

DNA polymerase-alpha (pol-alpha) is essential for eukaryotic replication but lacks proofreading activity. Its turnover is regulated by the E2 Ubc4 and the E3 Not4, which are known transcriptional regulators. This pathway likely prevents accumulation of the potential mutator pol-alpha to promote genome stability.
Abstract
The accurate duplication of chromosomal DNA is required to maintain genomic integrity. However, from an evolutionary point of view, a low mutation rate during DNA replication is desirable. One way to strike the right balance between accuracy and limited mutagenesis is to use a DNA polymerase that lacks proofreading activity but contributes to DNA replication in a very restricted manner. DNA polymerase-α fits this purpose exactly, but little is known about its regulation at the replication fork. Minichromosome maintenance protein (Mcm) 10 regulates the stability of the catalytic subunit of pol-α in budding yeast and human cells. Cdc17, the catalytic subunit of pol-α in yeast, is rapidly degraded after depletion of Mcm10. Here we show that Ubc4 and Not4 are required for Cdc17 destabilization. Disruption of Cdc17 turnover resulted in sensitivity to hydroxyurea, suggesting that this pathway is important for DNA replication. Furthermore, overexpression of Cdc17 in ubc4 and not4 mutants caused slow growth and synthetic dosage lethality, respectively. Our data suggest that Cdc17 levels are very tightly regulated through the opposing forces of Ubc4 and Not4 (destabilization) and Mcm10 (stabilization). We conclude that regular turnover of Cdc17 via Ubc4 and Not4 is required for proper cell proliferation.
INTRODUCTION
The accurate duplication of the genome is crucial for the prolonged health of eukaryotic organisms. Inaccurate DNA replication and/or replication of any portion of the genome more than once can result in genomic instability, which is a consistently observed hallmark of cancer cells (Vaziri et al., 2003; Venkatesan et al., 2007). It is crucial, therefore, to understand the entire process of DNA replication. The initiation of DNA replication requires the coordinated recruitment of several proteins. Prereplicative complexes (Diffley et al., 1994), including the core helicase Mcm2–7 (Bochman and Schwacha, 2008), form at origins of replication, are converted to pre-initiation complexes (Zou and Stillman, 1998), and DNA is subsequently unwound (reviewed in Bell and Dutta, 2002). Once the DNA is unwound, the accuracy of DNA replication depends in large part upon DNA polymerases (pol)-α, -δ, and -ε, all of which coordinate to synthesize the nascent copy of DNA during replication in eukaryotes (Burgers, 2009). Highlighting the importance of accurate DNA replication, 43% of mice homozygous for a proofreading-deficient allele of pol-δ develop cancer, primarily lymphoma, but also squamous-cell carcinoma (Goldsby et al., 2001). Interestingly, pol-α, the only enzyme that can synthesize DNA de novo, naturally lacks proofreading activity (Morrison et al., 1991). Unlike pol-δ, pol-α is foremost a replicative — and not a repair — polymerase (Wu et al., 2001; Wang et al., 2004). The lack of proofreading activity likely reflects one mechanism by which nature enforces evolution. However, this raises the question of how humans use a potentially mutagenic polymerase responsible for the initiation of more than 30 million Okazaki fragments without regularly developing cancer. Fortunately, pol-α incorporates only 12–20 nucleotides before turning over bulk DNA synthesis to either pol-δ (lagging strand) or pol-ε (leading strand), both of which are more processive and contain proofreading activity (Nethanel and Kaufmann, 1990; Morrison et al., 1991; Waga and Stillman, 1994; Garg et al., 2004; Pursell et al., 2007; Nick McElhinny et al., 2008).
The fact that pol-α is limited to synthesizing only 12–20 nucleotides of DNA implies a very tight regulation of this enzyme. Our laboratory and others have shown previously that minichromosome maintenance (Mcm) 10 interacts with the catalytic subunit of pol-α in budding and fission yeast and higher eukaryotes (Fien et al., 2004; Ricke and Bielinsky, 2004, 2006; Chattopadhyay and Bielinsky, 2007; Zhu et al., 2007; Robertson et al., 2008). Mcm10 is an essential DNA replication factor that is conserved from yeast to humans (Merchant et al., 1997; Aves et al., 1998; Homesley et al., 2000; Izumi et al., 2000). It associates with replication origins before replication initiates and is required for the stable association of pol-α with chromatin (Ricke and Bielinsky, 2004). In addition to interacting with the catalytic subunit of pol-α, Mcm10 also binds to proliferating cell nuclear antigen (PCNA), the processivity clamp for both DNA pol-δ and -ε. The interaction between Mcm10 and PCNA is essential in budding yeast and depends on diubiquitination of Mcm10 (Das-Bradoo et al., 2006). At the same time, this modification appears to inhibit the interaction between Mcm10 and pol-α (Das-Bradoo et al., 2006). These data suggest that Mcm10 plays an integral role in the switch from pol-α to pol-δ and pol-ε. As mentioned above, Mcm10 does not only interact with Cdc17 it also regulates its stability (Ricke and Bielinsky, 2004), and this function is evolutionarily conserved (Chattopadhyay and Bielinsky, 2007). The catalytic subunit of pol-α is rapidly degraded after depletion of Mcm10 from yeast and human cells (Ricke and Bielinsky, 2004; Chattopadhyay and Bielinsky, 2007). However, the pathway responsible for this degradation is not known.
The ubiquitin-mediated degradation system is a major proteolytic pathway in cells. In this system, ubiquitin, an 8-kDa peptide, is transferred from an ubiquitin activating (E1) enzyme to an ubiquitin conjugating (E2) enzyme, then to an ubiquitin ligase (E3) and finally, to the target protein (Hershko et al., 1983). This process is repeated several times to polyubiquitinate the protein and this serves as a signal for degradation by the proteasome, a large, multisubunit proteolytic machine (Varshavsky, 2005). The fact that Cdc17 is rapidly degraded in the absence of Mcm10 suggests that it might be a target of proteasomal degradation.
Here, we first sought to elucidate the mechanism of Cdc17 degradation in the absence of Mcm10. We found that Cdc17 was degraded via the proteasome and we identified ubiquitin conjugating enzyme (Ubc) 4 and negative regulator of transcription (Not) 4 as the E2 and E3 enzymes required for the destabilization of Cdc17. Although we were unable to detect ubiquitin conjugates of Cdc17, protein turnover clearly depended on ubiquitination. Moreover, overexpression of Cdc17 resulted in slow growth of ubc4Δ mutants and revealed synthetic dosage lethality (SDL) with a NOT4 deletion. Lastly, cooverexpression of Cdc17 and Mcm10 exhibited a similar slow growth phenotype and a significant increase in microsatellite-mediated DNA rearrangements. Our results suggest that Cdc17 levels are carefully regulated in the cell through Ubc4- and Not4-dependent destabilization counterbalanced by Mcm10-dependent stabilization. We propose that tipping this balance in favor of increasing Cdc17 and Mcm10 levels causes genomic instability.
MATERIALS AND METHODS
Strains and Plasmids
Strains are isogenic derivatives of either W303-1a or S288C. All strains carrying gene deletions were constructed using a PCR-based gene disruption method (Brachmann et al., 1998). The genotypes of the yeast strains used in this study are listed in Table 1. All plasmids were constructed using standard molecular cloning techniques. pRS316-NOT4 was constructed by cloning the entire NOT4 coding sequence as well as 497 base pairs of promoter sequence into pRS316. Site-directed mutagenesis was used to generate pRS316-not4-L35A from pRS316-NOT4. The 3HA tag was added by PCR. Strains ABy661 and ABy1051 were constructed by transforming plasmid YEp105, which harbors a synthetic Myc-tagged ubiquitin gene (Ellison and Hochstrasser, 1991), and YEp105-G75,76A (Das-Bradoo et al., 2010) into ABy527, respectively.
