


Abstract
Although the last few years have seen great progress in DNA sequence retrieval from fossil specimens, some of the characteristics of ancient DNA remain poorly understood. This is particularly true for blocking lesions, i.e. chemical alterations that cannot be bypassed by DNA polymerases and thus prevent amplification and subsequent sequencing of affected molecules. Some studies have concluded that the vast majority of ancient DNA molecules carry blocking lesions, suggesting that the removal, repair or bypass of blocking lesions might dramatically increase both the time depth and geographical range of specimens available for ancient DNA analysis. However, previous studies used very indirect detection methods that did not provide conclusive estimates on the frequency of blocking lesions in endogenous ancient DNA. We developed a new method, polymerase extension profiling (PEP), that directly reveals occurrences of polymerase stalling on DNA templates. By sequencing thousands of single primer extension products using PEP methodology, we have for the first time directly identified blocking lesions in ancient DNA on a single molecule level. Although we found clear evidence for blocking lesions in three out of four ancient samples, no more than 40% of the molecules were affected in any of the samples, indicating that such modifications are far less frequent in ancient DNA than previously thought.
INTRODUCTION
Improvements in sequencing technology and other methodological advances have greatly increased the scope of sequence retrieval from ancient DNA over the last years, enabling both whole genome sequencing (1–3) and large scale targeted re-sequencing (4–6) using degraded DNA from fossils. However, ancient DNA research is still hampered by DNA damage that accumulates after death of an organism. Such damage can be classified into three general categories: (i) strand breaks, which result in short DNA fragments, (ii) miscoding lesions, which lead to sequence errors by causing nucleotide misincorporations during DNA amplification, and (iii) blocking lesions, which are DNA modifications that prevent polymerase bypass, and thus amplification and sequencing.
Encouragingly, problems resulting from strand breaks and miscoding lesions have been greatly alleviated in recent years. High-throughput sequencing platforms have enabled rapid sequencing of short DNA fragments (7,8), and studies have identified that most miscoding lesions result from deamination of cytosine to uracil (9,10), which can be removed from DNA by enzymatic treatment (11). However, in contrast to strand breaks and miscoding lesions, which are revealed by fragment endpoints or errors in ancient DNA sequences, blocking lesions entirely prevent sequencing and are therefore less well understood. This is despite the fact that modifications likely to act as blocking lesions have long been presumed to exist in ancient DNA. For example, in the earliest study on the characteristics of ancient DNA in 1989 (12), it was found that ancient DNA is susceptible to alkali treatment and various DNA repair enzymes, arguing for the presence of modified base and sugar residues as well as abasic sites. In the same study, HPLC profiling indicated that up to 95% of all thymines might be modified or lost in ancient DNA. A later study identified several oxidative base modifications by gas chromatography and mass spectrometry (13), including hydantoins, the abundance of which was correlated with the inability to retrieve DNA sequences from some samples. A third study assayed the refractivity of ancient soil DNA from permafrost cores to heat denaturation (14) and identified inter-strand cross-links as the most abundant type of DNA damage, affecting on average every second base already in relatively young samples (19 000 years).
Since most of the above modifications are expected to act as blocking lesions for DNA polymerases, these studies point toward the existence of a large fraction of DNA molecules that are present but not amenable to amplification and sequencing. Rescue of these damaged molecules might allow more genetic data to be obtained from high-quality samples or even the retrieval of DNA sequences from particularly old or degraded samples that fail to yield DNA sequences using current techniques. This notion has spurred the engineering of new polymerases for the amplification of ancient DNA (15,16), which can bypass abasic sites and other base modifications. However, when actually applied to ancient DNA in one of the studies, gains in amplification success were small at best (15), indicating either insufficient performance of the enzymes or a low frequency of blocking lesions in ancient DNA. Thus, determining the types and frequencies of blocking lesions in ancient DNA more directly would be highly desirable for the development of more efficient rescue strategies. More importantly, the question of whether or not ancient DNA samples contain a substantial amount of currently unamplifiable material has important implications for future prospects or limits of ancient DNA research.
To determine the frequency of blocking lesions in ancient DNA, we have developed a new high-throughput sequencing-based method called polymerase extension profiling (PEP). PEP allows the direct detection of polymerase-blocking DNA damage on a single molecule level by revealing the places on sequence templates where polymerase extensions are aborted (Figure 1). First, an asymmetrical biotinylated adapter is ligated to both ends of ancient template molecules. After ligation, each template strand carries one adapter sequence at the 3′-end, which serves as the priming site for a subsequent primer extension reaction, and another short adapter sequence and a biotin at the 5′-end, which serves as an end-of-template recognition sequence. Primer extension proceeds either through the molecule to the end of the recognition sequence or until a blocking lesion or a nick is encountered on the template strand. Extension products are then captured on streptavidin beads and all products from ancient molecules with nicks or incompletely ligated adapters are washed away. All other products are bound to the beads, since they remain base-paired with biotinylated templates. Captured extension products are released from the beads by heat denaturation and then converted into a 454 sequencing library by single-strand ligation of a second adapter to their 3′-ends. Sequences from undamaged templates will carry the end-of-template recognition sequence, whereas the absence of the recognition sequence will indicate templates with blocking lesions. Similar to the standard 454 library preparation (7), PEP excludes all molecules with nicks or end modifications that prevent end-repair or ligation from sequencing. However, PEP recovers molecules with blocking lesions, such as abasic sites, hydantoins and inter-strand cross-links, which have previously been inaccessible to DNA sequencing.
Figure 1.

Workflow of PEP. Double-stranded template DNA is blunt-end repaired using T4 DNA polymerase and T4 polynucleotide kinase (not depicted). (I) Using T4 DNA ligase, biotinylated adapters are attached to both ends of the template molecules. The blunt end ligation reaction also produces adapter dimers, which are subsequently removed by size selective purification. (II) 5′-tailed primers carrying the 454 ‘B’ sequence (shown in blue) are hybridized to the overhanging 3′-ends of the adapters. Primer extension is carried out under reaction conditions optimal for the assayed polymerase. Unless second-strand synthesis stops prematurely, due to a blocking lesion, a nick or random polymerase stalling, the flanking adapter sequence (shown in red) is copied. (III) Primer extension products are captured on streptavidine beads to remove excess primers and extension products from nicked template strands. Extension products are released by heat denaturation. (IV) A 454 sequencing library is created by attaching single-stranded adapters with the 454 ‘A’ sequence (shown in green) to the 3′-ends. The sequencing library is converted to double-stranded form (not depicted) to allow for efficient removal of excess A-adapters. The 454 sequencing is initiated from the A-adapter. If primer extensions were complete, sequences will start with an 8-bp adapter sequence, which serves as the end-of-template recognition sequence (framed by rectangles).
