


Abstract
Transfer ribonucleic acids (tRNAs) are challenging to identify and quantify from unseparated mixtures. Our lab previously developed the signature digestion approach for identifying tRNAs without specific separation. Here we describe the combination of relative quantification via enzyme-mediated isotope labeling with this signature digestion approach for the relative quantification of tRNAs. These quantitative signature digestion products were characterized using liquid chromatography mass spectrometry (LC-MS), and we find that up to 5-fold changes in tRNA abundance can be quantified from sub-microgram amounts of total tRNA. Quantitative tRNA signature digestion products must (i) incorporate an isotopic label during enzymatic digestion; (ii) have no m/z interferences from other signature digestion products in the sample and (iii) yield a linear response during LC-MS analysis. Under these experimental conditions, the RNase T1, A and U2 signature digestion products that potentially could be used for the relative quantification of Escherichia coli tRNAs were identified, and the linearity and sequence identify of RNase T1 signature digestion products were experimentally confirmed. These RNase T1 quantitative signature digestion products were then used in proof-of-principle experiments to quantify changes arising due to different culturing media to 17 tRNA families. This method enables new experiments where information regarding tRNA identity and changes in abundance are desired.
INTRODUCTION
Ribonucleic acids (RNAs) play a critical role in the expression, transmission, and processing of genetic information. In particular, transfer RNAs (tRNAs) are a necessary component of the protein translation process. Because tRNAs are involved in protein translation, there is a high correlation between the relative abundance of an individual tRNA and use of the corresponding codon in the gene sequence (1). Codon usage is the frequency that a codon is translated per unit time in the cell; codon usage bias is thought to have evolved based on several factors including tRNA availability as well as codon–anticodon pairing. High-usage or preferred codons are used most often, specifically in highly expressed genes, and the tRNAs for these preferred codons are hypothesized to be in higher abundance (2,3). On the other hand, low-usage or non-preferred codons are used rarely or infrequently by poorly expressed genes; non-preferred codons are thought to be decoded by low-abundant tRNAs. As tRNA abundances are thought to change based on codon usage, the analysis of tRNA provides insight into changes in ncRNA expression levels based on experimental and environmental changes (1). The analysis of tRNA abundances in light of codon usage is especially important considering these preferences are not readily tracked through protein expression.
There are few experimental approaches that can be used to measure the relative abundances of tRNAs. One approach involves separating a mixture of intact tRNAs by two-dimensional polyacrylamide gel electrophoresis (2D PAGE) (4). This approach identifies individual tRNAs through sequence specific probes; quantification of tRNAs is performed through radioactive labeling with 3H, 32P and/or 14C. A more recent approach has been the development of microarrays with probes specific to individual tRNAs (5–7). Hybridization probes are used for tRNA identification, and tRNAs are labeled with either Cy3 or Cy5 fluorescent probes for detection and quantification. Although both of the methods are capable of quantification and detection of individual tRNAs, the laborious nature of the 2D PAGE approach has limited its further use by additional laboratories.
We have been interested in developing mass spectrometry (MS) methods that can be used for the identification and relative quantification of RNAs (8–11). Typically, characterization of nucleic acids by MS is performed by RNase mapping followed by sequencing via tandem mass spectrometry (MS/MS) (12). Initially, purified RNA is digested with endonucleases prior to on-line separation and analysis using liquid chromatography electrospray ionization mass spectrometry (LC-ESI-MS). Using endonucleases with different cleavage specificity, a variety of oligonucleotides are produced. The MS/MS analysis can provide sequence information on each oligonucleotide. By combining the sequences derived from multiple endonucleases, the full sequence can be mapped for the entire RNA. This basic approach allows for the identification of RNAs through database searches (13), de novo sequencing (14), and has been used to identify RNAs present in ribonucleoprotein complexes (15,16).
Although LC-MS has been a popular technique for the identification and sequencing of oligonucleotides and nucleic acids (13,15–22), there are no published methods on the use of LC-MS for the quantitative analysis of mixtures of tRNAs. Prior work by our lab has demonstrated the use of matrix-assisted laser desorption/ionization (MALDI) mass spectrometry to analyze and quantify isotopically labeled RNAs (23,24). Similar samples are enzymatically digested in 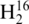 O or
O or 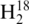 O. Only one 16O or 18O atom from the surrounding solvent is incorporated into the oligonucleotide digestion product, thus labeling the 3′ terminal phosphate. These labeled digestion products are combined prior to analysis yielding isotope pairs, which differ by 2 Da in their mass spectra. From measured experimental ratios of these isotope pairs, the relative quantities of the original RNAs can be calculated.
O. Only one 16O or 18O atom from the surrounding solvent is incorporated into the oligonucleotide digestion product, thus labeling the 3′ terminal phosphate. These labeled digestion products are combined prior to analysis yielding isotope pairs, which differ by 2 Da in their mass spectra. From measured experimental ratios of these isotope pairs, the relative quantities of the original RNAs can be calculated.
More recently, we developed the signature digestion product (SDP) approach for tRNA identification (10,11). This approach involves the enzymatic digestion of tRNAs and takes advantage of unique mass values that arise after tRNA digestion with specific endonucleases. Detection of these unique mass values enables the identification of specific tRNAs from an unseparated mixture.
Because both the isotope labeling approach for RNA relative quantification and the SDP approach for tRNA identification first require endonuclease digestion of the sample of RNAs, we present here the combination of these two approaches and develop a new LC-MS method for the quantitative analysis of individual tRNAs. The criteria for establishing quantitative signature digestion products, the analytical figures of merit for this LC-MS method, and a proof-of-concept application of this approach to the relative quantification of Escherichia coli tRNAs from two different culturing conditions are presented.
MATERIALS AND METHODS
Materials
Escherichia coli strain K12 was purchased from American Type Culture Collection (ATCC, Manassas, VA). Triethylamine (TEA), 1,1,1,3,3,3-hexafluoroisopropanol (HFIP), magnesium acetate, ammonium chloride, β-mercaptoethanol, Tri-Reagent, lysozyme chloride from chicken egg white, chloroform, 2-propanol, absolute ethanol, sodium chloride and individual E. coli tRNAs were used as received from Sigma-Aldrich (St. Louis, MO). HPLC-grade methanol and acetonitrile were obtained from the Tedia Company Inc. (Fairfield, OH). Ammonium acetate, potassium chloride and magnesium chloride were purchased from Fisher Scientific (Fairlawn, NJ). Molecular biology grade tris-hydrochloride was purchased from Promega (Madison, WI). Bacto yeast extract and bacto tryptone were used as received from Becton, Dickinson, & Company (Sparks, MD). Sodium citrate was purchased from Mallinckrodt Baker, Inc. (Paris, KY). UltraPure agarose was purchased from Invitrogen Corporation (Carlsbad, CA). RNase T1 was purchased from Roche Molecular Biochemicals (Indianapolis, IN). Sep-Pak C18 cartridges were obtained from Waters (Milford, MA). 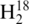 O (95% purity) was used as purchased from Isotec (Miamisburg, OH). Nanopure water (18 MOhms) from a Barnstead (Dubuque, IA) nanopure system was used as a mobile phase solvent or autoclaved before use in enzymatic digestions.
O (95% purity) was used as purchased from Isotec (Miamisburg, OH). Nanopure water (18 MOhms) from a Barnstead (Dubuque, IA) nanopure system was used as a mobile phase solvent or autoclaved before use in enzymatic digestions.
