of H89. Cells were transfected with 50 nM miR-372 mimics, siRNA against CREB, HULC, or Prkacb (GenePharma Co. Ltd, Shanghai China) using Lipofectamine 2000 (Invitrogen Corp., Carlsbad, CA, USA) according to the manufacturer’s instructions for indicated period of time after replacement of transfection culture medium in the DMEM with 5% FBS. Incubation with 50 μg/ml α-amanitin was performed for 1 day.
5′-RLM-RACE
The GeneRacer system (Invitrogen), based on RNA ligase-mediated and oligo-capping rapid amplification of cDNA, was carried out according to the manufacturer’s instructions. The kit ensures the amplification of only full-length transcripts by eliminating truncated messages from the amplification process. HULC-specific primers located in the first exon were used for amplification by PCR using the LaTaq GC-Rich PCR system (Takara, Inc., Dalian, China).
Chromatin accessibility analysis of chromatin structure
Accessibility of DNA to digestion with DNase I and restriction enzymes (All from Takara, except Bra I from New England Biolabs, Beverly, MA, USA) analyzed using chromatin accessibility by real-time PCR (CHART-PCR) as described previously (16,17). Briefly, nuclear pellets were re-suspended in 3 ml of buffer A (10 mM Tris–HCl, pH 7.4, 10 mM NaCl, 3 mM MgCl2, 0.3 M sucrose). Aliquots of nuclei were digested with increasing concentrations of DNase I (0, 1, 3, 7 U) in a 100 μl final volume with 1 mM CaCl2 for 5 min at room temperature. The reaction was stopped by adding 100 μl of stop buffer (50 mM Tris–HCl, pH 7.5, 150 mM NaCl, 50 mM EDTA, 0.3% SDS) and incubated with 10 U RNaseA (Takara) at 37°C for 2 h. The samples were extracted once with phenol–chloroform–isoamyl alcohol (25 : 24 : 1) and once with chloroform. Nucleic acids were precipitated with 0.7 volumes of isopropanol, washed with 70% ethanol and resuspended in TE buffer. For the restriction enzyme accessibility assays, nuclei were isolated as described above and digested with 25–150 U of restriction enzyme in 100 μl of the corresponding reaction buffer for 30 min at 37°C Reactions were terminated and DNA was extracted as described earlier. After purification, 0.1 μg DNA from nuclease-digested or non-digested control cells was used in real-time PCR with the primer sets listed in Supplementary Data. Percent protection was calculated as the amount of DNA recovered from the digested− cells relative to the control cells.
ChIP and Re-ChIP
Chromatin immunoprecipitation (ChIP) assays were performed according to the manufacturer’s instructions (Active Motif, Carlsbad, CA, USA). Briefly, 2 × 106 cells were fixed with 1% formaldehyde, washed with cold phosphate-buffered saline, and lysed in buffer. Nuclei were sonicated to shear DNA, and the lysates were pelleted and precleared. The protein–DNA complexes were incubated with 4 μg antibodies overnight and then incubated with protein A beads followed by elution in 1% SDS/0.1 M NaHCO3 and cross-links were reversed at 65°C. At last, DNA was subjected to quantitative PCR analysis after recovered. In the Re-ChIP experiments, complexes were eluted by incubation for 30 min at 37°C in 25 ml 10 mM DTT. After centrifugation, the supernatant was diluted 20 times with Re-ChIP buffer (1% Triton X-100, 2 mM EDTA, 150 mM NaCl, 20 mM Tris–HCl, [pH 8.1]) and subjected again to the ChIP procedure. Real-time PCR reactions were performed in triplicate with 1 μl of precipitated DNA. DNA recovered from samples containing an antibody was compared with no antibody negative controls performed on aliquots from the same chromatin preparation. Data are presented as the amount of DNA recovered relative to the appropriate negative control antibody provided by the manufacturer. In this way, differences between chromatin preparations are normalized. The results were expressed as the means ± SDs of three independent experiments. The PCR primers are available in Supplementary Data.
Reverse transcription PCR
Total RNA was extracted by Trizol reagent (Invitrogen). Reaction mixture (20 μl) containing 1 μg of total RNA was reverse transcribed to cDNA using PrimeScript RT-polymerase (Takara). Quantitative PCR was performed on the cDNA using primers specific for HULC, CREB, Prkacb. Specifically, stem-loop reverse transcriptase polymerase chain reaction (RT-PCR) for mature miR-372 was performed as described previously (18). RNA input was normalized to the level of β-actin, both for genes and miRNA analysis (19). All reaction were carried out using SYBR Green Mix (Takara), and the PCR conditions for quantitative RT-PCR were as follows: activation of enzyme at 94°C or 5 min, 40 cycles of denaturation at 94°C for 30 s, annealing at 55°C for 30 s, and extension at 72°C for 20 s. qRT-PCR was carried out using a Mx3000P real-time PCR System (Stratagene, La Jolla, CA, USA). The fluorescence of each sample was determined after every cycle. The fluorescence of samples was continuously traced during this period. Relative expression levels were calculated as ratios normalized against those of β-actin. The data from qRT-PCR were analyzed by the ΔCt method, and the ΔCt value was determined by subtracting the β-actin Ct value from the target gene Ct value. The ΔCt of the stimulated cells (ΔCts) was subtracted from the ΔCt of the untreated cells (ΔCtu) (ΔΔCt = ΔCts−ΔCtu), and the expression level for a target gene in the stimulated cells compared with the level in the untreated cells was calculated as follows: x-fold of unstimulated control = 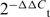 . All results are expressed as the means ± SD of three independent experiments. Primers can be found in the Supplementary Data.
. All results are expressed as the means ± SD of three independent experiments. Primers can be found in the Supplementary Data.
Construction of reporter and expression plasmids and mutagenesis
PCR was performed using sets of oligonucleotide primers specific for the Hulc promoter, of which the forward primer was Mlu I-site-linked and the reverse primer Xho I-site-linked. Hep3B or HL-7702 genomic DNA was used as the template. These PCR products were digested with Mlu I and Xho I, and cloned into the Mlu I/Xho I sites of the pGL3-Basic vector (Promega, Madison, WI, USA), yielding the promoter reporter plasmid. HULC, Prkacb 3′-untranslated regions (UTR), and flank sequences around pre-miRNA, which containing miRNA putative target sites were amplified from Hep3B or HL-7702 genomic DNA, and then were cloned into the Xho I/Not I sites of psiCHECK vector (promega). For over-expression studies, the complete cDNA sequence of HULC was obtained from Hep3B cells by RT-PCR and then cloned into EcoR I and Xho I sites of pcDNA3.1(+) expression vector (Invitrogen). Substitution mutation, deletion, and 2 × or 3 × CREB binding sites connection constructs were generated by a PCR-based site-directed mutagenesis kit (Takara), using corresponding plasmid as the template. For eight-base substitutions, we introduced the GCGCGCGC into the substitution sites.
Luciferase assay
In promoter assay, promoter reporter plasmid DNA was transiently transfected into Hep3B and HL-7702 cells using Lipofectamine 2000 (Invitrogen). pRL-TK (Promega) was co-transfected as an internal control for transfection efficiency. In miRNA sensor reporter assay, recombinant psiCHECK plasmids (including both firefly and Renilla luciferase reporter genes, with the former gene as an internal control) containing possible miRNA target sites were co-trasnsfected with miR-372 mimics with or without miR-372 inhibitor as described earlier. After further cultivation for 24 h, the transfected cells were harvested, lysed, centrifuged and the pellet subjected to luciferase assay. Luciferase activity was measured as chemiluminescence in a luminometer (PerkinElmer Life Sciences, Boston, MA, USA) using the Dual-Luciferase reporter assay system (Promega) according to the manufacturer’s protocol. All transfections were performed in triplicate, and the results were expressed as the means ± SD of three independent experiments.
Electrophoretic mobility shift assays
Electrophoretic mobility shift assay (EMSAs) were performed using the gel shift kit (Promega). Nuclear extract proteins (5.0 μg) were incubated in 10 μl of reaction containing 4% glycerol, 1 mM MgCl2, 0.5 mM dithiothreitol, 0.5 mM EDTA, 50 mM NaCl, 10 mM Tris–HCl, pH 7.5 and 2.0 μg poly(dI–dC) with or without 30-fold excess of unlabeled DNA competitors on ice for 10 min followed by the addition of the radio labeled probe. For supershift assays, antibody against CERB or phospho-CREB was added to the reaction mixture 10 min before the addition of the probe. All DNA–protein complexes were resolved by electrophoresis on 5% native polyacrylamide.