In contrast to previous studies of blocking lesions in ancient DNA (12–14), which used analytic chemistry or enzymatic reactions to obtain composite data from millions of molecules, PEP is sequencing-based and hence provides a single data point for each molecule. This is particularly important for ancient DNA samples, which are usually dominated by contaminating environmental DNA that may greatly differ from the endogenous DNA in age and degree of damage. The source organism of each PEP sequence can be identified from sequence alignments and the presence or absence of the recognition sequence determines whether the original template molecule carried a blocking lesion.
MATERIALS AND METHODS
Template preparation
To obtain artificial test templates, 12 75-bp PCR products were created by amplifying two short regions of pUC19 plasmid DNA in a standard PCR assay using AmpliTaq Gold polymerase (Applied Biosystems). Modified primers were used to introduce a uracil into one strand of each PCR product. In addition, six different 5′-barcodes were attached to all primers to allow for efficient use of the sequencing capacity through pooling (17). All PCR products were purified using the MinElute PCR purification kit (Qiagen) and quantified on a Nanodrop ND-1000 spectrophotometer (Nanodrop Technologies). Products with identical barcodes were pooled, and half of the pools were incubated with 20 U uracil DNA glycosylase (UDG) for 30 min at 37°C in 100 µl reactions containing 500 ng DNA and 1× UDG buffer (NEB) in order to convert uracils into abasic sites. The reactions were terminated by purification with the MinElute PCR purification kit (Qiagen). DNA was eluted in a 0.1× TE buffer (1 mM Tris−HCl, 0.1 mM EDTA, pH 8.0), quantified and pooled further. See Supplementary Table S1 for all primer sequences and the pooling scheme.
Genomic horse DNA was extracted from fresh blood using the Genfind v2 DNA purification kit (Agencourt). Thirty micrograms of DNA were enzymatically fragmented in three sets of ten 100 µl reactions, each containing 1 µg of DNA, 0.02 U DNAse I (Fermentas), 1× DNAse buffer and 10 mM MnCl2. Each set was incubated at 15°C for 6, 8 and 10 min, respectively. Reactions were terminated by the addition of 500 µl PBI buffer and purified using the MinElute PCR purification kit (Qiagen). Subsequently, the fragmented DNA from all reactions was pooled, concentrated with a Microcon YM-10 column (Millipore) and separated on a preparative 2% agarose gel. DNA was visualized on a Dark Reader Transilluminator (Clare Chemical Research) to avoid damage from UV irradiation. Two narrow slices, below and above 100 bp, were cut from the gel and dissolved at room temperature in the QC buffer (Qiagen). DNA was extracted using the QIAquick gel extraction kit (Qiagen).
UV-damaged genomic DNA was obtained by exposing 4 ng of fragmented horse DNA (fraction 1, <100 bp), diluted in 7 mM MgCl2, to a dose of 120 mJ/cm2 UV-light (254 nm) in a CL-1000 UV-Crosslinker (Clare Chemical Research) in an open tube. The DNA was then purified using the MinElute PCR purification kit (Qiagen) and eluted in 0.1× TE.
Ancient DNA extracts from four samples were, alongside with mock extraction controls, prepared in a dedicated clean room using a silica-based method (18). See Supplementary Table S2 for sample information. Extracts were stored at −20°C and used within 2 weeks after extraction.
PEP assays and sequencing
For blunt end repair, ∼15 ng of PCR product pool, 2 ng of fragmented horse DNA, 4 ng of UV-irradiated horse DNA, 10 µl ancient DNA extract or a water sample were incubated for 15 min at 12°C and 15 min at 25°C in a 40 µl reaction containing in final concentrations 1× Tango buffer, 0.1 U/µl T4 DNA polymerase, 0.5 U/µl T4 polynucleotide kinase (all Fermentas), 1 mM ATP and 0.1 mM dNTP. Reactions were purified using the MinElute PCR Purification kit. DNA was eluted in 20 µl EB (Qiagen).
An adapter (B-BL) was generated by annealing two partially complementary oligonucleotides (B-BL1 and B-BL2; see Supplementary Table S3 for the sequences) in a reaction containing 1× T4 ligase buffer (Fermentas) and 200 µM of each oligonucleotide. The reaction was incubated for 10 s at 95°C and slowly cooled to 8°C decreasing the temperature by 0.1°C/s. Adapter B-BL was subsequently ligated to both ends of the template in a 40 µl reaction containing 0.5 µl B-BL adapter, 20 µl end-repaired DNA and in final concentrations 1× T4 Ligase buffer, 5% PEG-4000 and 0.1 U/µl T4 ligase (Fermentas). The reaction was incubated for 1 h at 22°C.
Since both template and adapter molecules are 5′-phosphorylated, ligation generates an excess of adapter dimers in addition to the desired adapter-ligated templates. Adapter dimers were removed by repeated size selective purification with SPRI beads (19) using the AMPure PCR purification kit (Agencourt). The manufacturer’s instructions were followed, except that the bead suspension/sample volume ratio was changed to 3.5. As experimentally determined, this ratio retains adapter-ligated templates of 60 bp, while adapter-ligated 40-bp templates are removed. Thus, this treatment excludes double-stranded starting molecules shorter than 40−60 bp from sequencing. DNA was eluted in 20 µl 0.1× TET (0.1× TE, 0.05% Tween-20).
Single-primer extensions were carried out using different polymerase/buffer systems in 25 µl reactions, containing 10 µl adapter-ligated DNA and 200 nM of the extension primer EP (see Supplementary Table S4 for detailed reaction conditions).
To remove excess primers and extension products from nicked or not fully adapter-ligated templates, the single-primer extension products were purified using streptavidin beads. To avoid non-specific binding of DNA to tube walls or beads, we used siliconized tubes and added Tween-20 to all wash buffers. Twenty-five microliters of Dynabeads MyOne Streptavidin C1 (Invitrogen) were washed twice in a 2× BWT buffer (2 M NaCl, 10 mM Tris−HCl pH 8.0, 1 mM EDTA, 0.05% Tween-20) and resuspended in a 25 µl 2× BWT buffer. After adding the single-primer extension reaction, the mixture was incubated for 15 min at room temperature. Subsequently, the beads were separated using a magnet and the supernatant was discarded. The beads were then four times washed in a 1× PCR buffer II (Applied Biosystems) with 2.5 mM MgCl2 and incubated at 60°C for 3 min after each resuspension. To elute the bound DNA, the beads were resuspended in a 12 µl 1× TET and heated to 95°C for 30 s in a thermal cycler. Using a magnet, the supernatant was separated and purified using the nucleotide removal kit (Qiagen). The DNA was eluted in 10 µl 0.1× TE. Prior to performing PEP-assays, we tested this purification method using olignucleotides of different lengths as test templates. We did not detect length-dependent differences in the recovery of oligonucleotides longer than 20 bases, which is shorter than the primer (39 bases) that was used in the single-primer extension reaction. Thus, the recovery of extension products should be independent of their length.