Table 1.
List of yeast strains used in this study
| Strain name | Relevant genotype | Source |
|---|---|---|
| Strains derived from W303-1a (MATa ura3-1 ade 2-1 his3-11,-15 leu2-3,-112 can1-100 trp1-1) | ||
| ABy013 | mcm10-1, CDC17::3HA-TRP1 | Ricke and Bielinsky, 2004 |
| ABy342 | mcm10-1, ubc4::LEU2, CDC17::3HA-TRP1 | This study |
| ABy455 | mcm10-1, UBC4::NES-3HA-URA3, CDC17::3HA-TRP1 | This study |
| ABy529 | mcm10-1, not4::LEU2, CDC17::3HA-TRP1 | This study |
| ABy363 | mcm10-1, ubc5::LEU2, CDC17::3HA-TRP1 | This study |
| ABy359 | mcm10-1, rad6::LEU2, CDC17::3HA-TRP1 | This study |
| ABy360 | mcm10-1, ubc13::LEU2, CDC17::3HA-TRP1 | This study |
| ABy361 | mcm10-1, mms2::LEU2, CDC17::3HA-TRP1 | This study |
| ABy781 | mcm10-1, CDC17::3HA-TRP1, pRS316 | This study |
| ABy762 | mcm10-1, not4::LEU2, CDC17::3HA-TRP1, pRS316 | This study |
| ABy738 | mcm10-1, not4::LEU2, CDC17::3HA-TRP1, pRS316-NOT4 | This study |
| ABy739 | mcm10-1, not4::LEU2, CDC17::3HA-TRP1, pRS316-not4-L35A | This study |
| ABy596 | mcm10-1, CDC17::3HA-TRP1, NOT4::3HA-URA3 | This study |
| ABy740 | mcm10-1, not4::LEU2, CDC17::3HA-TRP1, pRS316-NOT4-3HA | This study |
| ABy741 | mcm10-1, not4::LEU2, CDC17::3HA-TRP1, pRS316-not4-L35A-3HA | This study |
| ABy448 | CDC17::3HA-TRP1 | This study |
| ABy607 | ubc4::LEU2, CDC17::3HA-TRP1 | This study |
| ABy597 | not4::LEU2, CDC17::3HA-TRP1 | This study |
| ABy662 | UBC4::NES-3HA-URA3, CDC17::3HA-TRP1 | This study |
| ABy764 | ubc5::LEU2, CDC17::3HA-TRP1 | This study |
| ABy605 | mcm10-1, UBC4::GFP-HIS3 | This study |
| ABy660 | mcm10-1, UBC4::NES-GFP-HIS3 | This study |
| ABy527 | mcm10-1, pdr5::LEU2, rpn4::HIS3, CDC17::2HA-URA3 | This study |
| ABy661 | mcm10-1, pdr5::LEU2, rpn4::HIS3, CDC17::2HA-URA3, YEp105 | This study |
| ABy1051 | mcm10-1, pdr5::LEU2, rpn4::HIS3, CDC17::2HA-URA3, YEp105-G75,76A | This study |
| ABy640 | pSH44, pRS423gal, pRS425gal | This study |
| ABy641 | pSH44, pRS423gal, pRS425gal-CDC17-2HA | This study |
| ABy642 | pSH44, pRS423gal-MCM10-3HA, pRS425gal | This study |
| ABy643 | pSH44, pRS423gal-MCM10-3HA, pRS425gal-CDC17-2HA | This study |
| ABy695 | pRS423gal, pRS424gal, pRS425gal, pRS426gal | This study |
| ABy694 | pRS423gal, pRS424gal, pRS425gal, pRS426gal-CDC17-2HA | This study |
| ABy696 | pRS423gal-MCM10-3HA, pRS424gal, pRS425gal, pRS426gal | This study |
| ABy705 | pRS423gal-MCM10-3HA, pRS424gal, pRS425gal, pRS426gal-CDC17-2HA | This study |
| ABy732 | pRS423gal-MCM10-3HA/POL12-3HA, pRS424gal-PRI1-3HA/PRI2-3HA, pRS425gal, pRS426gal-CDC17-3HA | This study |
| ABy962 | pRS423gal | This study |
| ABy963 | pRS423gal-CDC17-2HA | This study |
| ABy964 | pRS423gal-MCM10-3HA | This study |
| ABy965 | ubc4::LEU2, pRS423gal | This study |
| ABy966 | ubc4::LEU2, pRS423gal-CDC17-2HA | This study |
| ABy967 | ubc4::LEU2, pRS423gal-MCM10-3HA | This study |
| ABy968 | ubc5::LEU2, pRS423gal | this study |
| ABy969 | ubc5::LEU2, pRS423gal-CDC17-2HA | This study |
| ABy970 | ubc5::LEU2, pRS423gal-MCM10-3HA | This study |
| ABy971 | ubc4::LEU2, ubc5::TRP1, pRS423gal | This study |
| ABy972 | ubc4::LEU2, ubc5::TRP1, pRS423gal-CDC17-2HA | This study |
| ABy973 | ubc4::LEU2, ubc5::TRP1, pRS423gal-MCM10-3HA | This study |
| ABy974 | not4::LEU2, pRS423gal | This study |
| ABy975 | not4::LEU2, pRS423gal-CDC17-2HA | This study |
| ABy976 | not4::LEU2, pRS423gal-MCM10-3HA | This study |
| ABy977 | pRS316-TRP1, pRS423gal, pRS425gal | This study |
| ABy978 | pRS316-TRP1, pRS423gal, pRS425gal-CDC17-2HA | This study |
| ABy979 | pRS316-TRP1, pRS423gal-MCM10-3HA, pRS425gal | This study |
| ABy980 | pRS316-TRP1, pRS423gal-MCM10-3HA, pRS425gal-CDC17-2HA | This study |
| ABy831 | ubc4::LEU2, CDC17::3HA-TRP1, pRS426gal-CDC17-2HA | This study |
| ABy837 | not4::LEU2, CDC17::3HA-TRP1, pRS426gal-CDC17-2HA | This study |
| ABy981 | CDC17::3HA-TRP1, pRS426gal-CDC17-2HA | This study |
| Strains derived from S288C (MATa hisΔ1 leu2Δ0 met15Δ0 ura3Δ0) | ||
| ABy150 | MATa hisΔ1 leu2Δ0 met15Δ0 ura3Δ0 | Mortimer and Johnston, 1986 |
| ABy152 | ubc4::KANMX | This study |
| ABy575 | not4::LEU2 | This study |
| ABy574 | UBC4::NES-3HA-URA3 | This study |
| ABy577 | ubc5::LEU2, UBC4::NES-3HA-URA3 | This study |
| GAP510 | ubc4::HIS3, ubc5::LEU2 | Trotter et. al., 2001 |
| ABy258 | pRS426gal-CDC17-2HA | This study |
| ABy259 | rad6::KANMX, pRS426gal-CDC17-2HA | This study |
| ABy260 | ubc4::KANMX, pRS426gal-CDC17-2HA | This study |
| ABy261 | ubc5::KANMX, pRS426gal-CDC17-2HA | This study |
| ABy262 | ubc7::KANMX, pRS426gal-CDC17-2HA | This study |
| ABy263 | ubc8::KANMX, pRS426gal-CDC17-2HA | This study |
| ABy264 | ubc10::KANMX, pRS426gal-CDC17-2HA | This study |
| ABy265 | ubc13::KANMX, pRS426gal-CDC17-2HA | This study |
| ABy266 | mms2::KANMX, pRS426gal-CDC17-2HA | This study |
| ABy267 | rtt101::KANMX, pRS426gal-CDC17-2HA | This study |
| ABy268 | cul3::KANMX, pRS426gal-CDC17-2HA | This study |
| ABy563 | pRS426gal-CDC17-2HA | This study |
| ABy564 | pRS423gal-MCM10-3HA | This study |
| ABy565 | pRS423gal-MCM10-3HA, pRS426gal-CDC17-2HA | This study |
Protein Preparation and Western Blot Analysis
Total protein extracts were prepared as described previously by trichloroacetic acid extraction (Ricke and Bielinsky, 2006). Proteins were separated by SDS-PAGE, transferred to a nitrocellulose membrane, and probed by Western blot with anti-HA (16B12; Covance, Princeton, NJ) for HA-tagged proteins and anti–α-tubulin (B512; Covance). Alexa Fluor 680 (Invitrogen, Carlsbad, CA) secondary antibody was used to visualize proteins with Odyssey v3.0 software on an Odyssey scanner from LI-COR Biosciences (Lincoln, NE).