Isolation of tRNAs
Escherichia coli strain K12 was cultured in house with enriched MOPS media and minimal MOPS media as described (4). The washed cells were distributed into polypropylene tubes with ∼6.5–7 g of cells per tube. After adding 1.5 ml of lysozyme buffer (16 mg/ml of lysozyme in 25 mM Tris–HCl, 60 mM KCl and 10 mM MgCl2), the cells were allowed to incubate for 10 min at 4°C. After incubation, 10 ml of Tri Reagent were added and the mixture was vortexed. The mixture was allowed to incubate for another 5 min at room temperature. The mixture was vortexed again prior to adding 5.2 ml of chloroform. This mixture was incubated at room temperature for 5 min; the polypropylene tubes were then centrifuged at 12 000 rpm for 15 min in a SS-34 rotor with the Sorvall RC5C centrifuge. The aqueous phase (∼36 ml) was collected and placed in fresh polypropylene tubes. To the aqueous phase, 5 ml of 2-propanol and 5 ml of sodium chloride/sodium citrate solution [1.2 M NaCl, 0.8 M Na3C3H5O(CO2)3] was added. The tube was inverted five to six times and allowed to incubate at room temperature for 10 min. The tubes were then centrifuged at 12 000 rpm for 10 min in a 50.2 Ti rotor with an Optima L-XP Ultracentrifuge to pellet the ribosomal RNA. The supernatant containing tRNA was removed and placed in Kimble HS tubes. To the supernatant, 0.7 volumes of 2-propanol were added to each tube. After incubating at room temperature for 10 min, the tubes were centrifuged at 12 000 rpm for 15 min producing three layers with 2-propanol at the top, a salt cushion on the bottom, and tRNA contained at the interface. The isolated tRNA was then washed with 10 ml of 75% ethanol. The washed pellet was vortexed and centrifuged at 12 000 rpm for 3 min. After centrifuging the pellet to the bottom of the tube, the supernatant was removed and discarded. The tRNA pellet was resuspended in 1 ml of autoclaved, nanopure water and stored at −20°C until utilized for analysis. The separation of tRNA from ribosomal RNA was verified with a 1% nondenaturing agarose gel. The purity and concentration of tRNA were determined by the A260/A280 absorbance ratio.
Enzyme purification and digestions
RNase T1 was precipitated from its original solution with acetone, resuspended and eluted in 1 ml of 75% aqueous acetonitrile from a Sep-Pak C18 cartridge. All solutions were then taken to dryness and resuspended in sterile  O or
O or 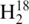 O. For digestion, 500 units of RNase T1 were added to 10 µg of tRNA and 5 µl of 220 mM ammonium acetate buffer. The reaction mixture was incubated in a 37°C water bath for 2 h.
O. For digestion, 500 units of RNase T1 were added to 10 µg of tRNA and 5 µl of 220 mM ammonium acetate buffer. The reaction mixture was incubated in a 37°C water bath for 2 h.
For experiments focused on monitoring changes in tRNA abundance from different cell media conditions, the six total cultures (three in enriched MOPS media and three in minimal MOPS media) were processed to isolate the tRNA as described above. All tRNA solutions were taken to dryness and resuspended in sterile 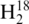 O or
O or 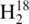 O. The three biological replicates of tRNA from the enriched MOPS media were isotopically labeled with 18O through enzymatic digestion; the three biological replicates from the minimal MOPS media were isotopically labeled with 16O through enzymatic digestion. Biological replicates from each media source were combined prior to LC-MS analysis to generate three isotopically labeled mixtures of tRNAs. Each sample solution was lyophilized and reconstituted in mobile phase A to a concentration of 0.5 µg/µl and then analyzed in triplicate from 10 µl (5 µg) injections.
O. The three biological replicates of tRNA from the enriched MOPS media were isotopically labeled with 18O through enzymatic digestion; the three biological replicates from the minimal MOPS media were isotopically labeled with 16O through enzymatic digestion. Biological replicates from each media source were combined prior to LC-MS analysis to generate three isotopically labeled mixtures of tRNAs. Each sample solution was lyophilized and reconstituted in mobile phase A to a concentration of 0.5 µg/µl and then analyzed in triplicate from 10 µl (5 µg) injections.
LC-MS
High-resolution LC-MS was performed using a Hitachi HPLC system (Hitachi High-Technologies America, San Jose, CA) comprised of an L-7100 solvent pump, an L-7400 UV-Vis detector, and a D-7000 system controller connected in-line to a Thermo LTQ-FTTM (Thermo Scientific, Waltham, MA) mass spectrometer equipped with an ESI source. Low-resolution LC-MS was performed using a MicroAS autosampler, Surveyor MS Pump Plus HPLC system and Thermo LTQ XLTM (Thermo Scientific, Waltham, MA) mass spectrometer equipped with an ESI source.
Reversed phase chromatography was performed on an Xterra MS C18 1.0 × 150 mm column, 3.5 µm particle size and 50 Å pore size (Waters, Milford, MS) coupled with a guard column at a flow rate of 40 µl/min. Mobile phase A consisted of 16.3 mM TEA/400 mM HFIP at pH 7.0 in water; equal amounts of mobile phase A and methanol were combined to produce mobile phase B. The mobile phase gradient starting at 5%B increased to 20%B at 5 min, 30%B at 7 min, and to 95%B at 50 min. The mobile phase composition was held at 95%B for 5 min prior to re-equilibrating the column for 15 min under initial mobile phase conditions before the next injection.
For high-resolution LC-MS, the ESI source was set to 325°C and 4.25 kV. Data from the FT-ICR cell, equipped with a 7.0 Tesla magnet, was acquired in scan event 1. The mass range was restricted to m/z 360–2000 to avoid interference from the HFIP dimer ion. Negative ion data was collected in full scan and profile data type with the resolution set to 100 000. For low-resolution LC-MS, the ESI source was set to 275°C and 4.50 kV. For general data collection and verification of standards, negative ion data was collected in full scan with a mass range of m/z 550–2000 and profile data type. For relative quantification of isotopically labeled digestion products, the negative ion data was collected in profile data type using a zoom scan with the mass range restricted to m/z 800–2000. For sequence verification using collision-induced dissociation (CID), negative ion MS/MS mass lists for the signature digestion products of interest were created to include no more than 22 average m/z values detected experimentally, a normalized collision energy of 30, and a retention time window of five min or more. Vendor supplied Xcalibur software was used for all data acquisition and processing.
Quantitative signature digestion products
tRNA sequences were obtained from the tRNA Sequence Database (25). Published sequences were theoretically digested using Mongo Oligo Mass Calculator (version 2.06; http://library.med.utah.edu/masspec/) with the parameters set to the appropriate endonuclease digest, monoisotopic mass, negative mode with 5′-phosphate and 3′-hydroxyl group for RNA. The mass values from CID for sequencing were obtained by calculating the CID fragments using monoisotopic mass and negative mode for the RNA sequences with 5′-hydroxyl groups and 3′-phosphates.
Data analysis
To establish analytical parameters of the current method, enzymatic digestions of tRNA were performed separately in the presence of 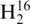 O and
O and  O. The digestion products were combined in various ratios and analyzed via LC-MS. Relative quantification was performed by first creating a selected ion chromatogram for a particular charge state of a specific oligonucleotide. Then, the mass spectra were summed over that particular peak. Relative quantification was performed on as many digestion products as available. Ion abundance ratios of the 18O-labeled and 16O-labeled digestion products were calculated by use of Equation (1):
O. The digestion products were combined in various ratios and analyzed via LC-MS. Relative quantification was performed by first creating a selected ion chromatogram for a particular charge state of a specific oligonucleotide. Then, the mass spectra were summed over that particular peak. Relative quantification was performed on as many digestion products as available. Ion abundance ratios of the 18O-labeled and 16O-labeled digestion products were calculated by use of Equation (1):
| (1) |
where IA represents the monoisotopic peak abundance of the 16O product, IA+2 represents the combination of the monoisotopic peak abundance of the 18O digestion product and the A+2 isotopic peak abundance of the 16O digestion product and b represents the A+2 isotopic peak abundance contribution from the 16O digestion product (23). All ratios were corrected for the 95% isotopic purity of 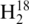 O.
O.