A second adapter, A-BL was ligated to the 3′-ends of the newly synthesized strands using an RNA-ligase with high efficiency for a single-strand DNA ligation (20). We adopted reaction conditions suggested in a previous study (21), which reported end-to-end ligation efficiencies close to 100%. Reactions were carried out in 40 µl reactions containing 10 µl purified DNA, 200 U CircLigase (Epicentre) and in final concentrations 500 nM adapter A-BL, 1× CircLigase buffer, 25 µM ATP, 2.5 mM MnCl2 and 20% PEG-6000. The reaction was incubated for 30 min at 60°C and then purified using the nucleotide removal kit. DNA was eluted in 10 µl 0.1× TE.
To remove excess A-BL adapters, the single-stranded DNA was converted into a double-stranded 454 sequencing library by second strand synthesis in a 25 µl reaction containing 10 µl sample, 2 U AmpliTaq Gold DNA polymerase and in final concentrations 500 nM primer emPCR-F, 1× PCR buffer II, 1.5 mM MgCl2 and 0.25 mM dNTP. The reaction profile comprised an activation step lasting 10 min at 95°C, followed by a primer annealing step at 60°C for 1 min and an elongation step at 72°C for 1 min. The product was then purified using SPRI beads with a bead suspension/sample volume ratio of 3.5. This ratio is not size selective. Thus, library molecules of the size of adapter dimers or larger are recovered with the same efficiency. The final 454 sequencing library was eluted in 10 µl 0.1× TE.
Sequencing libraries were quantified using quantitative PCR (22) with modifications to the original protocol described elsewhere (5). Quantity estimates derived from ancient DNA mock extracts were indistinguishable from no-template PEP assays, indicating the absence of contamination at a detectable level (see Supplementary Table S5).
All libraries were sequenced on small plate regions (1/16th) of the Genome Sequencer FLX (Roche/454) with the exception of libraries from PEP assays with PCR products, which were combined into two pools and sequenced on two plate regions each. Beads from different PEP-libraries were never loaded onto adjacent plate regions. Non-standard filter settings were used as described previously (22) to allow the retrieval of short sequence reads. Trimming of B-adapter sequences was turned off.
Quality filtering, artifact removal
Since the read length of the GS FLX platform (∼250 bp) is longer than the fragment size of PEP-templates (75 bp PCR products and highly fragmented modern and ancient genomic DNA), each read should start with the full sequence of a primer extension product (starting with the end-of-template recognition sequence) and end with the full 454-B-adapter sequence, unless reads were quality-trimmed by the 454 base-calling software. To exclude quality-trimmed reads, the first 25 bases of the B-adapter sequence were aligned to each read using the aligner described below. If the alignment failed (less than six bases aligned or any mismatches), the read was discarded. Otherwise the recognized B-adapter sequence was trimmed off. Since a modified B-adapter sequence was used for the PEP-experiments, this step also eliminated cross-contamination from neighboring plate regions, which can occur to a small extent if beads from several libraries are loaded onto the same sequencing plate.
While manually inspecting sequences, we found a small fraction of reads carrying fragments of the A-adapter sequence at the 5′-end. Upon inspection of oligonucleotide A-BL on a polyacrylamide gel, we found artifacts of higher molecular weight in addition to the expected band, which are presumably oligodimers and other artifacts from synthesis. To remove sequences with A-adapter fragments, we discarded all sequences that in their first 52 bases produced an exact match of 11 or more bases to the A-adapter sequence (∼2.7% of all reads).
Sequence analysis of PEP assays with PCR product templates
Since libraries from PEP-assays with different polymerases were pooled for sequencing, we first identified the barcode sequences at the 3′-ends of the sequence reads and discarded reads that did not perfectly match any of the six barcodes. Next, we used the three bases adjacent to the barcode to identify the original template molecule and its orientation (GCC → amplicon 1, forward strand; CGT → amplicon 1, reverse strand; AGG → amplicon 2, forward strand; TCG → amplicon 2, reverse strand), again removing all reads that did not produce perfect matches. After separating all reads according to the identified barcode and template strand, we used MIA (23) (http://sourceforge.net/projects/mia-assembler/) to generate multiple sequence alignments to the appropriate reference sequences, which were the basis of all subsequent analysis.
The extent of abasic-site-induced strand breakage was inferred from the sequence representation of the complementary template strands. To remove breakage-independent biases in strand representation from the data, we corrected for the ratio of unmodified (control) strands to uracil-containing modified strands found in the uracil experiments in order to determine the fraction of templates (X) that broke due to abasic sites: 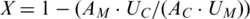 , where UC and AC represent sequences from the control strands and UM and AM sequences from the modified strands of the assays with uracils and abasic sites, respectively.
, where UC and AC represent sequences from the control strands and UM and AM sequences from the modified strands of the assays with uracils and abasic sites, respectively.
Data analysis for PEP assays with genomic DNA templates
After adapter trimming, we searched for the 8-bp end-of-template recognition sequence. If a read matched this sequence with not more than one mismatch, insertion or deletion, we classified the extension product as complete, otherwise as terminated. The recognition sequence was trimmed off, and the sequence was aligned to the appropriate reference genome using Blastn 2.2.14 (default settings, no repeat masking). As reference genomes we used horse (Broad Institute, assembly ‘equCab2′ September 2007), elephant (Broad Institute, assembly ‘loxAfr1′, May 2005) and dog (Broad Institute, assembly ‘canFam2′, May 2005) for the horse, mammoth and cave bear experiments, respectively. For every read that produced a highly significant alignment to the reference (Blastn E-value ≤ 10−6), the reference sequence around the alignment was cut out. We then re-aligned the query sequence and reference using a self-written semi-global aligner. This was necessary because Blastn produces local alignments and does not enforce a full-length alignment of query and reference. The semi-global aligner is based on the Smith–Waterman algorithm and scores +1 for a match and −2 for either a mismatch or gap. Small linear gap costs were chosen, because insertions and deletions are very frequent errors in 454 sequencing.