Protein Overexpression
Strains carrying plasmids for exogenous overexpression of Cdc17, Mcm10, Pol12, Pri1, and Pri2 were grown overnight in minimal medium with 2% raffinose. Proteins were overexpressed from a galactose promoter in the presence of 2% galactose or repressed in the presence of 2% glucose. Total protein was extracted using the trichloroacetic acid extraction protocol. Cdc17-2HA, Mcm10-3HA, Pol12-3HA, Pri1-3HA, Pri2-3HA, and α-tubulin were detected by Western blot using anti-HA (16B12) for HA-tagged proteins and anti–α-tubulin (B512).
Heat Shock Survival Assay
This assay was performed as described previously (Seufert and Jentsch, 1990). Briefly, cells were grown in YPD liquid medium to OD600 = 1.0. Before and after incubation at 52°C for 5 min, appropriate aliquots were spread on YPD plates and incubated at 30°C for 3 d. Colonies were counted and data presented as percentage of colonies formed after heat shock.
Microsatellite Instability Assay
Assay was performed as described previously (Gutierrez and Wang, 2003) except for the following. Seven individual colonies from each strain tested were grown in liquid SC-his-trp-leu minimal medium with 2% galactose to saturation. From each culture, 107 cells were spread on SC-trp plates containing 1 mg/ml 5-FOA and 100 cells were spread on SC-trp plates for a viable cell count. Plates were incubated at 30°C for 4–5 d and FOAR colonies were counted. The frequency of FOAR clones was calculated by dividing the number of FOAR colonies by the number of viable cells. To assess whether microsatellites were altered, pSH44 was recovered from FOAR clones and the microsatellite region was sequenced. For clones in which the microsatellite was unaltered, pSH44 was digested with HindIII to detect gross sequence changes. Sequencing of these plasmids indicated that all of them had lost the microsatellite region, which was replaced with LEU2 sequence that likely recombined with the upstream LEU2 promoter sequence that immediately precedes the microsatellite tract in pSH44 (Henderson and Petes, 1992). We constructed a control plasmid for this assay that had the same auxotrophic markers as pSH44 but lacked a microsatellite tract. To this end, TRP1 was inserted into the multicloning site of pRS316 with KpnI and SacI.
Microscopy
For nuclei staining, DAPI stain (10 μg/ml, Sigma, St. Louis, MO, D1380) was added to cultures for 30 min, washed twice with sterile water, air-dried on a microscope slide, and visualized using a Zeiss AxioplanII microscope with a DIC Alpha Plan-Fluar ×100/1.45 objective and DAPI filter. Ubc4-GFP and Ubc4-NES-GFP were visualized using a GFP filter. All images were captured using an Axiocam HRm camera and Axiovision software 4.0.
RNA Extraction and RT-PCR
Total RNA extraction was performed with the Qiagen RNeasy kit according to the manufacturer's protocol. Total RNA was digested with RNase-free DNase (Ambion, Austin, TX). Digested RNA (30, 6, and 3 ng) was subjected to a one-step RT-PCR reaction using the Superscript III One-Step RT-PCR kit (Invitrogen). The PCR cycle used was as follows: 52°C for 30 min followed by 94°C for 2 min and 30 cycles of 94°C for 15 s, 55°C for 30 s, and 68°C for 30 s. CDC17 was amplified with primer 5′ CDC17-3454 (5′-GCTCTAAGAATGCGTAAGGCTGGTAG-3′) and primer 3′ CDC17-3712 (5′-CAAACGCACCACGTTGAAGCTATC-3′) and ACT1 was amplified with primer 5′ ACT1-511 (5′-GTTACCCAATTGAACACGGTATTGTC-3′) and primer 3′ ACT1-836 (5′-CAAAATGGCGTGAGGTAGAGAGAAAC-3′).
RESULTS
Cdc17 Degradation Is Proteasome-Dependent
Our laboratory has previously shown that Cdc17 is rapidly degraded when Mcm10 is depleted from yeast cells (Ricke and Bielinsky, 2004, 2006). The fact that this pathway is conserved in human cells (Chattopadhyay and Bielinsky, 2007) implies that it serves an important function. Thus, we were interested in elucidating the mechanism of the Cdc17 degradation pathway. Because Cdc17 was degraded very rapidly, we hypothesized that its turnover might be mediated by the proteasome. To test this hypothesis, we inhibited the proteasome and monitored Cdc17 levels in the absence of Mcm10 using the temperature sensitive mcm10-1 mutant (Merchant et al., 1997; Sawyer et al., 2004). We deleted RPN4 and PDR5 to sensitize cells to treatment with the proteasome inhibitor MG132 (Rock et al., 1994; Fleming et al., 2002). As expected, addition of MG132 at 30°C had no effect (Figure 1). When cells were shifted to 37°C in the presence of DMSO, Cdc17 was degraded. In contrast, when cells were shifted to 37°C in the presence of MG132, Cdc17 was stabilized (Figure 1), suggesting that Cdc17 degradation is proteasome-dependent.
Figure 1.
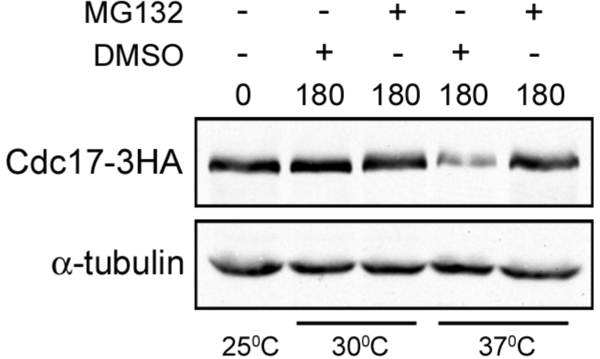
Cdc17 degradation is proteasome-dependent. ABy527 (mcm10-1, pdr5Δ, rpn4Δ) cells were grown in rich liquid medium at 25°C to midlog phase and split in four. MG132 was added to two of the cultures, DMSO was added to the other two, and cultures were shifted either to 30°C or 37°C for 180 min. Cdc17–3HA and α-tubulin were analyzed by Western blot.