Western blot analysis
Cells were harvested and lysed in 0.5 ml of lysis buffer (10 mM Tris–HCl, pH 7.6, 5 mM EDTA, 50 mM NaCl, 30 mM sodium pyrophosphate, 50 mM NaF, 0.1 mM Na3VO4, 1% Triton X-100, 1 mM phenylmethylsulfonyl fluoride and protease inhibitor mixture tablet (Roche Applied Science, Indianapolis, IN, USA). Lysates were clarified by centrifugation at 15 000g for 10 min. Protein (30 μg) was processed for SDS–PAGE, which was performed on 12% gels. The proteins were electrophoretically transferred to Immobilon P (Bio-rad, Hercules, CA, USA). The blots were blocked with 5% non-fat milk in Tris–buffered saline (TBS, pH 7.4) for 1 h and then incubated with antibodies in 5% non-fat milk in TBS. They were then washed with TBS and incubated with secondary antibodies conjugated with horseradish peroxidase in 5% non-fat milk in TBS. After washing with TBS, the bound antibodies were visualized by enhanced chemiluminescence (Pierce, Rockford, IL, USA) and recorded on X-ray films.
Statistical analysis
Statistical evaluations were conducted using the t-test. P < 0.05 were considered to be statistically significant.
RESULTS
HULC is up-regulated in HCC and liver cancer cell lines at transcription level
To validate early observation from previous report that HULC was up-regulated in HCC (14), expression of HULC in clinical specimens was assessed by quantitative real-time PCR. In a collection of 14 pairs of randomly chosen samples, HULC expression was up-regulated in all tumor tissues compared to the corresponding adjacent non-tumor liver specimens (Figure 1A). An analysis of HULC expression in 10 different liver cell lines showed it to be up-regulated in liver cancer cell lines (Huh-1, Huh-4, Hep3B, HepG2, Huh-7, SNU-449 and SNU-475) compared to that of normal liver cell lines (QSG-7701, Chang liver and HL-7702). The degree of HULC expression level was highest in Hep3B cells, whereas almost undetectable in HL-7702 cells (Figure 1B). Thus, Hep3B and HL-7702 cell lines were selected as a research pair represents liver cancer and normal cells in the following studies. To determine whether up-regulation of HULC in liver cancer was mainly at transcription level, α-amanitin, a RNA polymerase II inhibitor (20), was applied. Treatment of Hep3B and HL-7702 cells with α-amanitin in culture at a concentration of 50 μg/ml for 24 h resulted in the inhibition of HULC expression (Figure 1C), suggesting the up-regulation of HULC expression is dependent of transcription.
Figure 1.

Up-regulation of HULC in HCC and liver cancer cell lines. (A) Change in HULC expression between adjacent normal and HCC tissues was quantified by real-time PCR. (B) HULC expression levels in different liver cell lines by semi-qRT-PCR. β-actin was treated as internal control. (C) Hep3B and HL-7702 cells were stimulated with α-amanitin (50 μg/ml) for 24 h. All results are means ± SD of three independent experiments. The asterisk indicates statistical significance at the P < 0.01 level using t-test.
Open chromatin structure around the transcription start site
The transcription start site (TSS) of the Hulc gene was identified by 5′ RACE analysis and predicted to be ∼146 bp from our gene-specific nested primer (Figure 2A, left panel). Sequencing of PCR products indicated that the first base was C (Figure 2A, right panel), which is 60 bp downstream of the first base provided by the National Center for Biotechnology Information (Gene ID: 728655). It is generally accepted that the local chromatin structure affects gene transcription (21). We performed CHART-PCR assays, which evaluates the accessibility of genomic DNA to nuclease by comparing the quantity of intact DNA from a nuclease-treated sample to that of an untreated sample and is expressed inversely proportional to the protection level (17), in an attempt to reveal differences of regions of open chromatin around the Hulc gene promoter between liver cancer and normal cell lines, in order to provide cues in the search for the regulation sites at the promoter level. Nuclei were isolated and subjected to digestion with DNase I and restriction enzymes and the genomic fragments between −10 028 and +9756 bp (relative to the TSS) were analyzed. In addition to the regions around TSS (R2–R7, Figure 2B), another two representative regions were also included: R1 region is a distal region upstream the TSS and R8 region is a distal region downstream of the Hulc gene. Thus, we were able to evaluate the nuclease accessibility of the Hulc gene from 5′ distal region to the 3′ distal region. As shown in Figure 2C, the regions spanning the proximal promoter (R4–R7) were more sensitive to DNase I digestion, particularly, R7 (from −90 to +77) exhibited the lowest protection level of ∼8% in Hep3B cells, whereas ∼30% in HL-7702 cells. In comparison, higher levels of protection against DNase I digestion were seen at the R1 and the R8 region, furthermore, the protection levels of R1 and R8 were not affected as much as those of R6 and R7 when using increasing concentrations of DNase I (1, 3 and 7 U) in both Hep3B and HL-7702 cells. Thus, DNase I accessibility is limited to the proximal promoter region (especially to the R7 region), and the degree of accessibility of cancer cells is much greater than that of normal cells. To confirm the results performed by DNase I, a restriction enzyme accessibility assay was employed. Nuclei isolated from Hep3B or HL-7702 cells were subjected to limited digestion with a panel of restriction enzymes with recognition sites at different locations within the corresponding regions (Figure 2D, left panel). As shown in Figure 2D, in both cells, restriction sites of Hae III/R1 and Alu I/R8 were resistant to nuclease digestion, indicating their coverage by nucleosomes. In contrast, stronger cleavage with lower protection was detected for Alu I/R6, and Sac I/R7 (Sac I was stronger) in Hep3B cells than those of HL-7702 cells, suggesting that these sites were in two linker regions, with greater accessibility to restriction enzymes in a cancer dependent manner.
Figure 2.

Analysis of chromatin accessibility around the Hulc promoter. (A) 5′RACE PCR products suggest the putative TSS is ∼146 bp upstream of the HULC reverse 5′RACE primer (left panel). Mapping the TSS of Hulc in HCC (right panel). (B) Schematic representation of the DNA regions around Hulc promoter, which can be amplified by corresponding primer sets. (C) DNase I accessibility of Hulc gene in Hep3B and HL-7702 cells. Nuclei from two cell lines were harvested and treated with three units (left panel) or increasing amount (1–7 units, right panel) of DNase I for 5 min at room temperature. Then the genomic DNA was purified and quantitated relative to DNA from undigested nuclei using the primers described in (B) by quantitative PCR and listed as percent protected. (D) Restriction enzymes accessibility of Hulc gene in Hep3B and HL-7702 cells. Nuclei was treated with 25–150 units of restriction enzymes for 30 min at 37°C. Then the genomic DNA was purified and quantitated relative to DNA from undigested nuclei using the primers described in (B) by quantitative PCR and listed as percent protected. Chromatin from Hep3B and HL-7702 cells were harvested and precipitated with anti-diacetyl-H3 (E), anti-tetra-acetyl-H4 (F), anti-tri-methyl-H3-K4 (G) and anti-dimethyl-H3-K9 (H) antibodies. After DNA recovery, the precipitates were evaluated by real-time PCR for the level of enrichment over negative control antibody using primer sets described in (B). All results are the means ± SD of three independent experiments. The asterisk indicates statistical significance using t-test, *P < 0.05; **P < 0.01.