RESULTS
The enzymatic digestion of a tRNA by a specific endonuclease will produce a mixture of digestion products. For a tRNA of known sequence, the digestion products and their mass values can be calculated. Some digestion product base compositions are redundant, or shared with multiple tRNA sequences. Some digestion products and their mass values are unique to a specific tRNA sequence; these unique digestion products are known as signature digestion products. The mass spectrometric detection of these signature digestion products allows for the identification of tRNAs, even from unfractionated mixtures (10,11). Because both the generation of signature digestion products and relative quantification using isotope labeling are RNase-mediated approaches, it seemed that they could be combined to allow for the relative quantification of mixtures of tRNAs. To do so, we propose to designate quantitative signature digestion products (qSDPs) as signature digestion products that can be used for simultaneous identification and quantification of individual tRNAs.
Quantitative signature digestion product criteria
The following criteria have been established to designate those signature digestion products that can be used for quantification.
qSDPs are signature digestion products that must incorporate an 16O/18O label on the 3′-terminus of the oligonucleotide during endonuclease digestion.
qSDPs must differ by more than 2 Da from other known digestion peaks.
The sequences of qSDPs have been verified by MS/MS.
qSDPs must yield a linear response (calibration curve) spanning at least a 5-fold change in relative abundance.
These criteria were arrived at based on the anticipated application of qSDPs using LC-MS approaches. In particular, criterion (i) is necessary to ensure RNase-mediated incorporation of the appropriate 16O/18O isotope label. This criterion eliminates signature digestion products arising from the 3′-terminus of a tRNA as well as those with 2′-OH modifications on their 3′-terminal nucleotide. The second criterion eliminates potential mass spectral interferences that would inhibit accurately measuring the 16O/18O isotopic doublet. The third criterion simply ensures that signature digestion products generated using tRNA sequences obtained from databases or other resources are correct and do not reflect differences due to variations in the organism strain used or the culturing conditions. The last criterion was established to ensure the qSDP can accurately report changes in tRNA expression levels over biologically relevant dynamic ranges.
To generate a list of potential qSDPs, the signature digestion products of E. coli were determined as previously described (http://bearcatms.uc.edu/RNAccess) (10). Signature digestion products, their corresponding sequences, and tRNA of origin are then compared against criteria (i) and (ii) to identify potential qSDPs. To finalize the experimentally appropriate qSDPs, LC-MS results of known mixtures of SDPs are analyzed to verify sequences (by MS/MS), linearity and dynamic range. Once complete, this set of experimentally appropriate qSDPs for a particular organism’s tRNAs can then be used to monitor changes in individual tRNA expression levels.
Potential quantitative signature digestion products
To illustrate the process for identifying qSDPs that can used to monitor changes in individual tRNA expression levels, E. coli was used as the model system. RNase T1, which cleaves specifically at the 3′-end of unmodified guanosine residues, yields 105 E. coli signature digestion products. Application of criteria (i) and (ii) results in 64 signature digestion products that potentially can be used for relative quantification (Supplementary Table S1). These 64 potential quantitative signature digestion products would enable the characterization of 27 individual tRNAs and all tRNA families except threonine.
RNase A, which cleaves at the 3′-end of cytosine and uridine residues, yields 58 E. coli signature digestion products. Application of criteria (i) and (ii) results in 27 signature digestion products that potentially could be used for the relative quantification of tRNAs (Supplementary Table S2). The 27 potential quantitative signature digestion products would enable the characterization of 16 individual tRNAs and 15 different tRNA families.
RNase U2 or TA, which cleave at the 3′-terminus of unmodified adenosine residues under limiting digestion conditions, yields 168 signature digestion products from E. coli tRNAs. As before, application of criteria (i) and (ii) results in 79 E. coli signature digestion products that are potentially useful in the relative quantification of tRNAs (Supplementary Table S3). These digestion products could monitor changes in 33 individual tRNAs and all tRNA families except threonine.
Examining the data present in Supplementary Tables S1–S3, the use of more than one endonuclease will be required to characterize the complete set of E. coli tRNAs (11). Of note, all tRNA families can be analyzed if a combination of RNase T1, A and U2 are used, and ∼70% of the individual tRNAs could potentially be quantified using these three RNases (Table 1). These results represent the upper limit on the number of qSDPs that result from these RNases.
Table 1.
Summary of quantitative signature digestion products for RNase T1, A or U2 digestion of E. coli tRNAs that meet the first two criteria
| tRNA | RNase | tRNA | RNase |
|---|---|---|---|
| Ala 1 | T1 (0); A (1); U2 (2) | Lys | T1 (1); A (0); U2 (2) |
| Ala 2 | T1 (0); A (0); U2 (0) | Met f1 | T1 (0); A (1); U2 (1) |
| Ala 3 | T1 (0); A (1); U2 (2) | Met f2 | T1 (0); A (0); U2 (1) |
| Ala 1/2 | T1 (1); A (1); U2 (2) | Met f1/f2 | T1 (2); A (1); U2 (3) |
| Ala 2/3 | T1 (0); A (1); U2 (0) | Met m | T1 (3); A (1); U2 (3) |
| Arg 1 | T1 (0); A (0); U2 (0) | Phe | T1 (3); A (1); U2 (1) |
| Arg 2 | T1 (0); A (0); U2 (1) | Pro | T1 (3); A (2); U2 (3) |
| Arg 3 | T1 (2); A (0); U2 (2) | Sec6 | T1 (1); A (2); U2 (3) |
| Arg 4 | T1 (3); A (2); U2 (2) | Ser 1 | T1 (1); A (0); U2 (2) |
| Arg 5 | T1 (0); A (0); U2 (0) | Ser 2 | T1 (2); A (1); U2 (0) |
| Arg 1/2 | T1 (1); A (0); U2 (3) | Ser 3 | T1 (2); A (1); U2 (2) |
| Asn | T1 (3); A (0); U2 (3) | Ser 4 | T1 (0); A (0); U2 (0) |
| Asp | T1 (1); A (2); U2 (2) | Ser 5 | T1 (0); A (0); U2 (0) |
| Cys | T1 (3); A (0); U2 (2) | Ser 1/2 | T1 (1); A (0); U2 (0) |
| Gln 1 | T1 (1); A (0); U2 (2) | Ser 4/5 | T1 (1); A (0); U2 (0) |
| Gln 2 | T1 (0); A (1); U2 (1) | Ser 1/4/5 | T1 (1); A (0); U2 (0) |
| Gly 1 | T1 (1); A (0); U2 (1) | Ser 3/4/5 | T1 (0); A (0); U2 (1) |
| Gly 2 | T1 (2); A (0); U2 (1) | Thr 1 | T1 (0); A (0); U2 (0) |
| Gly 3 | T1 (3); A (0); U2 (1) | Thr 2 | T1 (0); A (0); U2 (0) |
| Glu 1 | T1 (0); A (0); U2 (0) | Thr 3 | T1 (0); A (0); U2 (0) |
| Glu 2 | T1 (0); A (0); U2 (0) | Thr 4 | T1 (0); A (0); U2 (0) |
| Glu 3 | T1 (0); A (0); U2 (0) | Thr 1/3 | T1 (0); A (1); U2 (0) |
| Glu 1/2/3 | T1 (3); A (0); U2 (2) | Trp | T1 (3); A (1); U2 (4) |
| His | T1 (4); A (0); U2 (3) | Tyr 1 | T1 (0); A (0); U2 (0) |
| Ile 1 | T1 (1); A (2); U2 (1) | Tyr 2 | T1 (0); A (0); U2 (0) |
| Ile 2 | T1 (2); A (0); U2 (2) | Tyr 1/2 | T1 (2); A (1); U2 (3) |
| Leu 1 | T1 (1); A (0); U2 (2) | Val 1 | T1 (1); A (1); U2 (2) |
| Leu 2 | T1 (0); A (0); U2 (1) | Val 2A | T1 (0); A (0); U2 (2) |
| Leu 3 | T1 (0); A (0); U2 (0) | Val 2B | T1 (1); A (0); U2 (2) |
| Leu 4 | T1 (0); A (0); U2 (0) | Val 2A/2B | T1 (0); A (1); U2 (0) |
| Leu 5 | T1 (0); A (0); U2 (0) | ||
| Leu 1/2 | T1 (1); A (0); U2 (1) | ||
| Leu 4/5 | T1 (2); A (0); U2 (3) | ||
| Leu 3/4/5 | T1 (0); A (0); U2 (2) |
LC-MS figures of merit
While the previous section focused on signature digestion products that potentially could be used for the relative quantification of tRNAs, the first two criteria do not account for the experimental behavior of these signature digestion products. Because relative quantification of RNAs by isotope labeling has only been described using MALDI-MS (23,24), it was necessary to determine first the LC-MS figures of merit for the analysis of isotopically labeled RNase digestion products. For all subsequent experimental studies, the analyses focused solely on RNase T1 generated digestion products. This RNase produces a large pool of signature digestion products, and the experimental data from this RNase should provide a reasonable illustration for identifying, characterizing and using qSDPs.