Ubc4 Is Required for Cdc17 Degradation
We next wanted to identify the ubiquitin conjugating enzyme (E2) and the ubiquitin ligase (E3) responsible for Cdc17 degradation. There are only 11 E2 enzymes in yeast and only one, Cdc34, is essential (Hochstrasser, 1996). We therefore screened a collection of yeast strains that each lack a different nonessential E2 enzyme by overexpressing Cdc17. In addition, we overexpressed Cdc17 in strains lacking MMS2 (Hofmann and Pickart, 1999), an E2 enzyme variant, and CUL3 and RTT101, both of which are components of separate E3 ligase complexes (Michel et al., 2003). Because overexpressed Cdc17 is unstable (Muzi Falconi et al., 1993), we reasoned that it would rapidly disappear in all strains except the one lacking the E2 enzyme required for its degradation. We overexpressed Cdc17 under the control of a galactose-inducible promoter from a high copy plasmid as described previously (Ricke and Bielinsky, 2006). To monitor Cdc17 stability, we grew cells in liquid medium to midlog phase, induced Cdc17 expression by adding galactose, then added glucose to inhibit Cdc17 expression and observed Cdc17 protein levels by Western blot over the course of three hours. Cdc17 was indeed unstable when overexpressed in our strain background. The protein was nearly undetectable 3 h after glucose addition (Figure 2). In most strains, overexpressed Cdc17 was degraded with kinetics very similar to those observed in wild-type cells with the notable exception of ubc4Δ mutants (Figure 2). Cdc17 was significantly stabilized in ubc4Δ cells compared with all other strains, including ubc5Δ mutants (Figure 2). We determined that Cdc17 overexpression levels in ubc4Δ mutants were ∼10-fold higher than in wild-type cells by diluting our protein extracts (data not shown). Although Ubc4 and Ubc5 are 93% identical and largely functionally redundant, we attribute this discrepancy to the differential regulation of Ubc4 and Ubc5. UBC5 mRNA is of low abundance in log phase cells (Seufert and Jentsch, 1990) and, although present at steady state levels similar to Ubc4, Ubc5 protein is much less stable (Panasenko et al., 2009). We concluded from this data that Ubc4 functions as the E2 enzyme in the ubiquitin-mediated degradation of overexpressed Cdc17 in cycling yeast cells.
Figure 2.

Overexpressed Cdc17 is stable in ubc4Δ cells. Strains were grown overnight in minimal medium with 2% raffinose. Cdc17-2HA was overexpressed from a galactose promoter in the presence of 2% galactose for 2 h. Cells were pelleted, resuspended in YPD, and samples were taken at 0, 60, 120, and 180 min after addition of glucose. Cdc17-2HA and α-tubulin, which served as a loading control, were analyzed by Western blot. The graphs show the quantification of Cdc17 relative to α-tubulin at each time point in each mutant strain relative to wild-type. The wild-type data in each graph are from the same experiment.
Because Ubc4 has been implicated in the degradation of misfolded proteins (Seufert and Jentsch, 1990), it was possible that overexpressed Cdc17 is aberrantly folded and the requirement of Ubc4 for the degradation of overexpressed Cdc17 is not physiologically relevant. Therefore, we determined whether degradation of endogenous Cdc17 also required Ubc4. We deleted UBC4 in mcm10-1 mutants in which Cdc17 is tagged with three hemagglutinin (HA) epitopes and monitored Cdc17 stability in the absence of Mcm10. On Mcm10 depletion at 37°C, Cdc17 was degraded in the presence of Ubc4, as expected (Figure 3, A and B; Ricke and Bielinsky, 2004). Conversely, when Mcm10 was depleted from mcm10-1 ubc4Δ cells, Cdc17 remained stable (Figure 3, A and B). To ensure that Cdc17 stability was specific to mcm10-1 ubc4Δ cells, we monitored Cdc17 stability in mcm10-1 ubc5Δ, mcm10-1 rad6Δ, mcm10-1 ubc13Δ, and mcm10-1 mms2Δ double mutants. None of the other gene deletions had a significant effect (Figure 3, A and B). We observed a slight but reproducible mobility shift in Cdc17–3HA when it was degraded. Whether this shift was due to protein modification is unclear. Notably, this shift was absent in ubc4Δ cells (Figure 3A). We concluded that endogenous Cdc17 degradation following depletion of Mcm10 is Ubc4-dependent.
Figure 3.

Ubc4 is the E2 enzyme required for endogenous Cdc17 degradation in the absence of Mcm10. (A) Asynchronous cultures of ABy448 (W303-1a), ABy013 (mcm10-1), ABy342 (mcm10-1, ubc4Δ), ABy363 (mcm10-1, ubc5Δ), and ABy359 (mcm10-1, rad6Δ) grown at 25°C were shifted to 37°C for 180 min. Cdc17–3HA and α-tubulin were analyzed by Western blot. (B) The graph shows Cdc17/tubulin ratios at each time point for each strain relative to wild-type at time 0 (average of 3 separate experiments, bars represent mean ± SD).
Not4 Is the E3 Ligase Responsible for Cdc17 Destabilization
To identify the E3 ligase that cooperates with Ubc4 in Cdc17 destabilization, we took a candidate approach. Contrary to the small number of E2 enzymes in yeast, there are ∼100 E3 ligases in budding yeast (Kostova et al., 2007). Although it would have been possible to screen 100 deletion strains, we took advantage of the fact that cells lacking Ubc4 along with Ubc5 have several characteristic phenotypes. For example, ubc4Δ ubc5Δ double mutants are resistant to heat shock and translational inhibitors (Seufert and Jentsch, 1990; Chuang and Madura, 2005). To identify candidate E3 ligases, we searched the literature for E3 ligase-deletion strains that phenocopied the heat shock resistance of ubc4Δ ubc5Δ cells. The E3 ligase Not4 fulfilled this criterion (Mulder et al., 2007). Furthermore, Not4 binds Ubc4, has Ubc4-dependent ubiqutination activity in vitro, and associates with the proteasome (Panasenko et al., 2006; Laribee et al., 2007; Mulder et al., 2007). Thus, we deleted NOT4 in mcm10-1 mutants and tested whether this gene is involved in Cdc17 degradation. When we shifted mcm10-1 not4Δ cells to the nonpermissive temperature of 37°C to deplete Mcm10, Cdc17 remained stable in the absence of Mcm10, to the same extent as in mcm10-1 ubc4Δ double mutants (Figure 4, A and B). To ensure that the observed Cdc17 stability was due to the E3 ligase activity of Not4 we constructed not4-L35A, which is unable to bind Ubc4 (Mulder et al., 2007). We performed a temperature shift experiment as described above with mcm10-1 mutants, mcm10-1 not4Δ double mutants, and mcm10-1 not4Δ double mutants supplemented with either pRS316-NOT4 or pRS316-not4-L35A. Cdc17 degradation was largely rescued in cells in which NOT4 was added back, but not in cells with not4-L35A, suggesting that Not4's E3 ligase activity was indeed required for the destabilization of Cdc17 (Figure 4, C and D). The incomplete rescue of Cdc17 degradation was likely due to decreased expression of Not4 protein from the plasmid (Figure 4E, compare middle lane to left lane).
Figure 4.