Determining the per-base frequency of blocking lesions
The proportion of molecules carrying one or more blocking lesions (Mblocked) was estimated as described in Results. The proportion of intact molecules (Mintact) can be described as 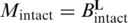 , where Bintact represents the probability that a base is intact and L represents the average fragment size. Since Mintact = 1 − Mblocked and Bintact = 1 − Bblocked, where Bblocked is the per-base frequency of blocking lesions, it follows that
, where Bintact represents the probability that a base is intact and L represents the average fragment size. Since Mintact = 1 − Mblocked and Bintact = 1 − Bblocked, where Bblocked is the per-base frequency of blocking lesions, it follows that 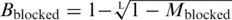 .
.
RESULTS
PEP on artificial templates
To evaluate the performance of PEP as a method for blocking lesion detection, we performed experiments with various polymerases on artificial templates containing modifications thought to occur in ancient DNA. These templates were short PCR products of 75 bp that were modified to contain either a uracil base or an abasic site in one of the two complementary strands. Three well-characterized polymerases were chosen to allow comparisons of the PEP results with those of previous studies. These polymerases were AmpliTaq Gold from Applied Biosystems, an enzyme derived from Thermus aquaticus (24); Pfu polymerase, a proof-reading enzyme derived from Pyrococcus furiosus (25); and Sso-Dpo4, a Y polymerase from Sulfolobus solfataricus with high capability for trans-lesion synthesis (26). In addition we assayed three less well-characterized polymerases: Platinum Taq from Invitrogen; Bacillus stearothermophilus (Bst) polymerase (large fragment), which possesses strong strand-displacement activity (27); and Platinum Taq HiFi from Invitrogen, which is a blend of two polymerases. Two different PCR products of 75 bp were created and assayed for each polymerase and modification.
Analysis of the PEP results showed that irrespective of DNA polymerase, 92−100% of primer extensions on non-modified template strands continued to the end-of-template recognition sequence (Table 1 and Figure 2A). Thus, in the absence of modifications, premature termination occurred on only 8% or less of templates, which is consistent with the level of inefficiency observed in typical PCR assays (28). On uracil-containing template strands, all polymerases efficiently incorporated adenine opposite uracil and continued extension with the exception of Pfu polymerase, which terminated extension exactly four bases before the uracil (Figure 2B). At abasic sites, behavior strongly differed between the polymerases. Whereas AmpliTaq Gold, Pfu and Platinum Taq HiFi synthesized across 34% or less of abasic sites, Platinum Taq, Bst and Sso-Dpo4 synthesized across 77% or more (Supplementary Figure S1). In extensions apparently blocked by abasic sites, Pfu terminated extension before the site, while all other polymerases extended one or a few bases across the site before terminating.
Table 1.
Lesion-bypass efficiencies of different polymerases
| Polymerase | Modification | Template | Extension products sequenceda |
||
|---|---|---|---|---|---|
| All | Terminated | Terminated (%) | |||
| AmpliTaq Gold | Uracil | Amplicon 1 BC2 | 1508 (1310) | 117 (104) | 7.8 (7.9) |
| Amplicon 2 BC2 | 735 (1375) | 54 (68) | 7.3 (4.9) | ||
| Abasic site | Amplicon 1 BC1 | 608 (1167) | 523 (82) | 86.0 (7.0) | |
| Amplicon 2 BC1 | 180 (883) | 118 (34) | 65.5 (3.9) | ||
| Pfu | Uracil | Amplicon 1 BC4 | 1358 (1354) | 978 (109) | 72.0 (8.1) |
| Amplicon 2 BC4 | 795 (1188) | 792 (80) | 99.6 (6.7) | ||
| Abasic site | Amplicon 1 BC3 | 115 (1519) | 90 (119) | 78.3 (7.8) | |
| Amplicon 2 BC3 | 125 (1730) | 107 (79) | 85.6 (4.6) | ||
| Platinum Taq HiFi | Uracil | Amplicon 1 BC6 | 786 (773) | 42 (59) | 5.3 (7.6) |
| Amplicon 2 BC6 | 329 (886) | 26 (45) | 7.9 (5.1) | ||
| Abasic site | Amplicon 1 BC5 | 614 (1255) | 466 (106) | 75.9 (8.4) | |
| Amplicon 2 BC5 | 445 (1577) | 418 (79) | 93.9 (5.0) | ||
| Platinum Taq | Uracil | Amplicon 1 BC2 | 1237 (1207) | 80 (86) | 6.5 (7.1) |
| Amplicon 2 BC2 | 667 (1234) | 37 (48) | 5.5 (3.9) | ||
| Abasic site | Amplicon 1 BC1 | 554 (1079) | 127 (72) | 22.9 (6.7) | |
| Amplicon 2 BC1 | 280 (727) | 48 (78) | 17.1 (4.5) | ||
| Bst, large fragment | Uracil | Amplicon 1 BC4 | 1015 (1016) | 22 (25) | 2.2 (2.5) |
| Amplicon 2 BC4 | 599 (987) | 17 (36) | 2.8 (3.6) | ||
| Abasic site | Amplicon 1 BC3 | 904 (1045) | 58 (50) | 6.4 (4.8) | |
| Amplicon 2 BC3 | 662 (1246) | 85 (40) | 12.8 (3.2) | ||
| Sso-Dpo4 | Uracil | Amplicon 1 BC6 | 828 (917) | 55 (63) | 6.6 (6.9) |
| Amplicon 2 BC6 | 849 (1264) | 54 (48) | 6.4 (3.8) | ||
| Abasic site | Amplicon 1 BC5 | 568 (976) | 115 (79) | 20.2 (8.1) | |
| Amplicon 2 BC5 | 574 (1156) | 87 (60) | 15.2 (5.2) | ||
aNumbers for the non-modified strand are shown in parentheses.
Figure 2.

Exemplary extension termination patterns on artificial templates. Shown are the numbers of template bases that were copied before Pfu polymerase terminated primer extension on (A) a non-modified and (B) a uracil-containing template strand. The template strands were 83 bases in length (75 bases PCR product plus 8 bases recognition sequence). Extension was severely inhibited by uracil.
The sequence results also revealed which nucleotides were incorporated opposite abasic sites (Supplementary Figure S2). All polymerases preferentially incorporated adenines and—to a much smaller extent—guanines. However, Bst and Sso-Dpo4 frequently did not incorporate any base, causing deletions of one to three bases in the sequence reads (Supplementary Figure S3). Although AmpliTaq Gold and Platinum Taq HiFi occasionally incorporated guanines across abasic sites, continued extension was only observed if adenines were incorporated. A similar pattern was observed for all other polymerases with the exceptions of Bst polymerase and Sso-Dpo4. Despite these stable general trends, we observed a considerable level of sequence context dependence, both in the ability of polymerases to synthesize across DNA lesions as well as in the identities of the nucleotides incorporated across such sites. For example, for one of the abasic site-containing templates, primer extension with AmpliTaq Gold was blocked in 66% of the cases, whereas this figure rose to 86% for the second template.