The figures of merit were first determined using single isoaccepting tRNAs that were digested and isotopically labeled, producing a mixture of approximately 12 different RNase T1 digestion products per tRNA. The 18O- and 16O-labeled oligonucleotides were then mixed at known ratios and concentrations. Experiments were performed to define the analytical parameters and limitations, focusing individually on the chromatographic and mass spectrometric steps as well as the integrated method. Once these figures of merit were obtained, criteria (iii) and (4) were examined for tRNA RNase T1 digestion products from an E. coli cell lysate.
Co-elution of isotopically labeled oligonucleotides
In the previously established MALDI method, all oligonucleotide digestion products, i.e., both the 16O- and 18O-labeled oligonucleotides, are analyzed simultaneously. Similarly, in this LC-MS method, relative quantification can be implemented as 16O- and 18O-labeled oligonucleotides co-elute. For example, the selected ion chromatograms (SICs) for the 16O- and 18O-labeled E. coli
 RNase T1 digestion product CCCAGp in Figure 1A reveal co-eluting peaks at 21 min. As noted in the mass spectral data in Figure 1B, both labeled RNase T1 digestion products can be detected simultaneously, thus allowing for relative quantification of the parent RNA molecules from these differentially labeled RNase digestion products.
RNase T1 digestion product CCCAGp in Figure 1A reveal co-eluting peaks at 21 min. As noted in the mass spectral data in Figure 1B, both labeled RNase T1 digestion products can be detected simultaneously, thus allowing for relative quantification of the parent RNA molecules from these differentially labeled RNase digestion products.
Figure 1.

Representative selected ion chromatograms and mass spectra of digestion product CCCAGp in 1:1 16O:18O. The digestion products are detected in both the (A) mass spectrum and (B) selected ion chromatograms at 100 fmol, but only the 18O labeled product is seen in the (C) mass spectrum and (D) selected ion chromatograms at 50 fmol.
Limits of detection
The limits of detection were established by analyzing decreasing concentrations of a 1:1 18O:16O mixture generated from the RNase T1 digestion of E. coli
 . Analyzing the amount injected on column versus 16O ion abundance for several RNase T1 digestion products finds the limit of detection to be approximately 100 fmol of each tRNA digest injected on column. For example, Figure 1A contains SICs for both the 16O- and 18O-labeled oligonucleotides when 100 fmol of tRNA digest was loaded on column, with the mass spectrum obtained by summing across a peak eluting at 21 min shown in Figure 1B. By way of comparison, Figure 1C contains SICs for both the 16O- and 18O-labeled oligonucleotides when 50 fmol of the tRNA digest was loaded on column, with the resulting mass spectrum obtained by summing across a peak eluting at 21 min shown in Figure 1D. Although both the 16O- and 18O-labeled ions for CCCAGp are detected in Figure 1C, the lack of an A+1 isotope peak for the 16O-labeled oligonucleotide is evidence that 50 fmol loaded on column is below the limits of detection for this LC-MS instrument.
. Analyzing the amount injected on column versus 16O ion abundance for several RNase T1 digestion products finds the limit of detection to be approximately 100 fmol of each tRNA digest injected on column. For example, Figure 1A contains SICs for both the 16O- and 18O-labeled oligonucleotides when 100 fmol of tRNA digest was loaded on column, with the mass spectrum obtained by summing across a peak eluting at 21 min shown in Figure 1B. By way of comparison, Figure 1C contains SICs for both the 16O- and 18O-labeled oligonucleotides when 50 fmol of the tRNA digest was loaded on column, with the resulting mass spectrum obtained by summing across a peak eluting at 21 min shown in Figure 1D. Although both the 16O- and 18O-labeled ions for CCCAGp are detected in Figure 1C, the lack of an A+1 isotope peak for the 16O-labeled oligonucleotide is evidence that 50 fmol loaded on column is below the limits of detection for this LC-MS instrument.
Calculating I18/I16 ratios
To determine the best parameters to use for relative quantification, I18/I16 calculations were performed on the digestion product AU[s4U]AGp from 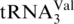 at a 18O/16O ratio of 1.00. SICs were created for both the 16O and 18O-labeled digestion products of interest. Comparisons were made between calculations using peak areas of the SICs, peak heights of the SICs, peak heights of the mass spectrum that were summed over the entire SIC, and peak heights of the mass spectrum that were summed over the highest 50% of the SIC. The experimentally calculated I18/I16 values from each approach were compared (data not shown) and it was found that calculations based on peak heights of the mass spectrum obtained after summing over the entire SIC were consistently closer to the expected value, and all subsequent I18/I16 values were obtained by that approach.
at a 18O/16O ratio of 1.00. SICs were created for both the 16O and 18O-labeled digestion products of interest. Comparisons were made between calculations using peak areas of the SICs, peak heights of the SICs, peak heights of the mass spectrum that were summed over the entire SIC, and peak heights of the mass spectrum that were summed over the highest 50% of the SIC. The experimentally calculated I18/I16 values from each approach were compared (data not shown) and it was found that calculations based on peak heights of the mass spectrum obtained after summing over the entire SIC were consistently closer to the expected value, and all subsequent I18/I16 values were obtained by that approach.
Effect of ion charge state on relative quantification
Electrospray ionization produces multiply charged species, thus investigating the effect that charge state has on relative quantification provides information regarding which charge states should be chosen, if any charge state in sufficient abundance can be used for quantification, or if all charge states should be used together for quantification. To identify whether the charge state of the oligonucleotide affects relative quantification, E. coli
 was digested with RNase T1 and isotopically labeled; these digestions were then combined at four different 18O:16O ratios for analysis by LC-MS. Relative quantification was performed three different ways. First, each charge state was analyzed independently. Second, the 18O:16O ratio from each charge state was averaged together. Lastly, each 18O:16O ratio at all charge states was summed together prior to relative quantification.
was digested with RNase T1 and isotopically labeled; these digestions were then combined at four different 18O:16O ratios for analysis by LC-MS. Relative quantification was performed three different ways. First, each charge state was analyzed independently. Second, the 18O:16O ratio from each charge state was averaged together. Lastly, each 18O:16O ratio at all charge states was summed together prior to relative quantification.
Table 2 contains the results obtained for representative RNase T1 digestion products of  using the three different approaches. It is evident from these data that, when the 16O:18O ratio is around 1, there is no significant difference in the method used for calculating relative amounts. Some differences are noted when this ratio is low (1:5 16O:18O) or high (5:1 16O:18O). However, when accounting for data obtained at low signal-to-noise ratios, any of these approaches are equally effective at determining 16O:18O labeling ratios. Such results should not be surprising, as ESI of oligonucleotides arises from deprotonation of the phosphodiester backbone in typical ESI buffer solutions (pH ∼7), rather than any effect due to the nucleobases present in the oligonucleotide (26). Unless otherwise noted, the most abundant charge state was used for relative quantification in all subsequent studies.
using the three different approaches. It is evident from these data that, when the 16O:18O ratio is around 1, there is no significant difference in the method used for calculating relative amounts. Some differences are noted when this ratio is low (1:5 16O:18O) or high (5:1 16O:18O). However, when accounting for data obtained at low signal-to-noise ratios, any of these approaches are equally effective at determining 16O:18O labeling ratios. Such results should not be surprising, as ESI of oligonucleotides arises from deprotonation of the phosphodiester backbone in typical ESI buffer solutions (pH ∼7), rather than any effect due to the nucleobases present in the oligonucleotide (26). Unless otherwise noted, the most abundant charge state was used for relative quantification in all subsequent studies.