Not4 is the E3 ligase required for endogenous Cdc17 degradation in the absence of Mcm10. (A) Asynchronous cultures of ABy013 (mcm10-1), ABy342 (mcm10-1, ubc4Δ), and ABy529 (mcm10-1, not4Δ) grown at 25°C were shifted to 37°C for 180 min. Cdc17–3HA and α-tubulin were analyzed by Western blot. (B) The graph shows Cdc17/tubulin ratios at each time point for each strain relative to mcm10-1 at time 0 (average of 3 separate experiments, bars represent mean ± SD). (C) Asynchronous cultures of ABy781 (mcm10-1, pRS316), ABy762 (mcm10-1, not4Δ, pRS316), ABy738 (mcm10-1, not4Δ, pRS316-NOT4), and ABy739 (mcm10-1, not4Δ, pRS316-not4-L35A) grown at 25°C were shifted to 37°C for 180 or 360 min. Cdc17–3HA and α-tubulin were analyzed by Western blot. (D) The graph shows Cdc17/tubulin ratios at each time point for each strain relative to ABy781 at time 0 (average of 3 separate experiments, bars represent mean ± SD). (E) Asynchronous cultures of ABy596 (mcm10-1, NOT4-3HA), ABy740 (mcm10-1, not4Δ, pRS316-NOT4-3HA), and ABy741 (mcm10-1, not4Δ, pRS316-not4-L35A-3HA) were grown in SC-ura liquid medium at 25°C to midlog phase. Not4-3HA and α-tubulin were analyzed by Western blot. (F) ABy661 (mcm10-1, pdr5Δ, rpn4Δ, YEp105) and ABy1051 (mcm10-1, pdr5Δ, rpn4Δ, YEp105-G75,76A) were grown in rich medium to OD600 = 0.6 at 25°C and shifted to 37°C for 3 h. Total protein was precipitated with trichloroacetic acid and analyzed by Western blot using anti-HA and anti-tubulin antibodies.
Because Cdc17 was stabilized in mcm10-1 cells in the presence of MG132 (Figure 1), we attempted to detect ubiquitinated Cdc17. Unfortunately, we were unable to detect ubiquitin-protein conjugates, although we performed immunoprecipitation experiments under both native and denaturing conditions (data not shown). To address whether ubiquitination was necessary for Cdc17 destabilization, we overexpressed synthetic wild-type or mutant ubiquitin in mcm10-1 pdr5Δ rpn4Δ cells. Clearly, expression of the G75,76A ubiquitin mutant (Das-Bradoo et al., 2010), which is incompetent for isopeptide bond formation blocked Cdc17 degradation, whereas wild-type ubiquitin had no effect (Figure 4F). Based on the results presented in Figure 4, we argue that Ubc4 acts in concert with the E3 ligase Not4 to destabilize Cdc17 in the absence of Mcm10. However, it remains unclear whether Cdc17 is a direct target of ubiquitination.
Cdc17 Degradation Occurs in the Nucleus
Because Ubc4 and Not4 localize to the cytoplasm as well as the nucleus, we addressed where in the cell Cdc17 is degraded. Although Cdc17 is localized to the nucleus, it is possible that the protein is translocated to the cytoplasm before degradation. To determine whether Cdc17 is degraded in the nucleus or the cytoplasm we fused a strong nuclear export signal (NES) (Wen et al., 1995) along with 3 HA epitopes to the C terminus of Ubc4 to restrict its localization to the cytoplasm. We confirmed that Ubc4-NES-3HA retained its function by testing the allele in a heat shock assay (Seufert and Jentsch, 1990). As mentioned above, ubc4Δ ubc5Δ cells are resistant to heat shock (Seufert and Jentsch, 1990). If Ubc4-NES-3HA is not a functional enzyme, then UBC4-NES-3HA ubc5Δ double mutants should be resistant to heat shock. However, when we tested this particular strain, it was not resistant to heat shock, suggesting that Ubc4-NES-3HA retained E2 enzyme function (Figure 5A). We also confirmed that Ubc4-NES-3HA was primarily cytoplasmic by fusing GFP to its C-terminus and observing its cellular localization by fluorescence microscopy (Figure 5B). Having verified that Ubc4-NES-3HA is a functional cytoplasmic enzyme, we then determined whether Cdc17 was degraded in the nucleus or the cytoplasm. If Cdc17 degradation occurred in the nucleus, then Cdc17 should have remained stable in mcm10-1 UBC4-NES-3HA double mutants. Conversely, if Cdc17 was degraded in the cytoplasm, then Cdc17 should have been degraded after Mcm10 depletion from mcm10-1 UBC4-NES-3HA cells. Our experiments showed that the former was the case, although Cdc17 was not as stable in UBC4-NES-3HA cells as in ubc4Δ mutants (Figure 5, C and D). We attribute this difference in stability to the fact that a small portion of mcm10-1 UBC4-NES-3HA cells may not have efficiently excluded Ubc4 from the nucleus. Thus, the cells with Ubc4 in the nucleus were able to degrade Cdc17 and this resulted in overall lower levels of Cdc17 in the entire population of cells compared with ubc4Δ mutants. These data strongly suggested that Cdc17 is degraded primarily in the nucleus in an Ubc4- and Not4-dependent manner.
Figure 5.

Cdc17 is degraded primarily in the nucleus. (A) ABy150 (S288C), ABy152 (ubc4Δ), ABy574 (UBC4-NES-3HA), ABy577 (ubc5Δ, UBC4-NES-3HA), and GAP510 (ubc4Δ, ubc5Δ) were heat shocked as described in the Materials and Methods, aliquots were grown on YPD plates for 3 d at 30°C, and colonies were counted to determine the percentage of cells that were viable following heat shock. (B) ABy605 (mcm10-1, UBC4-GFP) and ABy660 (mcm10-1, UBC4-NES-GFP) were examined using fluorescence microscopy. Merge represents GFP and DAPI. Yellow dots represent colocalization of Ubc4-GFP and DAPI-stained nuclei. Red dots represent DAPI-stained nuclei in which Ubc4-GFP is not present. Black circles represent vacuoles. (C) Asynchronous cultures of ABy013 (mcm10-1), ABy342 (mcm10-1, ubc4Δ), ABy455 (mcm10-1, UBC4-NES-3HA), ABy363 (mcm10-1, ubc5Δ), and ABy359 (mcm10-1, rad6Δ) grown at 25°C were shifted to 37°C for 180 min. Cdc17–3HA and α-tubulin were analyzed by Western blot. (D) The graph shows Cdc17/tubulin ratios at each time point for each strain relative to mcm10-1 at time 0 (average of 3 separate experiments, bars represent mean ± SD).
In the light of the fact that Not4 has been strongly implicated in transcriptional regulation and has thus far not been linked to the turnover of replication factors, we wondered whether Cdc17 transcript levels were affected in the various mutants that we analyzed in this study. To this end, we isolated total RNA and performed semiquantitative RT-PCR. We did not observe any significant changes in Cdc17 transcript levels (Figure 6), arguing that Ubc4/Not4 do not affect Cdc17 transcription.
Figure 6.

CDC17 mRNA levels are unchanged in not4Δ mutants. ABy448 (W303-1a), ABy013 (mcm10-1), ABy342 (mcm10-1, ubc4Δ), ABy455 (mcm10-1, UBC4-NES), ABy529 (mcm10-1, not4Δ), ABy363 (mcm10-1, ubc5Δ), ABy359 (mcm10-1, rad6Δ), ABy360 (mcm10-1, ubc13Δ), and ABy361 (mcm10-1, mms2Δ) were grown in rich medium to midlog phase at 25°C and shifted to 37°C for 180 min. Total RNA was extracted from cells taken just before the shift to 37°C and after 180 min at 37°C. Semiquantitative RT-PCR was performed on each sample using 30, 6, and 3 ng total RNA with primers specific for either CDC17 or ACT1.