Histone modification around the promoter
Next, we evaluated whether HULC expression is enhanced by epigenetic changes to the promoter by ChIP assays. Acetylation has long been known to be associated with actively transcribed genes and open chromatin configurations (22,23). Antibodies specific to diacetylated H3 (K9 and K14) and tetra-acetylated H4 (K5, K8, K12 and K16) were used in ChIP analysis to determine the pattern of histone modification in a cancer specific manner. In HL-7702 cells, both H3 and H4 were acetylated at all the testing areas, with the highest level in the R7 region. In cancer cells, histone acetylation was significantly increased in R7 region (Figure 2E and F). As for histone methylation, trimethyl-H3K4 [an euchromatic marker (24)] was scored significantly higher in R4–R7 regions in cancer cells. As for the heterochromatin marker Dimethyl-H3K9, which serves as a signal for chromatin silencing by recruiting the HP1 protein (heterochromatin protein 1) and is mutually exclusive with H3-K9 acetylation (25), it was scored lower in R6 and R7 regions in cancer cells (Figure 2G and H). Taken together, these results indicate that transcription activation of HULC is correlated with an increase in the local recruitment of euchromatic markers and a decrease in that of the heterochromatic markers in the transcriptional activation of Hulc gene expression in liver cancer cells.
CREB-binding site contributes to the activation of the promoter
The existence of a highest nuclease sensitive site (R7 region) strongly indicates the binding of transcription factors as transcriptional regulators, prompting us to examine the potential role of the cis elements at this region. To examine the transcriptional activity of the sequences at the 5′ end of the Hulc gene, various fragments of the proximal sequence were cloned upstream of the firefly luciferase reporter gene. A 5′ deletion series with a fixed 3′ end at the +72 position (relative to the TSS) were generated through PCR amplification from genomic DNA of Hep3B or HL-7702 cells. The constructs were transfected into Hep3B and HL-7702 cells, respectively. All the constructs (containing −132/−48 fragment) tested were capable of inducing a significant increase in luciferase activity compared with that of a promoter-less vector (pGL3-Basic) (Figure 3A). This observation suggests that the most proximal 84 nt of the Hulc gene is capable of initiating transcription. The promoter reporter containing the shortest DNA fragment encompassing −132/−48 region is named Luc-HCP hereafter. Surprisingly, no significant difference of activity in Hep3B compare with HL-7702 cells was detected in any of the constructs. We then performed a search for possible transcription factor-binding sites using online software programs MatInspector (www.genomatix.de/online_help/help_matinspector/matinspector_help.html) and TFSEARCH (www.cbrc.jp/research/db/TFSEARCH.html) revealed a Lyf-1 and a CREB binding site within this region (Figure 3B). To confirm the roles of these two sites in transcription activity of the Hulc gene, deletion of either of these two sites was tested for promoter function in the cell lines. The results showed that when the CREB binding site is deleted, promoter activities were reduced to background levels (Figure 3C). However, no promoter activity change was found when the Lyf-1 binding site is deleted. To further test the possible CREB binding, we have generated a series of 8 bp mutations between −132 and −48 nt within Luc-HCP (−132/+72) construct through a PCR-based site-directed mutagenesis technique. Particularly, the mutation in M6, M7 and M13 (M13 overlaps M6 and M7), which disrupts the CREB binding site reduced promoter activity by >86%. No significant changes in promoter activity had been observed in other mutant constructs (Figure 3D). Moreover, additional one (2×) or two (3×) possible CREB-binding sites increased promoter activity of Luc-HCP (1×CREB), however, no significant increasing effects were found between 2× CREB and 3× CREB constructs, which may due to steric effects (Figure 3E). These results demonstrate that the CREB binding site is necessary for transcriptional initiation.
Figure 3.

CREB-binding site contributes to the activation of the promoter. (A) Hep3B and HL-7702 cells were transiently co-transfected with reporter plasmids containing truncated versions of the promoter region of the Hulc gene, as indicated, and pRL-TK. Luc-HCP is defined as the reporter containing the shortest promoter region (from −132 to +72), which core elements located in. (B) Transcription factor binding sites present in the core promoter region (from −132 to −48) were predicated by web software TFSEARCH and MatInspector, as indicated by dashed lines. (C) Cells were transiently transfected with wild-type Luc-HCP as well as Luc-HCP which Lyf-1 or CREB binding site was deleted. Lyf-1-binding site is indicated by circle, whereas CREB-binding site is indicated by rectangle; deleted Lyf-1 and CREB sites are shown as black cycle and rectangle, respectively. (D) Scanning mutational analysis of the fragment from −132 to 48. As shown on the upper panel, a series of 8-bp substitutions were made within the fragment from −132 to 48 of the reporter construct Luc-HCP (from −132 to +72). M6, M7 and M13 overlap the possible CREB-binding site. (E) Wild-type Luc-HCP and Luc-HCP containing additional one (2×) or two (3×) possible CREB-binding sites were co-transfected with pRL-TK into Hep3B and HL-7702 cells. Firefly luciferase activity was normalized to Renilla luciferase activity, and the relative luciferase activities are presented as fold increase over the promoter-less pGL3 basic vector. Horizontal column lengths represent the means ± SD of three independent experiments.
Binding of phospho-CREB to the core promoter in liver cancer
To test DNA–protein interactions in the promoter region, we performed EMSA using the DNA sequence from −73 to −46 bearing the CREB binding site. Strong DNA–protein complex was detected in both Hep3B and HL-7702 cells (Figure 4A, lanes 1 and 4). To determine whether the putative CREB binding site is involved in the formation of these complexes, a 30-fold excess of unlabeled oligonucleotide corresponding to −73 to −46 sequences was used to compete with the complexes. As shown in lanes 2 and 5, the competitor competed away the complexes possibly by sequestering the available transcription factors present in the nuclear extract. These data indicate that the putative CREB binding site has protein binding capacity. In contrast, the unlabeled mutated oligonucleotide bearing the disrupted CREB binding site was unable to compete for protein binding capacity (lanes 3 and 6). To test if CREB is involved in the formation of the protein–DNA complexes detected in the EMSA, we performed supershift assays by using the anti-CREB antibody. As illustrated in Figure 4B lane 2 and 4, a super shift band was formed by antibody in nuclear extracts from Hep3B and HL-7702 cells. These results suggest that CREB can specifically interact with the wild-type −73 to −46 sequence probe in both Hep3B and HL-7702 cells. It is well known that phosphorylated CREB binds to promoter region, allowing it to switch certain genes on or off (26,27). Thus, we next perform super shift assay again using anti-p-CREB antibody to conform whether phospho-CREB binds to the Hulc promoter in liver cancer cells. As expected, super shift band was formed only in Hep3B cells, no super shift band was detected in HL-7702 cells (Figure 4C, lanes 2 and 4). These results suggest that phospho-CREB forms DNA–protein complexes in liver cancer cells in vitro. We then performed ChIP assays to examine whether CREB and its co-activator, CBP (CREB binding protein) interact with the Hulc promoter in vivo. As shown in Figure 4D, anti-CREB antibody specifically enriched the R7 region in both two cell lines, which containing the CREB binding site. However, DNA fragments immunoprecipated by anti-CBP antibody led to amplification of a single band using R7 primer sets only in Hep3B cells. Collectively, cancer induced CREB phosphorylation may greatly enhance the increase in HULC expression.
Figure 4.

HULC expression is regulated by phospho-CREB through PKA pathway in liver cancer cells. (A) Upon interaction with the nuclear extract of Hep3B (left panel) and HL-7702 (right panel), wild probe generated specific band (lanes 1 and 4) which was self-competed by 30-fold excess of unlabeled probe (lanes 2 and 5). No competition was observed when using the same amounts of mutant probe (lanes 3 and 6). (B) Interaction with anti-CREB antibody resulted in a super shift band formation in both two cell lines (lanes 2 and 4). (C) Interaction with anti-phospho-CREB antibody resulted in a super shift band formation (lane 2) in Hep3B, but not detected in HL-7702 cell line (lane 4). (D) ChIP analysis was performed to qualitative confirm the interaction of CREB and CBP with the Hulc promoter in vivo in Hep3B (upper panel) and HL-7702 (lower panel) cells using primer sets R1, R5, R7 and R8 described in Figure 2B. PCR products from the ChIP assay were run on an agarose gel. As the negative controls, the protein–DNA complexes were incubated without antibodies or with non-specific control IgG. The input DNA represents one-fifth of the starting material. (E) Pre-incubation with H89 was performed 1 day before transiently co-transfected Luc-HCP and internal control pRL-TK plasmids into Hep3B and HL-7702 cells and further incubation for 1 day in the continued presence of indicated amount of H89. Firefly luciferase activity was normalized to Renilla luciferase activity. Results are shown as relative percentage to those of cells untreated H89. (F) Total RNA was extracted for measurement of HULC mRNA expression level after treatment by indicated amount of H89 by real-time PCR. *P <0.05; **P <0.01 versus the corresponding untreated cells (E and F). (G) Endogenous CREB mRNA was quantified by real-time PCR and normalized to β-actin RNA in Hep3B and HL-7702 cells (left panel). The whole lysates of Hep3B and HL-7702 cells were examined by immunoblotting with antibodies against CREB, p-CREB and β-actin (right panel).