Table 2.
Comparison of approaches for relative quantification obtained by measuring three replicates of  mixtures prepared at the heavy-to-light ratios noted
mixtures prepared at the heavy-to-light ratios noted
| Sequence | Expected | −1 charge state | −2 charge state | Average of charge states | Sum of charge states |
|---|---|---|---|---|---|
| [m5U]ΨCGp | 0.20 | 0.187 ± 0.004 | 0.141 ± 0.027 | 0.164 ± 0.028 | 0.164 ± 0.015 |
| 1.00 | 1.11 ± 0.03 | 1.09 ± 0.02 | 1.10 ± 0.03 | 1.10 ± 0.02 | |
| 5.00 | 5.13 ± 0.04 | 5.50 ± 0.14 | 5.31 ± 0.21 | 5.31 ± 0.06 | |
| [m7G]UCGp | 0.20 | 0.177 ± 0.003 | 0.155 ± 0.023 | 0.0894 ± 0.0883 | 0.0984 ± 0.0103 |
| 1.00 | 1.13 ± 0.12 | 1.08 ± 0.18 | 1.11 ± 0.14 | 1.10 ± 0.05 | |
| 5.00 | 5.64 ± 0.04 | 5.24 ± 0.24 | 5.44 ± 0.25 | 5.44 ± 0.15 | |
| CUCAGp | 0.20 | 0.153 ± 0.001 | 0.150 ± 0.006 | 0.151 ± 0.004 | 0.234 ± 0.004 |
| 1.00 | 1.03 ± 0.13 | 1.03 ± 0.04 | 1.03 ± 0.08 | 1.11 ± 0.06 | |
| 5.00 | 6.34 ± 0.46 | 4.56 ± 0.08 | 5.45 ± 0.93 | 5.38 ± 0.18 | |
| A[s4U]UAGp | 0.20 | 0.113 ± 0.015 | 0.116 ± 0.006 | 0.115 ± 0.010 | 0.256 ± 0.005 |
| 1.00 | 0.991 ± 0.129 | 0.959 ± 0.053 | 0.975 ± 0.090 | 1.11 ± 0.04 | |
| 5.00 | 4.56 ± 0.082 | 4.96 ± 0.34 | 4.76 ± 0.34 | 4.88 ± 0.35 |
Charge states were analyzed separately, averaged and summed together. Overall, no specific approach yielded more accurate results than any other approach.
Dynamic range of labeling
The dynamic range of labeling provides information on the range of differences in expression levels that can be monitored. The linear dynamic range for isotope labeling was determined by plotting the actual 16O:18O ratio versus the measured 16O:18O ratio, which was obtained by comparing mass spectral ion abundances of the most abundant charge state from the RNase T1 digestion of E. coli
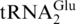 as shown in Supplementary Figure S1. The plot of light/heavy versus I16/I18 is linear from 1:1 to 5:1 (16O:18O), and the plot of heavy/light versus I18/I16 is linear from 1:1 to 10:1 (18O:16O). Thus, the dynamic range for labeling approximately is 5:1 to 1:10 16O:18O. This is comparable to the dynamic range of labeling for MALDI which was determined to be 10:1 to 1:10 18O:16O (23). With the exception of the RNase T1 digestion product 5′-ACACCGp-3′ (slope = 1.27), all digestion products analyzed yielded slopes of unity, within the experimental error of the measurements. Thus, this data does not reveal any biasing of the results until the heavy-to-light ratio exceeds 10:1. Above 10:1, the measured response for several RNase digestion products no longer reports the actual amount (i.e. slope ≠ 1). This dynamic range of labeling is within values previously reported for changes in tRNA abundance (4,6,7).
as shown in Supplementary Figure S1. The plot of light/heavy versus I16/I18 is linear from 1:1 to 5:1 (16O:18O), and the plot of heavy/light versus I18/I16 is linear from 1:1 to 10:1 (18O:16O). Thus, the dynamic range for labeling approximately is 5:1 to 1:10 16O:18O. This is comparable to the dynamic range of labeling for MALDI which was determined to be 10:1 to 1:10 18O:16O (23). With the exception of the RNase T1 digestion product 5′-ACACCGp-3′ (slope = 1.27), all digestion products analyzed yielded slopes of unity, within the experimental error of the measurements. Thus, this data does not reveal any biasing of the results until the heavy-to-light ratio exceeds 10:1. Above 10:1, the measured response for several RNase digestion products no longer reports the actual amount (i.e. slope ≠ 1). This dynamic range of labeling is within values previously reported for changes in tRNA abundance (4,6,7).
Accuracy and precision
Regardless of the ionization method and mass analyzer, experimental ratios are found to be more accurate and reproducible when the 18O-labeled digestion product is in excess of the 16O-labeled product. When the 16O-labeled product is present in large excess to the 18O-labeled product, the A+2 contribution of the 16O-labeled product will overlap with the A contribution of the 18O-labeled product, making it difficult to extract a representative quantity of the 18O-labeled digestion product from the peak at that m/z value.
For example, in Figure 2, the 16O-labeled digestion product, AU[s4U]AGp, is expected to be in five-fold excess to the 18O-labeled digestion product of the same sequence. The A+2 contribution of the 16O-labeled digestion product (m/z 823.589 for A) will be 77% of the doubly charged digestion product at m/z 824.591. In this example, the task requires accurately quantifying a peak of 25% relative abundance in a 75% background; analysis at such a high background increases the coefficients of variation and introduces more error into the analysis. In comparison, when the 18O-labeled digestion product is in excess, as in Figure 2, the A+2 contribution of the 16O-labeled digestion product has an insignificant effect (<4%) on the 18O-labeled digestion product ion abundance.
Figure 2.

Representative mass spectra of the [M−2H]2− ion from AU[s4U]AGp illustrating the limits for the dynamic range of isotope labeling. (A) Sample prepared at 5:1 16O:18O labeling ratio. (B) Sample prepared at 1:10 16O:18O labeling ratio.
To further confirm the dynamic range of labeling and the reproducibility of this method, RNase T1 digestion products of E. coli
 were prepared at various 18O:16O ratios and analyzed in triplicate. Representative results from these analyses obtained from RNase T1 products at all charge states detected are provided in Table 3. All RNase T1 digestion products yielded linear results over the dynamic range of 1:5 to 10:1 18O:16O, as noted earlier, and the results in Table 3 were obtained by use of Equation 1. Consistent, although not identical, results were obtained for the experimentally measured 18O:16O ratios, for almost all RNase T1 digestion products of
were prepared at various 18O:16O ratios and analyzed in triplicate. Representative results from these analyses obtained from RNase T1 products at all charge states detected are provided in Table 3. All RNase T1 digestion products yielded linear results over the dynamic range of 1:5 to 10:1 18O:16O, as noted earlier, and the results in Table 3 were obtained by use of Equation 1. Consistent, although not identical, results were obtained for the experimentally measured 18O:16O ratios, for almost all RNase T1 digestion products of  . Importantly, the charge state and oligonucleotide identity do not appear to have any significant effect on the application of this method, as the coefficients of variation for all oligonucleotides studied were less than 20%, with lower coefficients of variation for the charge states yielding higher signal-to-noise ratios.
. Importantly, the charge state and oligonucleotide identity do not appear to have any significant effect on the application of this method, as the coefficients of variation for all oligonucleotides studied were less than 20%, with lower coefficients of variation for the charge states yielding higher signal-to-noise ratios.
Table 3.