Since the PEP procedure starts with double-stranded templates, it is possible to measure if extension products from either strand are lost during the procedure by counting the representation of each strand in PEP results. For example, abasic sites are known to undergo thermal degradation followed by strand cleavage (29), and PEP excludes broken template strands from sequencing. We observed that sequences derived from strands containing abasic sites were indeed under-represented compared to sequences from the unmodified strands, indicating that between 15 and 90% of the abasic site-containing template strands broke at the elevated temperatures of the primer extension reactions (Supplementary Figure S4). As expected, the least breakage was observed with Bst polymerase, which, due to its strong strand-displacement activity, does not require initial heat denaturation of the template DNA.
PEP on modern genomic DNA
In order to enable side-by-side comparisons of modern genomic and ancient DNA, we next performed PEP on freshly extracted horse DNA. AmpliTaq Gold was used in this and all subsequent experiments, because it (i) is commonly used for ancient DNA amplification, (ii) synthesizes across uracils, which are known to occur in ancient DNA and (iii) shows high sensitivity to abasic sites, one of the candidates for blocking lesions in ancient DNA. To obtain a fragment-size distribution similar to ancient DNA, DNA was fragmented and two fractions with mean sizes below and above 100 bp were isolated. PEP results indicated that primer extensions on these two size fractions terminated at a rate of 16 and 14%, respectively (Table 2). These rates are slightly higher than the ones found in the experiments with PCR products, which may be due to differences in base composition or the on average longer fragment sizes of 91 and 124 bp compared to 75 bp of the PCR products.
Table 2.
PEP results on modern, UV-irradiated and ancient DNA
| Samplea | Sequence identity | Average fragment size (bp)b | Extension products sequenced |
Proportion of molecules with blocking lesions (%) / per-base frequency of blocking lesions (%) | ||
|---|---|---|---|---|---|---|
| All | Terminated | Term (%) | ||||
| Modern horse, fraction 1 | All sequences | 91.6 | 6419 | 1028 | 16.0 | |
| Horse (87.9%) | 91.5 | 5643 | 627 | 11.1 | 0/0 | |
| Modern horse, fraction 2 | All sequences | 124.6 | 8885 | 1228 | 13.8 | |
| Horse (94.1%) | 124.4 | 8360 | 980 | 11.7 | 0/0 | |
| Modern horse, fraction 1, UV | All sequences | 87.6 | 4526 | 1927 | 42.6 | |
| Horse (94.1%) | 88.9 | 3440 | 1091 | 31.7 | 26/0.3 | |
| Ancient horse 1 >47k YBP | All sequences | 154.9 | 2023 | 1151 | 56.9 | |
| Horse (3.8%) | 153.8 | 77 | 32 | 41.6 | 39/0.3 | |
| Ancient horse 2 27k YBP | All sequences | 120.7 | 9030 | 1982 | 21.9 | |
| Horse (91.6%) | 120.8 | 8314 | 1608 | 19.3 | 10/0.1 | |
| Mammoth 43k YBP | All sequences | 154.6 | 8458 | 2762 | 32.7 | |
| Elephant (2.1%) | 115.4 | 174 | 22 | 12.6 | 2/∼0.0 | |
| Cave bear 42k YBP | All sequences | 113.3 | 1542 | 822 | 53.3 | |
| Dog (1.2%) | 125.0 | 18 | 7 | 38.9 | 36/0.4 | |
aAge of ancient samples is denoted in ‘years before present’ (YBP) as determined by C14 dating.
bThe average fragment size was inferred from complete extension products only.
To test whether PEP can detect more substantial amounts of blocking lesions if they exist in natural DNA, we irradiated horse DNA with UV light prior to PEP. UV irradiation is known to create blocking lesions in DNA, predominantly pyrimidine dimers (30,31). PEP results from the irradiated DNA showed a strongly increased termination rate of 43%, clearly indicating the presence of blocking lesions after UV irradiation. To evaluate whether PEP can identify the sequence motifs of blocking lesions, we examined the base composition of the horse reference sequence around termination sites both in the UV-irradiated and control experiments (Figure 3). As expected, for the irradiated DNA but not the control DNA, we observed a strong elevation of extension terminations around pyrimidine dimers. Termination sites were not only particularly common in the middle of and in front of T/T dimers but were also elevated in the middle of T/C dimers and in front of C/T dimers. We conclude that PEP can not only detect the existence of blocking lesions in complex DNA but also the nucleotides or sequence motifs responsible for the lesions.
Figure 3.

Sequence motifs of blocking lesions in UV-irradiated DNA. Shown is the overall base composition of the horse reference sequence around extension termination sites (in a dinucleotide window and 3′→5′ orientation), as inferred by aligning all sequences without the recognition sequence (RS) against the horse genome. In UV-irradiated DNA, termination is strongly elevated around three pyrimidine dinucleotides (highlighted in black, red and blue; all other 13 dinucleotides are shown in gray). This pattern is absent in undamaged DNA.
Identifying blocking lesions in ancient DNA
To evaluate the presence of blocking lesions in ancient DNA, we performed PEP on DNA extracted from four different Pleistocene bone samples: three permafrost samples, including two horse bones and one mammoth bone, and a cave bear sample from a temperate cave site. PEP results from ancient samples differ from those of the previous experiments in that many sequences originated from environmental contaminants present in the bones. Therefore, endogenous DNA sequences must first be identified by successful alignment to reference genomes, in this case horse, elephant and dog. Unfortunately, very short sequences (<<30 bp) cannot usually be identified confidently as endogenous DNA (32). Consequently, primer extension products from ancient DNA may be too short to be recognized as endogenous, particularly if primer extension terminated prematurely, leading to undercounting of blocking lesions in ancient DNA. To quantify this effect, we calculated the rate of premature termination for the undamaged and UV-treated modern horse samples after aligning them to the horse reference genome and discarding sequences that did not produce a high confidence alignment to the reference. This filtering step causes a 1.2– to 1.4–fold (average 1.3–fold) reduction in the apparent rates of premature extension termination when compared with the results considering all reads (Table 2). Thus, to estimate the actual proportion of DNA molecules containing one or more blocking lesions, we first multiplied the observed rates of extension termination by a factor of 1.3 to correct for sequences that did not align to the reference. This correction factor is only a rough estimate, since the amount of divergence between the sample and reference genomes and the length distribution of fragments will also affect the extent of endogenous sequence undercounting. Nevertheless, this level of precision should be sufficient to roughly estimate the frequency of blocking lesions in ancient DNA. We then subtracted from the corrected termination rates the average termination rate from undamaged DNA (14.9%) to remove the background rate of random polymerase stalling.