PKA pathway up-regulates HULC expression
In previous studies (28–30), it was determined that the PKA pathway was responsible for immediate stimulation of CREB. The importance of the PKA pathway was examined by pretreatment of Hep3B and HL-7702 cells with H89, an inhibitor of PKA (31). Pre-incubation of indicated amount of H89 for 1 day and then transfected with Luc-HCP and further incubation for another 1 day in the continued presence of H89 blocked promoter activities in a dose-dependent manner (Figure 4E) through reducing phosphorylation of CREB (data not shown), and greatly inhibited the HULC expression (Figure 4F) in Hep3B cells. In contrary, no significant changes were detected both in promoter activity and HULC expression in HL-7702 cells (Figure 4E and F). To examine whether high HULC expression in liver cancer cells correlates with CREB expression, we examined Hep3B cells and HL-7702 cells for their expression patterns of CREB by real-time PCR and western blotting. Surprisingly, the CREB expression level in Hep3B cells was similar with that of HL-7702 cells at transcription level (Figure 4G, left panel), but was lower than that of the latter one at translation level (Figure 4G, right panel). However, phospho-CREB level was greatly enhanced in Hep3B cells compared with that of HL-7702 cells (Figure 4G, right panel). In addition, we sequenced cDNA of CREB, and found no changes from Hep3B and HL-7702 cells (data not shown), thus exclude possible sequences alteration caused HULC up-regulation. Taken together, phospho-CREB may involve in the up-regulation of the HULC expression through activation of PKA pathway.
Phospho-CREB recruits P300 and Brg I to the Hulc promoter in liver cancer cells
Knockdown studies were performed to further test the role of CREB in regulating Hulc promoter activation. To address this, we inhibited CREB expression using siRNA. Hep3B and HL-7702 cells were transfected with CREB targeted siRNA and a scrambled siRNA as control. The siRNA reduced the CREB mRNAs levels remarkably (reduction by 83 and 84% for Hep3B and HL-7702 cells, respectively) and resulted in inhibition of both promoter activation and expression levels of HULC (Figure 5A and B). Moreover, it was more obvious in Hep3B cells than that of HL-7702 cells. ChIP assays were next performed to monitor changes of histone acetylation. Histone deacetylation was observed after siRNA induction (Figure 5C and D). Consistent with the changes of HULC transcription (Figure 5A and B), RNA Polymerase II (pol II) was observed at high level under basal conditions, and decreased after knockdown of CREB (Figure 5E). It has been previously reported that transcription factors recruit the HAT P300 enzyme binds core promoter regions, and that its binding correlates with the presence of highly acetylated histones (21). So, we performed a Re-ChIP assay in order to confirm whether phospho-CREB and P300 are co-recruited onto the proximal promoter. In Re-ChIP analysis, we immunoprecipitated the phospho-CREB or P300-containing complexes with antibodies against P300 or phospho-CREB, respectively, and only those DNA sequences that are simultaneously bound by both proteins would be amplified in the subsequent PCR. Our results showed that P300 and phospho-CREB were co-recruited on the same DNA fragments (R7 region) (Figure 5F, upper panel), suggesting that the two factors co-occupy common target loci in Hep3B cells. We then performed knockdown experiments to verify whether reduction of CREB might dissociate the P300 enzyme away from Hulc promoter sequences. The ChIP results showed that P300 is present in the R7 region in untreated cells, a decrease was observed after siRNA induction in Hep3B cells (Figure 5G). Besides, we found that Brg1, the core component of the SWI–SNF remodeling complex, which can change the accessibility of nucleosome-packaged DNA (21), co-occupied with phospho-CREB in the same R7 region of the Hulc promoter (Figure 5F, upper panel), whereas, no significant bands were detected in HL-7702 cells (Figure 5F, lower panel). These results strongly support a direct role of phospho-CREB in P300 and Brg I recruitment to the Hulc promoter, leading to the activation of epigenetic markers and chromatin remodeling at the same location in liver cancer cells.
Figure 5.

Reduction of CREB results in changes of chromatin accessibility and histone acetylation. (A) RNAi for CREB reduced Hulc promoter activity in Hep3B and HL-7702 cells. CREB or negative control siRNAs were co-transfected with Luc-HCP into cells. Luciferase assays were performed 24 h after transfection. Firefly luciferase activity was normalized to Renilla luciferase activity. (B) Endogenous HULC mRNA was quantified by real-time PCR and normalized to β-actin RNA. Chromatin from Hep3B and HL-7702 cells treated with control or CREB specific siRNA was harvested and precipitated with anti-diacetyl-H3 (C), anti-tetra-acetyl-H4 (D) and anti-pol II (E) antibodies. After DNA recovery, the precipitates were evaluated by real-time PCR for the level of enrichment over negative control antibody. (F) ChIP and re-ChIP experiments performed with anti-phosph-CREB, anti-P300 and anti-Brg I antibodies on Hep3B and HL-7702 cells. (G) Chromatin from Hep3B cells treated with control or CREB specific siRNA was harvested and precipitated with anti-P300 antibody. All the results (A–E and G) are the means of three independent experiments ± SD. Results of cells transfected siRNA against CREB are normalized to those of cells treated by negative control siRNA. The asterisk indicates statistical significance at the P < 0.05 level using t-test.
HULC reduces both miR-372 expression and activity
Most recently, a report indicated that long ncRNA from Arabidopsis thaliana contains motif with sequence complementary to miRNA, and coin the term ‘target mimicry’ to define this mechanism of inhibition of miRNA activity (32). To examine whether HULC has a similar mechanism in liver cancer, prediction of miRNA target sites was performed by the online software MicroInspector (bioinfo.uni-plovdiv.bg/microinspector). As shown in Supplementary Figure S1, HULC RNA contains many elements complementary to various miRNA seed regions. To obtain a first insight into a possible role of HULC in deregulation of miRNAs, the effect of siRNA-mediated knockdown of HULC expression was investigated. Knockdown efficiency of 79% was obtained for HULC siRNA (data not shown). The expression profiling of seven randomly chosen miRNAs were measured in siRNA treated Hep3B cells by real-time PCR. Surprisingly, the expression levels of almost all the miRNAs were slightly down-regulated with exception of miR-613 (∼1-fold increase, Supplementary Figure S1) and miR-372 (over 3-fold increase, Figure 6A) in siRNA treated cells compared with those of cells treated with negative control siRNA, suggesting HULC does not affect miRNAs expression profiling in most cases. It may due to low expression level of HULC that miR-372 expression level had no significant change after siRNA treatment in HL-7702 cells (Figure 6A). To verify HULC can genuinely affect miRNAs activities, sensors were constructed (Figure 6B and Supplementary Figure S1) as described in figure legends. Reduction of Renilla luciferase activities suggests induction of miRNAs activities. As expected, the Renilla lucifease activities of all the chosen miRNAs sensors greatly decreased (lowest in miR-372) when cells treated with siRNA against HULC compared with those of negative control (Figure 6B and Supplementary Figure S1). These results indicated that HULC may act as endogenous sponge ‘antagomirs’ as described previously (33,34). To validate the observation that HULC can reduce miR-372 expression in HCC, expression of miR-372 in clinical specimens was assessed by real-time PCR. In a collection of 14 pairs of randomly chosen samples, which were used for detection of HULC expression levels described earlier, miR-372 was down-regulated in all tumor tissues compared to the corresponding adjacent non-tumor liver tissues (Figure 6C). In addition, expression levels of miR-372 significantly declined in liver cancer cell line, Hep3B compared to normal live cell line, HL-7702 (Figure 6D), which is consistent with the results in clinical samples (Figure 6C). Taken together, HULC reduces not only expression, but also activity of miR-372 in HCC.
Figure 6.