Accuracy and precision of the approach for relative quantification using LC-MS and 18O labeling as determined by measuring three replicates of  mixtures prepared at the heavy-to-light ratios noted
mixtures prepared at the heavy-to-light ratios noted
| Expected | Measured | CV (%) | |
|---|---|---|---|
| 5′-[m5U]ΨCGp-3′ | |||
| [M-H]− (m/z 1293.16) | 0.20 | 0.187 ± 0.0045 | 2.38 |
| 1.00 | 1.11 ± 0.029 | 2.61 | |
| 5.00 | 5.13 ± 0.0395 | 0.769 | |
| 10.00 | 9.16 ± 0.341 | 3.72 | |
| [M-2H]2- (m/z 646.074) | 0.20 | 0.141 ± 0.0266 | 18.8 |
| 1.00 | 1.09 ± 0.0249 | 2.28 | |
| 5.00 | 5.50 ± 0.142 | 2.58 | |
| 10.00 | 9.76 ± 0.370 | 3.79 | |
| 5′-A[s4U]UAGp-3′ | |||
| [M-H]− (m/z 1648.18) | 0.20 | 0.113 ± 0.0146 | 13.0 |
| 1.00 | 0.991 ± 0.130 | 13.1 | |
| 5.00 | 4.56 ± 0.285 | 6.24 | |
| 10.00 | 10.05 ± 0.919 | 9.14 | |
| [M-2H]2- (m/z 823.587) | 0.20 | 0.117 ± 0.00639 | 5.48 |
| 1.00 | 0.959 ± 0.0525 | 5.47 | |
| 5.00 | 4.96 ± 0.336 | 6.78 | |
| 10.00 | 9.21 ± 0.341 | 3.70 |
Quantification was done using the RNase T1 digestion products 5′-[m5U]ΨCGp-3′ and 5′-A[s4U]UAGp-3′, which were found to yield linear and consistent results. CV, coefficients of variation.
Analysis of quantitative signature digestion products of E. coli using RNase T1
With the figures of merit for LC-MS analysis of isotopically labeled RNase digestion products determined, the next step was to confirm that the potential quantitative signature digestion products meet criteria (iii) and (iv). Escherichia coli tRNAs were digested with RNase T1 and differentially labeled. Based on the dynamic range found above and on typical experimental variations in tRNA abundance (4,6), mixtures were prepared at 18O:16O ratios of 0.33, 1.00 and 3.00. These mixtures were analyzed in triplicate and the experimental isotopic ratios (measured at the 95% confidence level), were determined. In addition, for each RNase T1 16O:18O isotopic pair that yielded a linear response for these ratios, the RNase T1 digestion product was sequenced using CID to confirm the identity of the quantitative signature digestion product. As a result, a total of 29 RNase T1 signature digestion products were identified that meet all of the criteria for use in the relative quantification of E. coli tRNAs (Table 4).
Table 4.
Experimentally verified quantitative signature digestion products from an RNase T1 digest of E. coli tRNA
| tRNA | Sequence of qSDP | Mass |
|---|---|---|
| Ala 1, 2 | [m7G]UCUGp | 1639.21 |
| Arg 1, 2 | [m2A]ACCGp | 1645.26 |
| Asn | pUCCUCUGp | 2276.22 |
| Asp | CCUQUC[m2A]CGp | 3009.45 |
| Cys | CA[ms2i6A]AΨCCGp | 2685.41 |
| U[s4U]AACAAAGp | 2941.39 | |
| Glu 1, 2, 3 | UCCCCUUCGp | 2806.34 |
| AAUCCCCUAGp | 3182.43 | |
| CCCU[mnm5s2U]UC[m2A]CGp | 3208.42 | |
| Gly 1 | AUUCCCUUCGp | 3136.37 |
| Gly 2 | CCU[Um]CCAAGp | 2867.39 |
| Gly 3 | AAUAGp | 1656.24 |
| His | UU[m7G]UCGp | 1945.24 |
| AUUQUGp | 2081.30 | |
| [m2A]ΨΨCCAGp | 2257.31 | |
| AAUCCCAUUAGp | 3512.46 | |
| Ini 1, 2 | TΨCAAAUCCGp | 3197.43 |
| [Cm]UCAUAACCCGp | 3501.48 | |
| Leu 1 | UCCCCCCCCUCGp | 3720.48 |
| Phe | A[s4U]AGp | 1343.16 |
| U[m7G][acp3U]CCUUGp | 2657.35 | |
| AA[ms2i6A]AΨCCCCGp | 3319.51 | |
| Ser 1, 2 | A[ms2i6A]AACCGp | 2403.40 |
| Ser 1, 4, 5 | AAAGp | 1350.21 |
| Ser 3 | CUCCC[s2C]UGp | 2516.29 |
| Trp | UCUCUCCGp | 2501.30 |
| U[Cm]UCCA[ms2i6A]AACCGp | 3944.59 | |
| Val 1 | AU[s4U]AGp | 1649.19 |
| Tyr 1, 2 | ACUQUA[ms2i6A]AΨCUGp | 4098.61 |
These analyses revealed that the primary reason not all of the signature digestion products listed in Supplementary Table S1 were experimentally verified (Table 4) is due to the lack of linearity at various 18O:16O ratios. If a signature digestion product does not produce the expected I18/I16 value in a specific mixture, then that signature digestion product cannot be used to verify differences in an unknown system. The experimental I18/I16 values vary most from the theoretical primarily due to ions of low signal-to-noise ratio. Some digestion products, such as AAUCCCAUUAGp from tRNAHis, can be used for relative quantification at more than one charge state; however, other digestion products, such as UCUCUCCGp from tRNATrp, produce a linear response at only one charge state.
Application of quantitative signature digestion products
To illustrate the application of qSDPs for the characterization of a mixture of tRNAs, E. coli was cultured in two different media conditions: enriched MOPS media and minimal MOPS media. The growth curves contained in Supplementary Figure S2 show that E. coli grows faster in the enriched MOPS media than in the minimal MOPS media, as expected. All cultures in both media were harvested at mid-log phase between A600 of 0.50 to 0.70.
To monitor the changes in tRNA abundance based on different culturing media, the RNase T1 qSDPs were analyzed using isotopic labeling and LC-MS. As stated above, tRNAs from the enriched MOPS media were enzymatically digested and labeled with 18O; tRNAs from the minimal MOPS media were enzymatically digested and labeled with 16O. After digestion, these samples were combined and analyzed. Based on the previously described figures of merit, this qSDP approach is capable of detecting an increase in tRNA abundance of 30% or higher and a decrease in tRNA abundance of 10% or lower.
Table 5 summarizes the differences in tRNA abundances determined using RNase T1 qSDPs measured from three biological replicates. Two tRNA families were found to increase in relative abundance when E. coli was cultured in enriched MOPS media: Gly and Ser. Even more specific information could be obtained for the isoaccepting tRNAs of Glycine, where all three isoacceptors (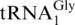 ,
,  and
and 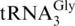 ) were found to increase in the enriched MOPS media relative to the minimal MOPS media. Two tRNA families were found to decrease in relative abundance when cultured in enriched versus minimal MOPS media, Cys and Tyr, although no information is available on differences at the isoacceptor level for Tyr. Six tRNA families were found to have no change in relative abundance between the two culturing conditions: Ala, Arg, Asn, Glu, Phe and Trp. More specific information at the level of individual isoacceptors was obtained from
) were found to increase in the enriched MOPS media relative to the minimal MOPS media. Two tRNA families were found to decrease in relative abundance when cultured in enriched versus minimal MOPS media, Cys and Tyr, although no information is available on differences at the isoacceptor level for Tyr. Six tRNA families were found to have no change in relative abundance between the two culturing conditions: Ala, Arg, Asn, Glu, Phe and Trp. More specific information at the level of individual isoacceptors was obtained from  ,
,
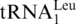 and
and 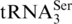 , where for each of these three isoacceptors no change in relative abundance between the two culturing conditions was detected.
, where for each of these three isoacceptors no change in relative abundance between the two culturing conditions was detected.
Table 5.