Cdc17 Levels Are Elevated in the Presence of Mcm10 in ubc4Δ and not4Δ Cells
Cdc17 is transcribed and translated at the G1/S transition of every cell cycle (Wahl et al., 1988), yet steady-state levels of Cdc17 remain unchanged throughout the cell cycle (Muzi Falconi et al., 1993). It is possible that Cdc17 levels remain stable via degradation of any excess Cdc17 by Ubc4 and Not4. Because Mcm10 stabilizes Cdc17 (Ricke and Bielinsky, 2004, 2006; Chattopadhyay and Bielinsky, 2007), we asked whether steady-state levels of the catalytic subunit of pol-α were elevated in the presence of Mcm10-1 when Cdc17 degradation was disrupted. We grew asynchronous cultures of ubc4Δ, UBC4-NES, not4Δ, ubc5Δ, rad6Δ, ubc13Δ, and mms2Δ cells (all in the mcm10-1 background) to midlog phase and analyzed the steady-state levels of Cdc17 by Western blot. The protein accumulated approximately twofold in ubc4Δ, UBC4-NES, and not4Δ cells (Figure 7, A and B), whereas no significant increase was observed in ubc5Δ, rad6Δ, ubc13Δ, or mms2Δ cells. We observed similar trends in ubc4Δ, not4Δ and ubc5Δ mutants that carried the wild-type gene for MCM10, although the effect was most pronounced in not4Δ cells (Figure 7C). We concluded from these data that cells keep Cdc17 levels tightly regulated not only through Ubc4- and Not4-mediated degradation, but also by limiting the amount of Mcm10 that is available to bind and stabilize Cdc17.
Figure 7.

Cdc17 levels are elevated twofold in the presence of Mcm10 in ubc4Δ, UBC4-NES-3HA, and not4Δ cells. (A) Asynchronous cultures of ABy013 (mcm10-1), ABy342 (mcm10-1, ubc4Δ), ABy455 (mcm10-1, UBC4-NES-3HA), ABy363 (mcm10-1, ubc5Δ), ABy359 (mcm10-1, rad6Δ), ABy360 (mcm10-1, ubc13Δ), ABy361 (mcm10-1, mms2Δ), and ABy529 (mcm10-1, not4Δ) were grown at 25°C to midlog phase. Cdc17–3HA and α-tubulin were analyzed by Western blot. (B) The graph shows the fold change in Cdc17 in each strain relative to mcm10-1 (average of 3 separate experiments, bars represent mean ± SD). (C) Asynchronous cultures of ABy448 (W303-1a), ABy607 (ubc4Δ), ABy597 (not4Δ), and ABy764 (ubc5Δ) were grown at 25°C to midlog phase. Cdc17–3HA and α-tubulin were analyzed by Western blot. The values represent the fold change in Cdc17 levels relative to wild-type.
Nuclear Ubc4 Is Important for Replication Stress Tolerance
Because cells keep Cdc17 tightly regulated, we hypothesized that elevation of Cdc17 steady-state levels may have functional consequences during DNA replication. It has been shown previously that not4Δ cells are extremely sensitive to hydroxyurea (HU), which correlates with Not4's role in the transcriptional upregulation of ribonucleotide reductase (Mulder et al., 2005). Cells lacking Ubc4 have been shown to be moderately sensitive to HU, but the reason for this sensitivity was not explained (Mulder et al., 2007), and we were curious as to whether it pertains to the nuclear function of Ubc4. Thus, we subjected UBC4-NES-3HA cells to HU treatment to determine whether Ubc4's nuclear localization is important for replication stress tolerance. Interestingly, UBC4-NES-3HA cells behaved identically to ubc4Δ cells (Figure 8A). Whereas ubc4Δ mutants were moderately sensitive to HU, not4Δ cells were highly sensitive to the drug, as seen previously (Figure 8A). Cells lacking Ubc5 were no more sensitive to HU than wild-type cells. These results suggested that Ubc4 has a function in the nucleus that is important for replication stress tolerance.
Figure 8.

Ubc4 is required in the nucleus for tolerance to hydroxyurea. (A) Successive 10-fold dilutions of ABy448 (W303-1a), ABy607 (ubc4Δ), ABy662 (UBC4-NES-3HA), ABy597 (not4Δ), ABy764 (ubc5Δ), ABy013 (mcm10-1), ABy342 (mcm10-1, ubc4Δ), ABy455 (mcm10-1, UBC4-NES-3HA), ABy529 (mcm10-1, not4Δ), and ABy363 (mcm10-1, ubc5Δ) were grown for 3 d at 30°C on YPD plates and YPD containing 50 mM, 100 mM, and 200 mM HU. (B) Asynchronous cultures of ABy597 (not4Δ) and ABy529 (mcm10-1, not4Δ) were grown to midlog phase. Cdc17–3HA and α-tubulin were analyzed by Western blot in undiluted extracts and extracts diluted 10-fold, 25-fold, and 50-fold.
To address whether Cdc17 plays a role in the observed HU sensitivity, we repeated the experiment in mcm10-1 mutants. Because the mcm10-1 mutation affects Cdc17 stability at 30°C (Ricke and Bielinsky, 2006), we predicted that the HU sensitivity should be alleviated in the double mutants, despite the fact that the mcm10-1 strain displays a higher HU sensitivity than wild-type cells (Figure 8A). This was indeed the case. Surprisingly, even the severe HU sensitivity of not4Δ mutants was suppressed by mcm10-1 (Figure 8A). This suppression went hand-in-hand with reduced Cdc17 levels in mcm10-1 not4Δ double mutants (Figure 8B; we estimate a 2.5- to fivefold reduction compared with not4Δ). Therefore, the level of Cdc17 expression correlates well with the degree of HU sensitivity in not4Δ cells although we cannot exclude the possibility that mcm10-1 ameliorates effects of replication stress in not4Δ mutants in a Cdc17-independent manner.
Overexpression of Cdc17 and Mcm10 Causes Slow Growth
We hypothesized that there may be other defects caused by elevated Cdc17 levels that occurred at low frequency, such that they would not be readily detected. To test this hypothesis we overexpressed Cdc17, along with its binding partner Mcm10, to exacerbate the effect of disrupting Cdc17 degradation. We overexpressed Cdc17 and Mcm10 from plasmids under the control of a galactose promoter (Figure 9A). To our surprise, 10-fold overexpression of Cdc17 and Mcm10 resulted in a significant growth defect in the absence of any exogenous stress (Figure 9B). According to our estimate, growth was inhibited ∼25-fold. Importantly, cells carrying the empty vector controls did not display any growth phenotype (Figure 9B). Overexpression of Cdc17 alone did not affect growth, likely because Mcm10 was not present at high enough levels to recruit excess Cdc17 to chromatin where it functions during DNA replication (Ricke and Bielinsky, 2006). Similarly, overexpression of Mcm10 alone had little effect, suggesting that the primary reason for the proliferation inhibition was the excess of Cdc17. It was possible that the growth defect we observed upon cooverexpression of Cdc17 and Mcm10 was due to sequestration of the rest of the pol-α/primase complex. To address this possibility, we overexpressed the remaining subunits of pol-α/primase, Pol12, Pri1, and Pri2, together with Mcm10 and Cdc17 (Figure 9A). However, this did not rescue the observed phenotype (Figure 9B). From these data we concluded that careful regulation of Cdc17 is required for cells to undergo normal proliferation.
Figure 9.