We found considerable variation in rates of prematurely terminated extensions, and hence frequencies of blocking lesions, among the different samples. While there was no evidence for a significant amount of blocking lesions in the mammoth DNA, we estimated that 39% of molecules contain blocking lesions in one of the permafrost horse samples and 10% in the other. In the only non-permafrost sample we tested, the cave bear, we estimated that ∼36% of fragments contain blocking lesions. Due to a low percentage of endogenous DNA in the sample and poor sequence yield, this last figure is based on only a small number of sequences. Nevertheless, it does not point toward an increased frequency of blocking lesions in the cave bear sample versus the permafrost samples. Interestingly, we also found no obvious correlation between the degree of DNA fragmentation and the frequency of blocking lesions (Table 2).
The experiments with UV-irradiated DNA demonstrated that analyzing the sequence context around termination sites can reveal insights about the chemical nature of blocking lesions. One of the permafrost horse samples yielded a sufficient number of endogenous sequences for such an analysis (Supplementary Figure S5). Since the proportion of blocked molecules is only around 10% in this sample, any ancient DNA-specific signal is hidden in a strong background of random extension termination. Nevertheless, in a direct comparison to modern DNA, there is an increased frequency of extension terminations at guanines in the ancient DNA sample. This elevation may represent a polymerase-blocking guanine derivative rather than an abasic site following guanine depurination, since the synthetic oligonucleotide experiments showed that AmpliTaq Gold mostly extends one or two bases past abasic sites. However, while the signal of elevated guanine is pronounced, it is not strong enough to account for all—or even the majority—of blocking lesions in this ancient horse sample. No additional signals point toward the nature of the remaining blocking lesions. This is probably due to a lack of power with the current sequence sample size to extract information from non-uniform termination patterns.
DISCUSSION
Despite more than two decades of ancient DNA research, the knowledge about blocking lesions has remained very scarce. Previous studies, which by means of analytical chemistry or molecular biology attempted to detect polymerase-blocking modifications in ancient DNA, used assays that could not distinguish endogenous from contaminating environmental DNA and were limited to few specific DNA modifications. In contrast, by sequencing thousands of individual primer extension products using PEP, blocking lesions can be detected regardless of their chemical nature, and specifically in DNA sequences from the organism of interest as opposed to environmental DNA contamination. While single-primer extension procedures have been used with ancient DNA before (10), a key feature of PEP is the end-of-template recognition sequence, which is necessary for distinguishing prematurely aborted primer extensions from extensions that reached the ends of templates.
Applying PEP to four ancient DNA samples, we observed considerable frequencies of blocking lesions in three of the samples, providing the first direct evidence of polymerase abortion at ancient DNA blocking lesions. Interestingly, the proportion of molecules carrying one or more blocking lesions did not exceed 40% in any of the samples, with per-base estimates for blocking lesion frequencies of 0.4% or less. This estimate is orders of magnitude lower than some studies have suggested (12,14). It has to be noted though that the frequency of blocking lesions depends on the polymerase that is used. These estimates therefore only refer to PEP assays with AmpliTaq Gold, which is very sensitive to abasic sites. If, in fact, abasic sites constituted a large proportion of blocking lesions in ancient DNA, as may be indicated by the fragmentation patterns of ancient DNA (9,33), two adverse effects would substantially influence our estimated blocking lesion frequencies. On the one hand, estimates would go down if a different polymerase was used that more efficiently extends across abasic sites, such as Platinum Taq or Bst polymerase. On the other hand, the heat lability of abasic sites would lead to an underestimate of blocking lesions due to strand breakage and loss of molecules, which is ∼60% in PEP assays with AmpliTaq Gold. Taking the latter number into account, it can be calculated that not more than ∼70% of the molecules could carry one or more blocking lesion in any of our samples, even if abasic sites were the only type of blocking lesion present. Our results therefore suggest that blocking lesions do exist in ancient DNA but are not as common as some have predicted. This may help to explain the lack of success that was achieved by breeding polymerases specifically for ancient DNA amplification (15).
In addition to estimating the frequency of blocking lesions, PEP potentially allows insight into their chemical nature by revealing sequence contexts where premature termination preferentially occurs. However, the properties of a polymerase must be understood before inferring the nature of unknown blocking lesions from PEP results. We used PEP to investigate the response of various polymerases to uracil bases and abasic sites. Most of our findings are in congruence with the results of previous studies. For example, our data confirm that Pfu polymerase stalls polymerization exactly four bases upstream of uracils in template strands (34), and that polymerases preferentially incorporate adenines across non-instructive lesions such as abasic sites (35). Due to the high resolution of PEP we also observed some previously undescribed polymerase features: for example, the capability of AmpliTaq and Platinum Taq HiFi to synthesize past an abasic site depends on the base incorporated opposite the site (only adenine incorporations lead to continued primer extension). We also found that Bst polymerase bypasses abasic sites with high efficiency, exhibiting a mutagenic mode of replication similar to that of Sso-Dpo4, a Y-family polymerase (36). Of the four ancient samples we tested with PEP, only one produced sufficient data for in-depth analysis of the chemical modifications underlying blocking lesions. In this sample, a slightly elevated signal of extension termination opposite guanines points toward a guanine modification accounting for at least some of the blocking lesions. Much higher numbers of sequences than generated here will be necessary to resolve the sequence contexts of extension termination in sufficient detail. Alternatively, further experiments that are easy to conceive could be performed. For example, by pre-treating the template DNA with AP-lyases or other enzymes involved in base excision repair, specific types of lesions could be removed and their effects on PEP results observed.