Analysis of RNase T1 quantitative signature digestion products from E. coli
| tRNA | qSDP sequence | Experimental I18/I16 | CV (%) |
|---|---|---|---|
| Decrease in relative abundance | |||
| Cys | CA[ms2i6A]AΨCCGp | 0.75 | 19 |
| Cys | U[s4U]AACAAAGp | 0.67 | 25 |
| Tyr 1, 2 | ACUQUA[ms2i6A]AΨCUGp | 0.60 | 18 |
| Increase in relative abundance | |||
| Gly 1 | AUUCCCUUCGp | 1.44 | 26 |
| Gly 2 | CCU[Um]CCAAGp | 1.28 | 24 |
| Gly 3 | AAUAGp | 1.88 | 13 |
| Ser 1, 4, 5 | AAAGp | 2.43 | 18 |
| Ser 1, 2 | A[ms2i6A]AACCGp | 1.42 | 10 |
| No change in relative abundance | |||
| Ala 1, 2 | [m7G]UCUGp | 1.17 | 15 |
| Arg 1, 2 | [m2A]ACCGp | 1.04 | 20 |
| Asn | UCCUCUGp | 1.21 | 15 |
| Glu 1, 2, 3 | AAUCCCCUAGp | 1.06 | 15 |
| Glu 1, 2, 3 | UCCCCUUCGp | 1.20 | 15 |
| Leu 1 | UCCCCCCCCUCGp | 1.09 | 15 |
| Phe | AA[ms2i6A]AΨCCCCGp | 1.31 | 13 |
| Phe | A[s4U]AGp-3′ | 1.22 | 25 |
| Phe | U[m7G][acp3U]CCUUGp | 1.21 | 25 |
| Ser 3 | CUCCC[s2C]UGp | 1.00 | 17 |
| Trp | UCUCUCCGp | 1.21 | 27 |
| Trp | U[Cm]UCCA[ms2i6A]AACCGp | 1.29 | 22 |
| Val 1 | AU[s4U]AGp | 0.86 | 16 |
| Indeterminate | |||
| His | UU[m7G]UCGp | 1.80 | 24 |
| His | AAUCCCAUUAGp | 1.36 | 21 |
| His | [m2A]ΨΨCCAGp | 1.00 | 27 |
| Ini 1, 2 | TΨCAAAUCCGp | 1.51 | 23 |
| Ini 1, 2 | [Cm]UCAUAACCCGp | 0.97 | 41 |
CV, coefficients of variation.
The results obtained from tRNAHis and tRNAIni were the only data that were inconsistent and could not allow a determination of the relative change in these tRNA families. Two qSDPs for His denoted an increase in relative abundance while one qSDP denoted no change in abundance with growth media. Similarly, one qSDP for Ini showed an increase in relative abundance while another qSDP showed no change in abundance with growth conditions. As all qSDPs were validated for linear response prior to this application, these results could suggest differences arising from post-transcriptional modifications within the tRNA. Overall, using only this minimal set of RNase T1 qSDPs, relative changes in tRNA abundance could be monitored for 14 of the 22 tRNA families. Changes in relative abundance ranged from a decrease of 40% (for Tyr) to an increase of 143% for Ser, and the coefficients of variation for three technical replicates each of the three biological samples ranged from 15 to 25%.
DISCUSSION
Accuracy and precision of the qSDP approach
The results above demonstrate that an LC-MS platform can be used for the relative quantification of tRNAs without requiring extensive fractionation or purification of individual tRNAs before analysis. The criteria for establishing qSDPs were evaluated using tRNA standards and tRNAs isolated from E. coli. During the course of the method development and validation steps, the relevant figures of merit for this approach have been determined. Not surprisingly, the analytical variability is lower than the biological variability, with biological variability ultimately determining the precision associated with measuring relative changes in tRNA amounts. As noted by the representative data in Table 3, the coefficients of variation for measuring isotope ratios are typically less than 10%, unless the 16O:18O ratio is significantly less than 1.00. Similarly, the accuracy of measuring isotope ratios is also greater when the ratio is more than 1.00, due to the overlap of the natural A+2 isotope contribution of the 16O-labeled oligonucleotide with the A isotope contribution of the 18O-labeled oligonucleotide. It was found that isotope ratios >1.00 can be accurately measured at the 95% confidence interval.
To evaluate the variability of this method for the characterization of tRNAs isolated from biological systems, six separate cultures of E. coli were grown. Three cultures were grown in minimal MOPS media and three cultures were grown in an enriched MOPS media. These six cultures resulted in three sets of biological replicates, where the tRNAs isolated from E. coli grown in minimal media were labeled with 16O and the tRNAs isolated from E. coli grown in enriched media were labeled with 18O. Each biological replicate was analyzed in triplicate by the LC-MS method developed here. The detected isotope ratios for the qSDPs of tRNAs from the three biological replicates were then evaluated using a paired t-test to identify any statistically significant differences in the experimental data. The results of those analyses found no significant differences in the isotope ratios, thus the values for all nine analyses (three technical replicates of the three biological replicates) were then evaluated to generate the results presented in Table 5. As noted above, the reproducibility of the isotope ratio measurements decreased when analyzing multiple biological replicates, with coefficients of variation in the range of 15–25%. Based on the accuracy and precision identified from these proof-of-principle studies, we propose that this LC-MS method can be used to measure relative changes in tRNA concentration, specifically an increase in tRNA abundance of 30% or higher and a decrease in tRNA abundance of 10% or lower
Coverage of tRNA families by the qSDP approach
The data above illustrate the process for identifying and utilizing qSDPs for the quantitative analysis of tRNAs. Although the number of RNase T1 qSDPs that were validated for use (29, Table 4) was about half of the number potentially available (64, Supplementary Table S1), experimental validation of qSDPs is limited, in large part, by the complexity of the sample mixture being analyzed. In many cases, multiple RNase T1 digestion products co-eluted, which leads to challenges in detecting qSDPs at a sufficient signal-to-noise ratio for quantitative purposes (criterion iv). That limitation should be possible to overcome through higher resolution chromatographic separations, such as UPLC, or through an initial fractionation of the sample via approaches such as affinity purification.
Although RNase T1 qSDPs yielded information from 14 of the 22 tRNA families, only a few isoaccepting tRNAs were specifically monitored. Greater coverage of all the tRNAs present in E. coli (or any organism) will require the application of qSDPs generated from multiple RNases. For example, from the potential qSDPs listed in Supplementary Tables S2 and S3, addition of RNases U2 and A would allow for all tRNA families from E. coli to be monitored, and would significantly increase the number of isoaccepting tRNAs that could be characterized. Furthermore, redundant coverage of tRNAs would provide multiple, independent measurements of changes in the relative abundance of specific tRNAs, allowing for improved precision when measuring changes in relative abundances.
Another approach that could be explored would be to examine the outcome of reverse labeling the samples. Here, to illustrate the process, we chose to label the minimal media culture with 16O and the enriched media culture with 18O. As noted above, accuracy improves when the 18O-labeled digestion products are in excess to the 16O-labeled digestion products. Because one can not necessarily predict a priori which tRNAs will increase in relative abundance with changing culturing conditions, it may prove advantageous to compare data where the labeling of different cultures (or control versus experimental samples) is performed both ways; that is, one analysis would involve labeling the control with 16O and a second analysis would involve labeling the control with 18O. While the additional step will increase the overall analysis time, the accuracy of the data should be improved.
Quantitative signature digestion products for determining changes in the relative abundance of specific tRNAs have been described. The incorporation of an isotopic label during the enzymatic hydrolysis of tRNAs allows for their relative quantification using LC-MS. The criteria for identifying qSDPs from the complete pool of signature digestion products have been presented, and this process was illustrated using RNase T1 digestion products from E. coli. With a lower limit of detection at 100 fmol and a dynamic range of labeling from 10:1 to 1:5 heavy-to-light ratio, this LC-MS approach has the ability to provide a large amount of information with a minimal amount of sample. After demonstrating the process for identifying qSDPs, a potential application of this approach was illustrated by comparing relative tRNA abundances for E. coli grown in enriched and minimal MOPS media. These first studies illustrate the quantitative characterization of 14 tRNA families using RNase T1 qSDPs. Importantly, these studies identify a number of improvements that are possible for the qSDP approach, which should allow for a more complete and accurate characterization, including individual tRNA modification status, of all the tRNAs present in the organism. Efforts to enhance this method are currently underway.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Science Foundation (CHE 0602413 cofunded by the MPS/CHE and BIO/IDBR Divisions, and by the MPS Office of Multidisciplinary Activities and CHE 0910751 cofunded by the MPS/CHE and BIO/MCB Divisions); National Institutes of Health (RR019900); University of Cincinnati. Funding for open access charge: National Science Foundation (CHE0910751).