HULC inhibits miR-372 expression and activity. (A) Expression profiling of miR-372 48 h after HULC siRNA treatment in Hep3B and HL-7702 cells. (B) Construction of miR-372 sensor (upper panel). Genomic sequences (∼200 bp) flanking pre-miRNAs were reverse inserted into the psiCHECK vector, placing the 3′UTR with the miRNA binding sites downstream of coding sequence of Renilla luciferase. Luciferase assays indicated that enhanced miRNAs’ activities lead to decreasing levels of Renilla luciferase after treatment by HULC siRNA in Hep3B and HL-7702 cells (lower panel). Renilla luciferase activities were normalized to firefly luciferase activities. *P < 0.01, versus the cells treated with negative control siRNA (A and B). (C) Change in miR-372 expression between adjacent normal and HCC tissues was quantified by real-time PCR. *P < 0.01, versus adjacent normal tissue. (D) Differences in miR-372 expression level between Hep3B and HL-7702 cell lines. *P < 0.01, versus Hep3B cells. All the results described above are means of three independent experiments ± SD.
Interaction between HULC and miR-372
As described above, H89 can reduce HULC expression. As shown in Figure 7A, H89 induced expression of miR-372 in a dose-dependent manner in Hep3B cells, while no significant change was detected in HL-7702 cells, suggesting reduction of miR-372 is associated with expression of HULC. To further validate the inhibitory effect of HULC on miR-372 activity, Hep3B and HL-7702 cells were incubated with indicated amount of H89 and transfected with miR-372 sensor. The results indicated that H89 induces miR-372 activity in liver cancer cells (Figure 7B). Based on a recently study descried the ability of miR-372 to overcome a p21-mediated cell cycle arrest in the testicular germ cell tumor (35), we performed cell cycle analysis. Results indicated that miR-372 regulate cell cycle progression by inducing the S accumulation, which is consistent with previous report. However, cells incubated with H89 or siRNA against HULC enhanced miR-372 induced effects (data not shown). Bioinformatics reveal HULC RNA contains one conserved target site of miR-372 (Figure 7C, left panel). To verify the interaction between HULC and miR-372, the HULC RNA was cloned into psiCHECK vector (pSi-HULC), as described in figure legends. The constructs were co-transfected into Hep3B and HL-7702 cells with miR-372 mimics. Luciferase assays revealed that over-expression of miR-372 could significantly reduce the Renilla luciferase activities from the reporter pSi-HULC (Figure 7C, right panel). To inhibit the function of miR-372, 2′-O-methyle antisense inhibitory oligoribonucleotides targeted toward miR-372 was co-transfected, relative luciferase activities were significantly up-regulated compared to that of transfected with miR-372 alone. To confirm this putative target site of miR-372, deletion assay was performed. Luciferase assays indicated that over-expression of miR-372 had no effectiveness on deleted HULC RNA compared to the wild-type. Furthermore, over-expression of HULC (HULC wild-type) resulted in down-regulation of both miR-372 expression and activities in a does-dependent manner (Figure 7D and E). However, no significant change was detected between cells transfected with pcDNA 3.1(+) empty vector and HULC without miR-372 target site (HULC-Del-372). It may due to higher expression level of miR-372 in HL-7702 cells, which caused more obvious change compared with that of in Hep3B cells when HULC was over-expressed. These results revealed HULC may inhibit both expression and activity of miR-372 by this putative binding site at post-transcription level in both liver normal and cancer cells.
Figure 7.

Interaction between HULC RNA and miR-372. (A) Relative fold change of miR-372 in the presence of indicated amount of H89 for 24 h. (B) Pre-incubation with H89 was performed 1 day before transiently transfected miR-372 sensor into Hep3B and HL-7702 cells and further incubation for 1 day in the continued presence of indicated amount of H89. *P < 0.05 and **P < 0.01, versus the cells untreated with H89 (A and B). (C) Sequence inspection by microInspector online software reveals HULC RNA contains a conserved element complementary to miR-372 (left panel). psiCHECK vectors containing HULC RNA with (pSi-HULC, wild-type) or without (pSi-HULC, deletion) predicted miR-372 binding site were co-transfected with miR-372 or both miR-372 and miR-372 inhibitor simultaneously (right panel). Luciferase assays were performed 24 h post-transfection. *P < 0.05. (D) The indicated amount of HULC-pcDNA3.1(+) with or without miR-372 binding site and empty vectors were transfected into Hep3B and HL-7702 cells grown on a six-well plate for 48 h. Then the total RNA was extracted for detection of miR-372. **P < 0.01. (E) The miR-372 sensor reporter plasmid was co-transfected with pcDNA3.1(+) vector as described in (D) and then cells were harvested, lysed, centrifuged and the pellet subjected to luciferase assay. *P < 0.05 and **P < 0.01, versus cells transfected with empty vectors. Renilla luciferase activity was normalized to Firefly luciferase activity (C and E). All results are shown as means of three independent experiments ± SD.
Interaction between Prkacb and miR-372
As described earlier, phospho-CREB stimulates expression of HULC; furthermore, the level of phospho-CREB is much higher in Hep3B cells than in HL-7702 cells (Figure 4G). In the contrary, expression level of miR-372 in Hep3B cells is much lower than that in HL-7702 cells (Figure 6D). We hypothesized that reduction of miR-372 may decrease repression to its mRNA targets, thus enhanced the phosphorylation of CREB in liver cancer. So, we did a search for miR-372 target mRNAs through online software: Tagetscan (www.targetscan.org), MicroInspector and RNA22 (cbcsrv.watson.ibm.com/rna22_targets.html) revealed a common target site in 3′UTR of Prkacb (cAMP-dependent protein kinase catalytic subunit beta) mRNA (Figure 8A, left panel), the protein product of which can translocate into nucleus and thus activate CREB (14,29). The Prkacb 3′UTR was then cloned into psiCHECK vector (pSi-Prkacb). Luciferase assays indicated that Prkacb mRNA is genuinely targeted by miR-372 (Figure 8A, right panel) according to the procedure described in Figure 7C. However, Prkacb mRNA obtained no significant change after over-expression of miR-372 or miR-372 inhibitor for 1 day revealed miR-372 without affecting Prkacb at the transcriptional level (data not shown). As previous studies described that translation repression is likely the predominant mechanism by which miRNAs exert their function (11), we next examine PRKACB by western blotting. As expected, after over-expression of miR-372 for 1 day, PRKACB was markedly decreased compare to control resulted in reduction of phospho-CREB (Figure 8B). Futhermore, the protein expression level is higher in Hep3B than that in HL-7702 cells (Figure 8C). Moreover, after over-expression of HULC in HL-7702 cells, PRKACB was apparently up-regulated resulted from down-regulation of both expression and activity of miR-372 (Figure 7D and E) at translation level (Figure 8D). After transfected siRNA against Prkacb, both promoter activity and expression of HULC declined compared to that of control in Hep3B cells, however, no significant change was detected in HL-7702 cells (Figure 8E and F). In conclusion, the above data clearly demonstrate that reduction of miR-372 mediated Prkacb inhibition results in facilitation of phosporylation of CREB in liver cancer cells.
Figure 8.

Interaction between Prkacb and miR-372. (A) Prkacb 3′UTR contains a miR-372 target site, as shown in the left panel, and confirmed by Luciferase assays (right panel). pSi-Prkacb contains Prkacb 3′UTR. Experimental procedure is similar with that described in Figure 7C. (B) Immunoblotting assay indicated that over-expression of miR-372 (50 nM) results in down-regulation of PRKACB, thus reduces phosphorylation of CREB in Hep3B cells. (C) Differences in PRKACB expression level between Hep3B and HL-7702 cell lines were measured by immunoblotting assay. (D) PRKACB expression level was detected by immunoblotting assay after transfection with pcDNA3.1(+) vector as indicated in HL-7702 cells. (E) Prkacb or negative control siRNAs were co-transfected with Luc-HCP into Hep3B and HL-7702 cells. Luciferase assays were performed 24 h after transfection. Firefly luciferase activities were normalized to Renilla luciferase activities. (F) Endogenous HULC mRNA was quantified by real-time PCR and normalized to β-actin RNA. Results of RNAi are shown as fold change to the control. *P < 0.01 versus corresponding control cells. All results are shown as means of three independent experiments ± SD.