In conclusion, our results indicate that blocking lesions occur at only low or moderate frequencies in typical ancient DNA samples, although many more samples, preserved under various conditions, need to be studied to determine the frequency spectrum of blocking lesions in ancient DNA more comprehensively. This has severe implications for the future of the field. If most molecules are already accessible with current amplification and sequencing methods, no future rescue and repair strategies can dramatically improve the retrieval of sequence information from ancient DNA. One exception to this may be the presence of nicks in DNA molecules. Nicks are not detectable by PEP but nevertheless may represent repairable polymerase-blocking damage in ancient DNA (37,38). In fact, a recent study of damage patterns in Neandertal DNA suggested a nick frequency of ∼2.4% per base (9), which is higher than the frequencies of blocking lesions we estimated. For reasonably preserved samples, nick repair should result in a measurable increase in the number of molecules amenable to sequencing. However, since closely spaced nicks lead to un-repairable double strand breaks, the potential of nick repair to improve sequence retrieval from highly degraded ancient DNA is probably very limited. There are undoubtedly additional losses in material during ancient DNA analyses that are unrelated to DNA damage, such as during extraction and library preparation. However, as library-based sequencing approaches are making shorter molecules accessible to sequencing (4,6) and extraction and library preparation methods are becoming more efficient (39,40), it seems that ancient DNA research will soon reach a hard limit at which no major unexplored potentials remain for the recovery of additional sequences. Thus, despite huge recent advances in ancient DNA techniques, there is little hope for ever expanding ancient DNA research to fossils that are much older than the ones currently accessible to genetic analysis.
FUNDING
Max Planck Society. Funding for open access charge: Max Planck Society.
Conflict of interest statement. None declared.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
ACKNOWLEDGEMENTS
We thank Svante Pääbo, Richard E. Green, Nadin Rohland and Sebastian Lippold for helpful discussions, Adam Wilkins for comments on the manuscript, Eske Willerslev and Jaco Weinstock for providing radiocarbon-dated horse samples, Knut Finstermeier for help with the figures and the Max-Planck-Society for financial support.
REFERENCES
- 1.Miller W, Drautz DI, Ratan A, Pusey B, Qi J, Lesk AM, Tomsho LP, Packard MD, Zhao F, Sher A, et al. Sequencing the nuclear genome of the extinct woolly mammoth. Nature. 2008;456:387–390. doi: 10.1038/nature07446. [DOI] [PubMed] [Google Scholar]
- 2.Rasmussen M, Li Y, Lindgreen S, Pedersen JS, Albrechtsen A, Moltke I, Metspalu M, Metspalu E, Kivisild T, Gupta R, et al. Ancient human genome sequence of an extinct Palaeo-Eskimo. Nature. 2010;463:757–762. doi: 10.1038/nature08835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Green RE, Krause J, Briggs AW, Maricic T, Stenzel U, Kircher M, Patterson N, Li H, Zhai W, Fritz MH, et al. A draft sequence of the Neandertal genome. Science. 2010;328:710–722. doi: 10.1126/science.1188021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Briggs AW, Good JM, Green RE, Krause J, Maricic T, Stenzel U, Lalueza-Fox C, Rudan P, Brajkovic D, Kucan Z, et al. Targeted retrieval and analysis of five Neandertal mtDNA genomes. Science. 2009;325:318–321. doi: 10.1126/science.1174462. [DOI] [PubMed] [Google Scholar]
- 5.Stiller M, Knapp M, Stenzel U, Hofreiter M, Meyer M. Direct multiplex sequencing (DMPS)—a novel method for targeted high-throughput sequencing of ancient and highly degraded DNA. Genome Res. 2009;19:1843–1848. doi: 10.1101/gr.095760.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Burbano HA, Hodges E, Green RE, Briggs AW, Krause J, Meyer M, Good JM, Maricic T, Johnson PL, Xuan Z, et al. Targeted investigation of the Neandertal genome by array-based sequence capture. Science. 2010;328:723–725. doi: 10.1126/science.1188046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Margulies M, Egholm M, Altman WE, Attiya S, Bader JS, Bemben LA, Berka J, Braverman MS, Chen YJ, Chen Z, et al. Genome sequencing in microfabricated high-density picolitre reactors. Nature. 2005;437:376–380. doi: 10.1038/nature03959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bentley DR, Balasubramanian S, Swerdlow HP, Smith GP, Milton J, Brown CG, Hall KP, Evers DJ, Barnes CL, Bignell HR, et al. Accurate whole human genome sequencing using reversible terminator chemistry. Nature. 2008;456:53–59. doi: 10.1038/nature07517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Briggs AW, Stenzel U, Johnson PL, Green RE, Kelso J, Prüfer K, Meyer M, Krause J, Ronan MT, Lachmann M, et al. Patterns of damage in genomic DNA sequences from a Neandertal. Proc. Natl Acad. Sci. USA. 2007;104:14616–14621. doi: 10.1073/pnas.0704665104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brotherton P, Endicott P, Sanchez JJ, Beaumont M, Barnett R, Austin J, Cooper A. Novel high-resolution characterization of ancient DNA reveals C > U-type base modification events as the sole cause of postmortem miscoding lesions. Nucleic Acids Res. 2007;35:5717–5728. doi: 10.1093/nar/gkm588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Briggs AW, Stenzel U, Meyer M, Krause J, Kircher M, Pääbo S. Removal of deaminated cytosines and detection of in vivo methylation in ancient DNA. Nucleic Acids Res. 2010;38:e87. doi: 10.1093/nar/gkp1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pääbo S. Ancient DNA: extraction, characterization, molecular cloning, and enzymatic amplification. Proc. Natl Acad. Sci. USA. 1989;86:1939–1943. doi: 10.1073/pnas.86.6.1939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Höss M, Jaruga P, Zastawny TH, Dizdaroglu M, Pääbo S. DNA damage and DNA sequence retrieval from ancient tissues. Nucleic Acids Res. 1996;24:1304–1307. doi: 10.1093/nar/24.7.1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hansen AJ, Mitchell DL, Wiuf C, Paniker L, Brand TB, Binladen J, Gilichinsky DA, Ronn R, Willerslev E. Crosslinks rather than strand breaks determine access to ancient DNA sequences from frozen sediments. Genetics. 2006;173:1175–1179. doi: 10.1534/genetics.106.057349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.d'Abbadie M, Hofreiter M, Vaisman A, Loakes D, Gasparutto D, Cadet J, Woodgate R, Pääbo S, Holliger P. Molecular breeding of polymerases for amplification of ancient DNA. Nat. Biotechnol. 2007;25:939–943. doi: 10.1038/nbt1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gloeckner C, Sauter KB, Marx A. Evolving a thermostable DNA polymerase that amplifies from highly damaged templates. Angew. Chem. Int. Ed. Engl. 2007;46:3115–3117. doi: 10.1002/anie.200603987. [DOI] [PubMed] [Google Scholar]