Overexpression of Cdc17 and Mcm10 causes slow growth. (A) Strains containing the indicated plasmids were grown in media containing 2% raffinose to midlog phase, either 2% galactose (GAL) or 2% glucose (GLU) was added, and cells were grown for an additional 2 h. Cdc17-2HA, Mcm10-3HA, Pol12–3HA, Pri1–3HA, and Pri2–3HA were analyzed by Western blot. (B) Successive 10-fold dilutions of ABy695 (empty vectors), ABy694 (GAL-CDC17-2HA), ABy696 (GAL-MCM10-3HA), ABy705 (GAL-CDC17-2HA, GAL-MCM10-3HA), and ABy732 (GAL-CDC17-2HA, GAL-MCM10-3HA, GAL-POL12–3HA, GAL-PRI1–3HA, GAL-PRI2–3HA) were grown for 3 d at 30°C on SC-his-trp-ura with 2% glucose plates and SC-his-trp-ura with 2% galactose plates.
Overexpression of Cdc17 and Mcm10 Induces Genome Instability
As mentioned earlier, Cdc17 lacks proofreading activity, making it a potentially mutagenic enzyme (Morrison et al., 1991). We therefore reasoned that higher levels of Cdc17 and Mcm10 might cause an elevated mutation rate in cells. To test this idea, we overexpressed either Cdc17 alone, Mcm10 alone, or Cdc17 with Mcm10 in wild-type cells and monitored the ability of these cells to acquire resistance to canavanine, a toxic amino acid analog (Whelan et al., 1979). Individual colonies of each strain were grown in liquid medium containing galactose (to induce protein overexpression) to saturation and then plated on solid medium containing canavanine. A dilution of each culture was also plated on rich solid medium for a viable cell count. The numbers of canavinine-resistant (CanR) colonies were counted after 3–4 d growth at 30°C. Cells overexpressing both Cdc17 and Mcm10 showed a fivefold increase in mutation rate, whereas cells overexpressing Cdc17 or Mcm10 alone showed no significant increase in mutation rate (Figure 10A). Cells containing overexpression vectors for Cdc17 and Mcm10 grown in glucose served as a negative control. We also tested ubc4Δ and not4Δ mutants and they displayed a 1.6- and 2.5-fold increase in CanR colonies (Figure 10A).
Figure 10.

Overexpression of Cdc17 and Mcm10 increases genome instability. (A) Seven individual colonies of ABy563 (pRS426gal-CDC17-2HA), ABy564 (pRS423gal-MCM10-3HA), and ABy565 (pRS423gal-MCM10-3HA, pRS426gal-CDC17-2HA) were grown to saturation in the presence of 2% galactose, and seven colonies of ABy565 were grown to saturation in the presence of 2% glucose. Cells were grown on -arg plates containing canavanine (60 mg/L) to select for mutants and on YPD plates for a viable cell count. Colonies were counted after 2–3 d at 30°C. (B) Strains containing the indicated plasmids were grown in media containing 2% raffinose to midlog phase, either 2% galactose (GAL) or 2% glucose (GLU) was added, and cells were grown for an additional 2 h. Cdc17-2HA and Mcm10-3HA were analyzed by Western blot. (C) Seven individual colonies each from ABy640, ABy641, ABy642, and ABy643 were grown in SC-his-trp-leu with 2% galactose to saturation at 30°C. From each culture, 100 cells were plated on SC-trp with 2% glucose medium and 107 cells were plated on SC-trp with 2% glucose and 1 mg/ml 5-FOA medium. Plates were incubated for 5 d at 30°C and colonies were counted. (D) pSH44 was isolated from 5-FOA-resistant clones and digested with HindIII, which produces a 4.3-kb fragment (backbone) and a 3.6-kb fragment [poly(GT)-URA3 coding sequence] in the wild type plasmid. Digested plasmids showed either no detectable size change (circles) or complex rearrangements (squares). (E) Same as in C, but with the addition of strains ABy977, ABy978, ABy979 and ABy980, which contain pRS316-TRP1 in place of pSH44.
Different pol-α mutants have also been shown to induce microsatellite instability in budding yeast (Gutierrez and Wang, 2003). We hypothesized that elevated levels of Cdc17 and Mcm10 might have the same effect because they interfere with normal proliferation (Figure 9B). To test this hypothesis, we used a plasmid-based assay to measure microsatellite instability (Henderson and Petes, 1992; Gutierrez and Wang, 2003). In this assay, a plasmid (pSH44) containing an in-frame insertion of a poly (GT) tract upstream of URA3 was transformed into cells that contained either empty vector, vectors with galactose-inducible CDC17 or MCM10 alone, or vectors with galactose-inducible CDC17 and MCM10, rendering these cells Ura+ (Henderson and Petes, 1992). Alterations in the poly (GT) tract that change the reading frame result in Ura− cells, and these cells can be selected for on 5-fluoroorotic acid (5-FOA)-containing medium (Boeke et al., 1984). We found that cells overexpressing both Cdc17 and Mcm10 disrupted the URA3 gene at an ∼20-fold higher rate than cells that carried empty vector controls (Figure 9, B and C). Conversely, cells overexpressing Cdc17 alone showed no increase in mutation rate, whereas cells overexpressing Mcm10 alone showed a fourfold increase in mutation rate (Figure 9, B and C). To verify that 5-FOA resistant (FOAR) cells carried mutations in the microsatellite tract we attempted to recover the plasmids of 30 colonies. Only 13 of these gave rise to ampicillin resistant bacteria colonies. DNA sequencing revealed that 5/13 had an intact poly (GT) tract, whereas 8/13, or ∼60%, had lost the microsatellite tract, indicating that the plasmid DNA was rearranged. To confirm this finding, we cleaved the plasmids with HindIII, which should yield a 3.6- and 4.3-kb fragment (Gutierrez and Wang, 2003). All plasmids that had lost the poly (GT) tract displayed DNA rearrangements (Figure 10D). To further ascertain that the rearrangements were dependent on the microsatellite tract, we compared the number of FOAR colonies between pSH44 and pRS316-TRP1, which is devoid of a poly (GT) tract. Whereas the former exhibited a 20-fold change, we only observed a fivefold increase in mutation rate for the latter in cells that cooverexpressed Cdc17 and Mcm10 (Figure 10E). Therefore, the majority of plasmid rearrangements must have been triggered by replication through the microsatellite tract. Together, these findings demonstrate that disregulation of Cdc17 and Mcm10 can induce microsatellite-mediated DNA recombination.
Overexpression of Cdc17 Results in Synthetic Dosage Lethality with not4Δ
Thus far, all of our functional assays used the cooverexpression of Cdc17 and Mcm10. However, if cells are defective for Cdc17 degradation, the protein should accumulate and ultimately interfere with cell viability or fitness independently of cooverexpression of Mcm10. To test this prediction, we overexpressed either Cdc17 or Mcm10 in ubc4Δ, ubc5Δ, ubc4Δ ubc5Δ, and not4Δ mutants. Because ubc4Δ ubc5Δ double and not4Δ single mutants grow more slowly than the other strains, we evaluated cell growth after 3 and 5 d, respectively. Figure 11A shows that Cdc17 and Mcm10 were induced at similar levels in all strains after 1 h in galactose-containing medium. As predicted, ubc4Δ mutants exhibited a significant growth defect when Cdc17, but not Mcm10, was overexpressed (Figure 11B). The effect was even more pronounced in ubc4Δ ubc5Δ double and not4Δ single mutants, which displayed SDL with CDC17 overexpression. In the case of ubc4Δ ubc5Δ double mutants, this was also true for MCM10 (Figure 11B). When we examined Cdc17 expression levels after overnight culture in galactose-containing medium, ubc4Δ and not4Δ cells had accumulated much higher levels of Cdc17 than the wild-type strain (Figure 11C). The amount of Cdc17 protein detected in the different strains correlated well with the observed growth defect. These results are consistent with our claim that Ubc4 and Not4 regulate the degradation of Cdc17 either directly or indirectly.