MiR-372 mediates dissociation of transcription factors to the Hulc core promoter in vivo
In contrast with the results of the luciferase reporter assays and western blotting that PRKACB was gradually decreased resulted in phospho-CREB declining after miR-372 induction, by which lead to reduction of promoter activity from Luc-HCP (Figure 9A and B), the ChIP results showed that 0.5 h after miR-372 induction, binding of CREB protein began to decrease with the bottom level reached at 4 h (Figure 9C), followed by declining of CBP protein began at 1 h with the bottom level at 24 h (Figure 9D). Consistent with the kinetics of HULC transcription (Figure 9E), an increase in nuclease protection level was observed with Sac I digestion using R7 primer sets as early as 1 h and gradually increased during the whole detection period (Figure 9F). Collectively, local dissociation of CBP by CREB protein under miR-372-induced conditions reduces the level of chromatin opening of the core promoter, and thus decreases expression of HULC.
Figure 9.

Over-expression of miR-372 inhibits Hulc promoter activity. (A) Luciferase assays indicated that miR-372 could reduce promoter activity of Luc-HCP in Hep3B cells compared to that of HL-7702 cells. Firefly luciferase activity was normalized to Renilla luciferase activity, and the relative luciferase activities are presented as fold increase over the promoter-less pGL3-basic vector. *P < 0.01 versus control cells. (B) Immunoblotting assays indicated that over-expression of miR-372 could gradually down-regulation of PRKACB, thus reduce phosphorylation of CREB in Hep3B cells. Chromatin from Hep3B and HL-7702 cells was harvested 0, 0.5, 1, 2, 3, 4 and 24 h and precipitated with anti-CREB (C) and anti-CBP (D) antibodies. After DNA recovery, the precipitates were evaluated by real-time PCR for the level of enrichment over the negative control antibody. (E) Down-regulation of HULC expression by miR-372 (50 nM) treatment in Hep3B cells compared to that of HL-7702 cells for the indicated periods of time after replacement of transfection medium containing 50 nM miR-372 mimics with 5% FBS. (F) Nuclei were harvested 0, 0.5, 1, 2 and 24 h from Hep3B and HL-7702 cells. Then digested with Sac I. Purified DNA was quantitated by real-time PCR and relative to DNA from undigested nuclei using the primers compassing R7 region and listed as percent protected. *P < 0.05, **P < 0.01 versus cells harvested at 0 h (A and C–F). All the results are the means of three independent experiments ± SD.
DISCUSSION
In our study, we first identified the mechanism by which CREB up-regulates the expression of lncRNA, HULC in a cancer-specific manner and found the specific interaction of CREB with the Hulc promoter through its binding site located between −67 and −53 nt (Figures 3 and 4). We show that in this core promoter (i) phospho-CREB binding at its binding site through PKA pathway is crucial for Hulc promoter activity in liver cancer and (ii) phospho-CREB binding may result in an open chromatin configuration associated with local histone. Therefore, the present study delineates the fundamental elements of a core promoter structure that will be helpful for future studies on the differential regulation of Hulc expression in liver.
CREB is a ubiquitous transcription factor that activates the transcriptional activity of various promoters through its binding site (36). A typical sequence of events is as follows: the catalytic subunit of activated protein kinase A, including PRKACB (the inactive holoenzyme of PKA is a tetramer composed of two regulatory and two catalytic subunits. cAMP causes the dissociation of the inactive holoenzyme into a dimer of regulatory subunits bound to four cAMP and two free monomeric catalytic subunits.) translocates to the cell nucleus, where it phosphorylated CREB (37). The activated CREB protein binds to a CRE (cAMP response element) region, and is then bound to by CBP, which coactivates it, allowing it to switch certain genes on or off (38,39). Additionally, CREB may create a core promoter architecture that is suitable for assembly of a pre-initiation complex (40). In our study, using PKA inhibitor, H89 could reduce both promoter activity and expression of HULC in liver cancer cells (Figure 4). Furthermore, knockdown of CREB expression resulted in an inhibition of the promoter activation as well as endogenous HULC transcription level (Figure 5). Therefore, we show directly that phospho-CREB is crucial for Hulc promoter activity in liver cancer cells. The association of phospho-CREB protein with its binding site contained in this region may result in the recruitment of histone acetyltransferases (such as P300 and CBP) to the promoter, leading to the acetylation of the histone tail and maintaining the open configuration of the local chromatin. Furthermore, such events occurring at the proximal promoter provide chromatin accessibility to polymerase II to initiate the transcription of the Hulc gene (Figure 5).
SWI/SNF complex has the ability of mobilizing nucleosomes and remodeling chromatin (21,41). In comparison to normal liver cells, the SWI/SNF complex was found to be associated specifically with the proximal promoter region in cancer cells (Figure 5). It is sufficient to yield a state of open configuration of the local chromatin through releasing nucleosome from the CREB binding site. This primes for an increase in the HULC transcription level by facilitating of CREB enrichment, and it can further recruit P300. The putative P300 and CREB complex may up-regulate the H3 and H4 acetylation levels that are responsible for the initiation and maintenance of enhanced local chromatin accessibility to the general transcription apparatus, thus up-regulates the HULC expression level (Figure 2).
In this study, when we tried to delineate the Hulc promoter in liver cancer and normal cell lines, a conventional method, luciferase reporter assay was used. After transient transfection of the luciferase reporter containing the proximal promoter, the reporter plasmids showed no significant increasing activity in cancer cell lines compare with normal cell lines (Figure 3). We think that, unlike stably transfected plasmids or episomal constructs, the chromatin structure of transiently transfected plasmids is usually incomplete or aberrant, and may vary with cell type and the transfection method used. We postulate that in our transient transfection experiment, the proximal promoter region had not been packaged into a chromatin-like structure, and may even have been in a ‘naked’ state, and as a result, CREB binding site and the TSS may not have been occupied by nucleosomes, leading to high accessibility to various nuclear factors. Hence, cancer status cannot exert its regulatory role through remodeling the chromatin structure, and up-regulated promoter activity was undetectable in our luciferase reporter assay. Therefore, it is important that the promoter activity is studied in its native chromatin context.
RNA is not only a messenger operating between DNA and protein. Although lncRNAs are among the least well-understood of transcript species, they cannot all be dismissed as merely transcriptional ‘noise’. At least for the exception of HULC that: (i) its expression varies from different cell lines (Figure 1B); and, (ii), its expression can be up-regulated in liver cancer (Figure 1A). As previous studies described that lncRNA sequences convey functions through binding to DNA or protein. However, in this work, we provide additional evidence of lncRNA as an inhibitor to short ncRNA, miRNAs in liver cancer, in other contexts; HULC can also exert its functions through interaction with other RNAs (Figure 7). In our hands, depletion of HULC by specific siRNA enhanced miRNAs activities not through up-regulating miRNAs expression but instead reducing its ability of sequestering miRNAs at most cases. Franco-Zorrilla et al. (32) reported that a non-protein coding RNA, IPS1 from A. thaliana contains a motif with sequence complementary to the phosphate (Pi) starvation-induced miRNA, miR-399, however, IPS1 RNA is not cleaved but instead sequesters miR-399. Thus, IPS1 over-expression results in increased accumulation of the miR-399 target PHO2 mRNA and, concomitantly, in reduced shoot Pi content. Ebert et al. (34) developed competitive miRNA inhibitors, termed ‘miRNA sponges’ containing multiple, tandem binding sites to miRNAs of interest, by which can block an entire miRNA seed family. By the information collected above, we think HULC may act as an endogenous ‘miRNA sponge’, which regulates miRNAs activities in liver cancer cells. Apart from the above mentioned, not only activities but also expression levels of miR-372 and miR-613 can be affected by HULC, indicates there exists another pathway involved in HULC inhibitory to miRNAs. Recently, a family of exoribonucleases encoded by the small RNA degrading nuclease genes degrade mature miRNAs were found in Arabidopsis (42). Besides, miRNAs that are partially complementary to the target can also speed up deadenylation, causing miRNAs to be degraded sooner. Binding of miR-372 and miR-613 to their seed regions in HULC may stimulate such nuclease or facilitate miRNA deadenylation results in degrading of miRNAs (43). Further studies are required to fully define such mechanism. Furthermore, we demonstrated that through pairing with partially complementary sites in 3′UTRs, miR-372 mediate post-transcriptional silencing of PRKACB, which finally affects phosphorylation of CREB in liver cancer.