- 17.Binladen J, Gilbert MT, Bollback JP, Panitz F, Bendixen C, Nielsen R, Willerslev E. The use of coded PCR primers enables high-throughput sequencing of multiple homolog amplification products by 454 parallel sequencing. PLoS ONE. 2007;2:e197. doi: 10.1371/journal.pone.0000197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rohland N, Hofreiter M. Ancient DNA extraction from bones and teeth. Nat. Protoc. 2007;2:1756–1762. doi: 10.1038/nprot.2007.247. [DOI] [PubMed] [Google Scholar]
- 19.DeAngelis MM, Wang DG, Hawkins TL. Solid-phase reversible immobilization for the isolation of PCR products. Nucleic Acids Res. 1995;23:4742–4743. doi: 10.1093/nar/23.22.4742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Blondal T, Hjorleifsdottir SH, Fridjonsson OF, Aevarsson A, Skirnisdottir S, Hermannsdottir AG, Hreggvidsson GO, Smith AV, Kristjansson JK. Discovery and characterization of a thermostable bacteriophage RNA ligase homologous to T4 RNA ligase 1. Nucleic Acids Res. 2003;31:7247–7254. doi: 10.1093/nar/gkg914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li TW, Weeks KM. Structure-independent and quantitative ligation of single-stranded DNA. Anal. Biochem. 2006;349:242–246. doi: 10.1016/j.ab.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 22.Meyer M, Briggs AW, Maricic T, Höber B, Höffner B, Krause J, Weihmann A, Pääbo S, Hofreiter M. From micrograms to picograms: quantitative PCR reduces the material demands of high-throughput sequencing. Nucleic Acids Res. 2008;36:e5. doi: 10.1093/nar/gkm1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Green RE, Malaspinas AS, Krause J, Briggs AW, Johnson PL, Uhler C, Meyer M, Good JM, Maricic T, Stenzel U, et al. A complete Neandertal mitochondrial genome sequence determined by high-throughput sequencing. Cell. 2008;134:416–426. doi: 10.1016/j.cell.2008.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lawyer FC, Stoffel S, Saiki RK, Myambo K, Drummond R, Gelfand DH. Isolation, characterization, and expression in Escherichia coli of the DNA polymerase gene from Thermus aquaticus. J. Biol. Chem. 1989;264:6427–6437. [PubMed] [Google Scholar]
- 25.Lundberg KS, Shoemaker DD, Adams MW, Short JM, Sorge JA, Mathur EJ. High-fidelity amplification using a thermostable DNA polymerase isolated from Pyrococcus furiosus. Gene. 1991;108:1–6. doi: 10.1016/0378-1119(91)90480-y. [DOI] [PubMed] [Google Scholar]
- 26.Boudsocq F, Iwai S, Hanaoka F, Woodgate R. Sulfolobus solfataricus P2 DNA polymerase IV (Dpo4): an archaeal DinB-like DNA polymerase with lesion-bypass properties akin to eukaryotic poleta. Nucleic Acids Res. 2001;29:4607–4616. doi: 10.1093/nar/29.22.4607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lu YY, Ye SY, Hong GF. Large fragment of DNA polymerase I from Bacillus stearothermophilus (Bst polymerase) is stable at ambient temperature. Biotechniques. 1991;11:464, 466. [PubMed] [Google Scholar]
- 28.Olsen DB, Eckstein F. Incomplete primer extension during in vitro DNA amplification catalyzed by Taq polymerase: exploitation for DNA sequencing. Nucleic Acids Res. 1989;17:9613–9620. doi: 10.1093/nar/17.23.9613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lindahl T, Andersson A. Rate of chain breakage at apurinic sites in double-stranded deoxyribonucleic acid. Biochemistry. 1972;11:3618–3623. doi: 10.1021/bi00769a019. [DOI] [PubMed] [Google Scholar]
- 30.Wellinger RE, Thoma F. Taq DNA polymerase blockage at pyrimidine dimers. Nucleic Acids Res. 1996;24:1578–1579. doi: 10.1093/nar/24.8.1578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pfeifer GP. Formation and processing of UV photoproducts: effects of DNA sequence and chromatin environment. Photochem. Photobiol. 1997;65:270–283. doi: 10.1111/j.1751-1097.1997.tb08560.x. [DOI] [PubMed] [Google Scholar]
- 32.Prüfer K, Stenzel U, Hofreiter M, Pääbo S, Kelso J, Green R. Computational challenges in the analysis of ancient DNA. Genome Biol. 2010;11:R47. doi: 10.1186/gb-2010-11-5-r47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Krause J, Briggs AW, Kircher M, Maricic T, Zwyns N, Derevianko A, Pääbo S. A complete mtDNA genome of an early modern human from Kostenki, Russia. Curr. Biol. 2010;20:231–236. doi: 10.1016/j.cub.2009.11.068. [DOI] [PubMed] [Google Scholar]
- 34.Greagg MA, Fogg MJ, Panayotou G, Evans SJ, Connolly BA, Pearl LH. A read-ahead function in archaeal DNA polymerases detects promutagenic template-strand uracil. Proc. Natl Acad. Sci. USA. 1999;96:9045–9050. doi: 10.1073/pnas.96.16.9045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Strauss BS. The “A” rule revisited: polymerases as determinants of mutational specificity. DNA Repair (Amsterdam) 2002;1:125–135. doi: 10.1016/s1568-7864(01)00014-3. [DOI] [PubMed] [Google Scholar]
- 36.Fiala KA, Suo Z. Sloppy bypass of an abasic lesion catalyzed by a Y-family DNA polymerase. J. Biol. Chem. 2007;282:8199–8206. doi: 10.1074/jbc.M610719200. [DOI] [PubMed] [Google Scholar]
- 37.Pusch CM, Giddings I, Scholz M. Repair of degraded duplex DNA from prehistoric samples using Escherichia coli DNA polymerase I and T4 DNA ligase. Nucleic Acids Res. 1998;26:857–859. doi: 10.1093/nar/26.3.857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Di Bernardo G, Del Gaudio S, Cammarota M, Galderisi U, Cascino A, Cipollaro M. Enzymatic repair of selected cross-linked homoduplex molecules enhances nuclear gene rescue from Pompeii and Herculaneum remains. Nucleic Acids Res. 2002;30:e16. doi: 10.1093/nar/30.4.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rohland N, Hofreiter M. Comparison and optimization of ancient DNA extraction. Biotechniques. 2007;42:343–352. doi: 10.2144/000112383. [DOI] [PubMed] [Google Scholar]
- 40.Maricic T, Pääbo S. Optimization of 454 sequencing library preparation from small amounts of DNA permits sequence determination of both DNA strands. Biotechniques. 2009;46:51–52, 54–57. doi: 10.2144/000113042. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.