Conflict of interest statement. None declared.
Supplementary Material
REFERENCES
- 1.Chen D, Texada DE. Low-usage codons and rare codons of Escherichia coli. Gene Ther. Mol. Biol. 2006;10:1–12. [Google Scholar]
- 2.Ikemura T. Correlation between the abundance of Escherichia coli transfer RNAs and the occurrence of the respective codons in its protein genes. J. Mol. Biol. 1981;146:1–21. doi: 10.1016/0022-2836(81)90363-6. [DOI] [PubMed] [Google Scholar]
- 3.Ikemura T. Correlation between the abundance of Escherichia coli transfer RNAs and the occurence of the respective codons in its protein genes: a proposal for a synonymous codon choice that is optimal for the E. coli translational system. J. Mol. Biol. 1981;151:389–409. doi: 10.1016/0022-2836(81)90003-6. [DOI] [PubMed] [Google Scholar]
- 4.Dong H, Nilsson L, Kurland CG. Co-variation of tRNA abundance and codon usage in Escherichia coli at different growth rates. J. Mol. Biol. 1996;260:649–663. doi: 10.1006/jmbi.1996.0428. [DOI] [PubMed] [Google Scholar]
- 5.Dittmar KA, Mobley EM, Radek AJ, Pan T. Exploring the regulation of tRNA distribution on the genomic scale. J. Mol. Biol. 2004;337:31–47. doi: 10.1016/j.jmb.2004.01.024. [DOI] [PubMed] [Google Scholar]
- 6.Dittmar KA, Sørensen MA, Elf J, Ehrenberg M, Pan T. Selective charging of tRNA isoacceptors induced by amino-acid starvation. EMBO Rep. 2005;6:151–157. doi: 10.1038/sj.embor.7400341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pavon-Eternod M, Gomes S, Geslain R, Dai Q, Rosner MR, Pan T. tRNA over-expression in breast cancer and functional consequences. Nucleic Acids Res. 2009;37:7268–7280. doi: 10.1093/nar/gkp787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Meng Z, Limbach PA. RNase mapping of intact nucleic acids by electrospray ionization Fourier transform ion cyclotron resonance mass spectrometry (ESI-FTICRMS) and 18O labeling. Int. J. Mass Spectrom. 2004;234:37–44. [Google Scholar]
- 9.Meng Z, Limbach PA. Quantitation of ribonucleic acids using 18O labeling and mass spectrometry. Anal. Chem. 2005;77:1891–1895. doi: 10.1021/ac048801y. [DOI] [PubMed] [Google Scholar]
- 10.Hossain M, Limbach PA. Mass spectrometry-based detection of transfer RNAs by their signature endonuclease digestion products. RNA. 2007;13:295–303. doi: 10.1261/rna.272507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hossain M, Limbach PA. Multiple endonucleases improve MALDI-MS signature digestion product detection of bacterial transfer RNAs. Anal. Bioanal. Chem. 2009;394:1125–1135. doi: 10.1007/s00216-008-2562-2. [DOI] [PubMed] [Google Scholar]
- 12.Kowalak JA, Pomerantz SC, Crain PF, McCloskey JA. A novel method for the determination of posttranscriptional modifications in RNA by mass spectrometry. Nucleic Acids Res. 1993;21:4577–4585. doi: 10.1093/nar/21.19.4577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Matthiesen R, Kirpekar F. Identification of RNA molecules by specific enzyme digestion and mass spectrometry: software for and implementation of RNA mass mapping. Nucleic Acids Res. 2009;37:e48. doi: 10.1093/nar/gkp139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nakayama H, Akiyama M, Taoka M, Yamauchi Y, Nobe Y, Ishikawa H, Takahashi N, Isobe T. Ariadne: a database search engine for identification and chemical analysis of RNA using tandem mass spectrometry data. Nucleic Acids Res. 2009;37:e47. doi: 10.1093/nar/gkp099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lenz C, Kühn-Hölsken E, Urlaub H. Detection of protein-RNA cross-links by NanoLC-ESI-MS/MS using precursor ion scanning and multiple reaction monitoring (MRM) experiments. J. Am. Soc. Mass Spectrom. 2007;18:869–881. doi: 10.1016/j.jasms.2007.01.013. [DOI] [PubMed] [Google Scholar]
- 16.Taoka M, Yamauchi Y, Nobe Y, Masaki S, Nakayama H, Ishikawa H, Takahashi N, Isobe T. An analytical platform for mass spectrometry-based identification and chemical analysis of RNA in ribonucleoprotein complexes. Nucleic Acids Res. 2009;37:e140. doi: 10.1093/nar/gkp732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Apffel A, Chakel JA, Fischer S, Lichtenwalter K, Hancock WS. Analysis of oligonucleotides by HPLC-electrospray ionization mass spectrometry. Anal. Chem. 1997;69:1320–1325. doi: 10.1021/ac960916h. [DOI] [PubMed] [Google Scholar]
- 18.Oberacher H, Niederstatter H, Casetta B, Parson W. Detection of DNA sequence variations in homo- and heterozygous samples via molecular mass measurements by electrospray ionization time-of-flight mass spectrometry. Anal. Chem. 2005;77:4999–5008. doi: 10.1021/ac050399f. [DOI] [PubMed] [Google Scholar]
- 19.Oberacher H, Niederstatter H, Casetta B, Parson W. Some guidelines for the analysis of genomic DNA by PCR-LC-ESI-MS. J. Am. Soc. Mass Spectrom. 2006;17:124–129. doi: 10.1016/j.jasms.2005.09.012. [DOI] [PubMed] [Google Scholar]
- 20.Oberacher H, Niederstatter H, Parson W. Characterization of synthetic nucleic acids by electrospray ionization quadrupole time-of-flight mass spectrometry. J. Mass Spectrom. 2005;40:932–945. doi: 10.1002/jms.870. [DOI] [PubMed] [Google Scholar]
- 21.Pomerantz SC, McCloskey JA. Detection of the common RNA nucleoside pseudouridine in mixtures of oligonucleotides by mass spectrometry. Anal. Chem. 2005;77:4687–4697. doi: 10.1021/ac058023p. [DOI] [PubMed] [Google Scholar]
- 22.Fountain KJ, Gilar M, Gebler JC. Electrospray ionization mass spectrometric analysis of nucleic acids using high-throughput on-line desalting. Rapid Commun. Mass Spectrom. 2004;18:1295–1302. doi: 10.1002/rcm.1481. [DOI] [PubMed] [Google Scholar]
- 23.Meng Z, Limbach PA. Quantitation of ribonucleic acids using 18O labeling and mass spectrometry. Anal. Chem. 2005;77:1891–1895. doi: 10.1021/ac048801y. [DOI] [PubMed] [Google Scholar]
- 24.Berhane BT, Limbach PA. Stable isotope labeling for matrix-assisted laser desorption/ionization mass spectrometry and post-source decay of analysis of ribonucleic acids. J. Mass Spectrom. 2003;38:872–878. doi: 10.1002/jms.504. [DOI] [PubMed] [Google Scholar]
- 25.Sprinzl M, Vassilenko KS. Compilation of tRNA sequences and sequences of tRNA genes. Nucleic Acid Res. 2005;33:D139–D140. doi: 10.1093/nar/gki012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Limbach PA, Crain PF, McCloskey JA. Characterization of oligonucleotides and nucleic acids by mass spectrometry. Curr. Opin. Biotechnol. 1995;6:96–102. doi: 10.1016/0958-1669(95)80015-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.