Upon miR-372 treatment, gradually reduction of PRKACB through its miR-372 binding site in the 3′UTR accompanied with dephosphorylation of CREB (Figure 9). However, the PRKACB level was transiently up-regulated followed by induced phosphorylation of CREB 2 h after induction, but still lower than the basal level (Figure 9B), may due to compensation of the loss of PRKACB. Because PRKACB plays an important role in maintaining the normal function of cells (14), transient up-regulating the level of PRKACB may get more chances of survival. A decrease in CREB and CBP binding to the promoter is apparent at 30 min after miR-372 treatment (Figure 9C and D), which makes the chromatin from the proximal promoter region begin to close after 1 h of induction (Figure 9F). Such dissociation is slowly, gradually decreasing during the entire detection time—it is sufficient to yield a state of close configuration of the local chromatin through recruiting nucleosome to the TSS. This primes for a gradual decrease in the HULC transcription level by masking the CREB-binding sites, and in turn further reduces CREB and CBP binding to the promoter.
The regulatory circuitry described here provides another example of how gene reprogramming during tumorigenesis relies on fine modulation of gene expression. It is also interesting to note that fine tuning of HULC expression is part of an autoregulatory loop in which it’s inhibitory to expression and activity of miR-372 allows the establishment of the lncRNA up-regulated expression in liver cancer. CREB may be the link for up-regulation of HULC (Figure 1A) and down-regulation of miR-372 (Figure 6C) in HCC. HULC inhibitory to miR-372 reduces miRNA mediated translational repression of PRKACB, which can induce phosphorylation of CREB, in turn, stimulates HULC expression (Figure 10). Notably, the relevant function of HULC up-regulation would be not only increase of PRKACB mediated functions, which appear quite crucial, but also other putative substrates not yet tested, possibly important for cell-lineage specification. Besides, it is also important to mention that the activation of CREB, to a certain extent, may be the initial event of up-regulating HULC expression, because HULC is widely expressed, including liver cancer and normal cells (Figure 1B), however, HULC does not exert its function of inhibitory to miR-372 in normal cells as obvious as that in cancer cells. Environments in cancer cells may different from that in normal cells. Stimuli involved in the PKA pathway that can induce phosphorylation of CREB have been excellently reviewed elsewhere (44). Once HULC is up-regulated, miR-372 can be rapidly absorbed to the putative binding site in HULC and then may be cleared, by such event, enhances HULC expression and further inhibition to miR-372. Collectively, we think reduction of miR-372 is not only the result, but also a driving factor for greater suppression of HULC to other miRNAs, although more details need to be elucidated.
Figure 10.

Possible molecular mechanism underlying interaction between HULC and miR-372.
In conclusion, in this work we demonstrated that CREB up-regulates the HULC expression level by interaction with miR-372, which in turn initiates a cascade of molecular events to increase the chromatin accessibility for the general transcription in liver cancer.
FUNDING
Natural Science Foundation of Shanghai (grant numbers 0752NM028 and 09DZ1950100) and National Science Foundation of China (grant numbers 30600328 and 30740030). Funding for open access charge: Natural Science Foundation of Shanghai (grant number 09DZ1950100).
Conflict of interest statement. None declared.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
ACKNOWLEDGEMENTS
We sincerely thank Dr Xinmin Zheng for critical comments on the manuscript and Mr Zanlin Yu for carefully language editing the manuscript.
REFERENCES
- 1.Ponting CP, Oliver PL, Reik W. Evolution and functions of long nonocoding RNAs. Cell. 2009;136:629–641. doi: 10.1016/j.cell.2009.02.006. [DOI] [PubMed] [Google Scholar]
- 2.Wang J, Zhang J, Zheng H, Li J, Liu D, Li H, Samudrala R, Yu J, Wong GK. Mouse transcriptome: neutral evolution of ‘non-coding’ complementary DNAs. Nature. 2004;431:757–761. [PubMed] [Google Scholar]
- 3.Lau NC, Lim LP, Weinstein EG, Bartel DP. An abundant class of tiny RNAs with probable regulatory roles in Caenorhabditis elegans. Science. 2001;294:858–862. doi: 10.1126/science.1065062. [DOI] [PubMed] [Google Scholar]
- 4.Lagos-Quintana M, Rauhut R, Lendeckel W, Tuschl T. Identification of novel genes coding for small expressed RNAs. Science. 2001;294:853–858. doi: 10.1126/science.1064921. [DOI] [PubMed] [Google Scholar]
- 5.Lee RC, Ambros V. An extensive class of small RNAs in Caenorhabditis elegans. Science. 2001;294:862–864. doi: 10.1126/science.1065329. [DOI] [PubMed] [Google Scholar]
- 6.Mattick JS. Non-coding RNAs: the architects of eukaryotic complexity. EMBO Rep. 2001;2:986–991. doi: 10.1093/embo-reports/kve230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Costa FF. Non-codig RNAs: new players in eukaryotic biology. Gene. 2005;357:83–94. doi: 10.1016/j.gene.2005.06.019. [DOI] [PubMed] [Google Scholar]
- 8.Lu J, Getz G, Miska EA, Alvarez-Saavedra E, Lamb J, Peck D, Sweet-Cordero A, Ebert BL, Mak RH, Ferrando AA, et al. MicroRNA expression profiles classify human cancers. Nature. 2005;435:834–838. doi: 10.1038/nature03702. [DOI] [PubMed] [Google Scholar]
- 9.Bentwich I, Avniel A, Karov Y, Aharonov R, Gilad S, Barad O, Barzilai A, Einat P, Einav U, Meiri E, et al. Identification of hundreds of conserved and nonconserved human microRNAs. Nat. Genet. 2005;37:766–770. doi: 10.1038/ng1590. [DOI] [PubMed] [Google Scholar]
- 10.Jopling CL, Yi M, Lancaster AM, Lemon SM, Sarnow P. Modulation of hepatitis C virus RNA abundance by a liver-specific MicroRNA. Science. 2005;309:1577–1581. doi: 10.1126/science.1113329. [DOI] [PubMed] [Google Scholar]
- 11.Sun F, Wang J, Pan Q, Yu Y, Zhang Y, Wan Y, Wang J, Li X, Hong A. Characterization of function and regulation of miR-24-1 and miR-31. Biochem. Biophys. Res. Commun. 2009;380:660–665. doi: 10.1016/j.bbrc.2009.01.161. [DOI] [PubMed] [Google Scholar]
- 12.Bosch FX, Ribes J, Cléries R, Díaz M. Epidemiology of hepatocellular carcinoma. Clin. Liver Dis. 2005;9:191–211. doi: 10.1016/j.cld.2004.12.009. [DOI] [PubMed] [Google Scholar]
- 13.Panzitt K, Tschernatsch MM, Guelly C, Moustafa T, Stradner M, Strohmaier HM, Buck CR, Denk H, Schroeder R, Trauner M, et al. Characterization of HULC, a novel gene with striking up-regulation in hepatocellular carcinoma, as noncoding RNA. Gastroenterology. 2007;132:330–342. doi: 10.1053/j.gastro.2006.08.026. [DOI] [PubMed] [Google Scholar]
- 14.Matouk IJ, Abbasi I, Hochberg A, Galun E, Dweik H, Akkawi M. Highly upregulated in liver cancer noncoding RNA is overexpressed in hepatic colorectal metastasis. Eur. J. Gastroenterol. Hepatol. 2009;21:688–692. doi: 10.1097/meg.0b013e328306a3a2. [DOI] [PubMed] [Google Scholar]
- 15.Wu KJ, Mattioli M, Morse HC, Dalla-Favera R. c-MYC activates protein kinase A (PKA) by direct transcriptional activation of the PKA catalytic subunit beta (PKA-C beta) gene. Oncogene. 2002;21:7872–7882. doi: 10.1038/sj.onc.1205986. [DOI] [PubMed] [Google Scholar]
- 16.Rao S, Procko E, Shannon MF. Chromatin remodeling, measured by a novel real-time polymerase chain reaction assay, across the proximal promoter region of the IL-2 Gene. J. Immunol. 2001;167:4494–4503. doi: 10.4049/jimmunol.167.8.4494. [DOI] [PubMed] [Google Scholar]
- 17.Sun F, Xie Q, Ma J, Yang S, Chen Q, Hong A. Nuclear factor Y is required for basal activation and chromatin accessibility of fibroblast growth factor receptor 2 promoter in osteoblast-like cells. J. Biol. Chem. 2009;284:3136–3147. doi: 10.1074/jbc.M808992200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xue X, Sun J, Zhang Q, Wang Z, Huang Y, Pan W. Identification and characterization of novel microRNAs from Schistosoma japonicum. PLoS ONE. 2008;3:e4034. doi: 10.1371/journal.pone.0004034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hurteau GJ, Carlson JA, Spivack SD, Brock GJ. Overexpression of the microRNA hsa-miR-200c leads to reduced expression of transcription factor 8 and increased expression of E-cadherin. Cancer Res. 2007;67:7972–7976. doi: 10.1158/0008-5472.CAN-07-1058. [DOI] [PubMed] [Google Scholar]
- 20.Lee Y, Kim M, Han J, Yeom KH, Lee S, Baek SH, Kim KN. MicroRNA genes are transcribed by RNA polymerase II. EMBO J. 2004;23:4051–4060. doi: 10.1038/sj.emboj.7600385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sun F, Chen Q, Yang S, Pan Q, Ma J, Wan Y, Chang CH, Hong A. Remodeling of chromatin structure within the promoter is important for bmp-2-induced fgfr3 expression. Nucleic Acids Res. 2009;37:3897–3911. doi: 10.1093/nar/gkp261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Spencer VA, Davie JR. Role of covalent modifications of histones in regulating gene expression. Gene. 1999;240:1–12. doi: 10.1016/s0378-1119(99)00405-9. [DOI] [PubMed] [Google Scholar]
- 23.Tse C, Sera T, Wolffe AP, Hansen JC. Disruption of higher-order folding by core histone acetylation dramatically enhances transcription of nucleosomal arrays by RNA polymerase III. Mol. Cell Biol. 1998;18:4629–4638. doi: 10.1128/mcb.18.8.4629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shilatifard A. Molecular implementation and physiological roles for histone H3 lysine 4 (H3K4) methylation. Curr. Opin. Cell Biol. 2008;20:341–348. doi: 10.1016/j.ceb.2008.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vakoc C, Mandat S, Olenchock B, Blobel G. Histone H3 lysine 9 methylation and HP1γ are associated with transcription elongation through mammalian chromatin. Mol. Cell. 2005;19:381–391. doi: 10.1016/j.molcel.2005.06.011. [DOI] [PubMed] [Google Scholar]
- 26.Du K, Montminy M. CREB is a regulatory target for the protein kinase Akt/PKB. J. Biol. Chem. 1998;273:32377–32379. doi: 10.1074/jbc.273.49.32377. [DOI] [PubMed] [Google Scholar]
- 27.Chrivia JC, Kwok RP, Lamb N, Hagiwara M, Montminy MR, Goodman RH. Phosphorylated CREB binds specifically to the nuclear protein CBP. Nature. 1993;365:855–859. doi: 10.1038/365855a0. [DOI] [PubMed] [Google Scholar]
- 28.Gonzalez GA, Montminy MR. Cyclic AMP stimulates somatostatin gene transcription by phosphorylation of CREB at Serine 133. Cell. 1989;59:675–680. doi: 10.1016/0092-8674(89)90013-5. [DOI] [PubMed] [Google Scholar]
- 29.Chen AE, Ginty DD, Fan CM. Protein kinase A signalling via CREB controls myogenesis induced by Wnt proteins. Nature. 2005;433:317–322. doi: 10.1038/nature03126. [DOI] [PubMed] [Google Scholar]
- 30.Li Y, Yin W, Wang X, Zhu W, Huang Y, Yan G. Cholera toxin induces malignant glioma cell differentiation via the PKA/CREB pathway. Proc. Natl Acad. Sci. USA. 2007;104:13438–13443. doi: 10.1073/pnas.0701990104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Leemhuis J, Boutillier S, Schmidt G, Meyer DK. The protein kinase A inhibitor H89 acts on cell morphology by inhibiting Rho kinase. J. Pharmacol. Exp. Ther. 2002;300:1000–1007. doi: 10.1124/jpet.300.3.1000. [DOI] [PubMed] [Google Scholar]
- 32.Franco-Zorrilla JM, Valli A, Todesco M, Mateos I, Puga MI, Rubio-Somoza I, Leyva A, Weigel D, García JA, Paz-Ares J. Target mimicry provides a new mechanism for regulation of microRNA activity. Nat. Genet. 2007;39:1033–1037. doi: 10.1038/ng2079. [DOI] [PubMed] [Google Scholar]
- 33.Krutzfeldt J, Rajewsky N, Braich R, Rajeev KG, Tuschl T, Manoharan M, Stoffel M. Silencing of microRNAs in vivo with ‘antagomirs’. Nature. 2005;438:685–689. doi: 10.1038/nature04303. [DOI] [PubMed] [Google Scholar]
- 34.Ebert MS, Neilson JR, Sharp PA. MicroRNA sponges: competitive inhibitors of small RNAs in mammalian cells. Nat. Methods. 2007;4:721–726. doi: 10.1038/nmeth1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Voorhoeve P, le Sage C, Schrier M, Gillis A, Stoop H, Nagel R, Liu Y, van Duijse J, Drost J, Griekspoor A. A Genetic screen implicates miRNA-372 and miRNA-373 as oncogenes in testicular germ cell tumors. Cell. 2006;124:1169–1181. doi: 10.1016/j.cell.2006.02.037. [DOI] [PubMed] [Google Scholar]
- 36.Mayr B, Montminy M. Transcriptional regulation by the phosphorylation-dependent factor CREB. Nat. Rev. Mol. Cell Biol. 2001;2:599–609. doi: 10.1038/35085068. [DOI] [PubMed] [Google Scholar]
- 37.Delghandi MP, Johannessen M, Moens U. The cAMP signalling pathway activates CREB through PKA, p38 and MSK1 in NIH 3T3 cells. Cell Signal. 2005;17:1343–1351. doi: 10.1016/j.cellsig.2005.02.003. [DOI] [PubMed] [Google Scholar]
- 38.Du K, Montminy M. CREB is a regulatory target for the protein kinase Akt/PKB. J. Biol. Chem. 1998;273:32377–32379. doi: 10.1074/jbc.273.49.32377. [DOI] [PubMed] [Google Scholar]
- 39.Chrivia JC, Kwok RP, Lamb N, Hagiwara M, Montminy MR, Goodman RH. Phosphorylated CREB binds specifically to the nuclear protein CBP. Nature. 1993;365:855–859. doi: 10.1038/365855a0. [DOI] [PubMed] [Google Scholar]
- 40.Narayan S, Beard WA, Wilson SH. DNA damage-induced transcriptional activation of a human DNA polymerase beta chimeric promoter: recruitment of preinitiation complex in vitro by ATF/CREB. Biochemistry. 1995;34:73–80. doi: 10.1021/bi00001a009. [DOI] [PubMed] [Google Scholar]
- 41.Wang X, Sansam CG, Thom CS, Metzger D, Evans JA, Nguyen PT, Roberts CW. Oncogenesis caused by loss of the SNF5 tumor suppressor is dependent on activity of BRG1, the ATPase of the SWI/SNF chromatin remodeling complex. Cancer Res. 2009;69:8094–8101. doi: 10.1158/0008-5472.CAN-09-0733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ramachandran V, Chen X. Degradation of microRNAs by a family of exoribonucleases in Arabidopsis. Science. 2008;5895:1490–1492. doi: 10.1126/science.1163728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lu S, Sun YH, Chiang VL. Adenylation of plant miRNAs. Nucleic Acids Res. 2009;37:1878–1885. doi: 10.1093/nar/gkp031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Johannessen M, Delghandi MP, Moens U. What turns CREB on? Cell Signal. 2004;16:1211–1227. doi: 10.1016/j.cellsig.2004.05.001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.