Figure 11.

Overexpression of Cdc17 in ubc4Δubc5Δ and not4Δ mutants causes synthetic dosage lethality. (A) ABy962 (pRS423gal), ABy963 (GAL-CDC17-2HA), ABy964 (GAL-MCM10-3HA), ABy965 (ubc4Δ, pRS423gal), ABy966 (ubc4Δ, GAL-CDC17-2HA), ABy967 (ubc4Δ, GAL-MCM10-3HA), ABy968 (ubc5Δ, pRS423gal), ABy969 (ubc5Δ, GAL-CDC17-2HA), ABy970 (ubc5Δ, GAL-MCM10-3HA), ABy971 (ubc4Δubc5Δ, pRS423gal), ABy972 (ubc4Δubc5Δ, GAL-CDC17-2HA), ABy973 (ubc4Δubc5Δ, GAL-MCM10-3HA), ABy974 (not4Δ, pRS423gal), ABy975 (not4Δ, GAL-CDC17-2HA), and ABy976 (not4Δ, GAL-MCM10-3HA) were grown in media containing 2% raffinose to midlog phase, either 2% galactose (GAL) or 2% glucose (GLU) was added, and cells were grown for an additional hr. Cdc17-2HA, Mcm10-3HA, and α-tubulin were analyzed by Western blot. The asterisk indicates a nonspecific band recognized by the HA antibody. (B) Successive 10-fold dilutions of the strains in A were grown for the indicated time at 30°C on SC-his-trp-ura with 2% glucose plates and SC-his-trp-ura with 2% galactose plates. (C) ABy981 (CDC17-3HA, pRS426gal-CDC17-2HA), ABy831 (ubc4Δ, CDC17–3HA, pRS426gal-CDC17-2HA), and ABy837 (not4Δ, CDC17–3HA, pRS426gal-CDC17-2HA) were grown overnight in SC-ura medium to midlog phase. Cdc17 and α-tubulin were analyzed by Western blot.
DISCUSSION
In this study, we demonstrate that Cdc17 turnover is dependent on Ubc4 and Not4. This was unexpected because Not4 was originally isolated as a transcriptional regulator (Collart and Struhl, 1994). Not4 has also been shown to possess E3 ubiquitin ligase activity in vitro (Panasenko et al., 2006; Mulder et al., 2007). It is part of the nine-subunit Ccr4-Not complex that regulates trimethylation of histone H3 lysine 4 (Chen et al., 2001; Laribee et al., 2007). Importantly, we show that Cdc17 is not modulated at the transcript level (Figure 6) but rather through ubiquitin-mediated proteasomal degradation (Figures 1 and 4). This degradation is likely triggered when Mcm10 is ubiquitinated and no longer interacts with pol-α (Das-Bradoo et al., 2006) or the replisome becomes unstable (Lee et al., 2010). Our results suggest that yeast cells stabilize only enough Cdc17 to efficiently initiate replication. We propose that increased Cdc17 protein levels above a defined molecular threshold are detrimental to the cell and are therefore rapidly eliminated.
We have identified Cdc17 as a novel target of Not4-mediated degradation. Recently, Jhd2 (JmjC domain-containing histone demethylase) was reported to be a direct target of Not4 (Mersman et al., 2009). In addition, Not4 may have a role in the degradation of Egd2, which is part of the ribosome-associated EGD (enhancer of Gal4 DNA binding) complex (Panasenko et al., 2006). However, none of these proteins has a role in DNA replication. Cdc17 is stable in the absence of Mcm10 in not4Δ cells (Figure 4A) and accumulates in the presence of Mcm10 when NOT4 is deleted (Figure 7C). We take this as evidence that Not4 is required for regular turnover of Cdc17. This is supported by the observation that Cdc17 destabilization is clearly dependent on ubiquitin conjugation (Figure 4F), although it remains elusive whether Cdc17 is a direct target of ubiquitination. In this context, it is worthwhile to note that precedence exists for ubiquitin-independent proteasomal degradation (Schrader et al., 2009). In addition, we excluded that accumulation of Cdc17 in mcm10-1 not4Δ cells (Figure 4A) was due to a failure to degrade Mcm10-1 protein at the nonpermissive temperature (data not shown). Another striking observation supporting the claim that Not4 regulates Cdc17 turnover in conjunction with Ubc4 is provided by the SDL tests (Figure 11). SDL screens have been used to identify potential targets for protein kinases (Makhnevych et al., 2009; Zou et al., 2009) and might also be useful in revealing potential targets for E3 ligases. One question such a screen could address is whether Cdc17 is the only replication factor regulated by Not4 or if Not4 controls the turnover of other replication proteins as well. Mcm10 does not appear to be a potential target as its overexpression had little effect on the viability of not4Δ cells (Figure 11B). Nevertheless, a mutation in MCM10 almost completely alleviated the severe HU-sensitivity of not4Δ cells (Figure 8). Because mcm10-1 is HU-sensitive by itself, one might have expected that it exacerbates the drug sensitivity of the not4Δ strain. We believe that these results are most easily explained by the destabilizing effect that mcm10-1 has on Cdc17 (Ricke and Bielinsky, 2006; Lee et al., 2010).
The data presented here raise the possibility that ubiquitin-mediated degradation of Cdc17 occurs at or in close proximity of the replication fork. There have been other hints that the ubiquitination machinery operates at the replication fork, but direct evidence is lacking to date. For example, Dia2, an F-box protein, has been shown to bind origins of replication and promote genome stability, but no degradation target of Dia2 has been identified (Blake et al., 2006; Koepp et al., 2006; Mimura et al., 2009).
We propose that Ubc4 and Not4 may have a role in degrading Cdc17 to ensure efficient polymerase switching (pol-ε on the leading strand [Pursell et al., 2007] and pol-δ on the lagging strand [Nick McElhinny et al., 2008]). In support of this model, we demonstrated earlier that ubiquitinated Mcm10 binds PCNA, the processivity factor for pol-δ and pol-ε, but not pol-α (Das-Bradoo et al., 2006). Therefore, it is possible that after pol-α synthesizes a short DNA primer, Mcm10 becomes ubiquitinated and interacts with PCNA, causing Cdc17 to be released from Mcm10. Once released, Cdc17 might then be targeted for proteasomal degradation (Figure 12). We stress that a direct interaction between Cdc17 and Not4/Ubc4 is strictly hypothetical at present. Regardless of whether the interaction with Cdc17 is direct or indirect, Not4/Ubc4 could ensure that Cdc17, a potentially mutagenic enzyme, is not loaded back onto the DNA to further extend the RNA/DNA primer. The fivefold elevated mutation rate of cells overexpressing Cdc17 and Mcm10 (Figure 10A) is consistent with a mechanism by which Mcm10 “redelivers” pol-α to chromatin, thus interfering with the switch from pol-α to pol-δ (Figure 12). As a consequence, pol-α would further extend the RNA/DNA primer, which could account for the increased frequency of mutations (Figure 10A). Tight regulation of Mcm10 by ubiquitination is likely a prerequisite for regular turnover of Cdc17. Therefore, Mcm10 has a dual role, not only stabilizing but also providing timely degradation to inhibit the accumulation of Cdc17.
Figure 12.

Overexpression of Mcm10 and Cdc17 may hinder polymerase switching. After pol-α extends the RNA primer (red line) by 12–20 nucleotides, Mcm10 is ubiquitinated at two distinct lysines (yellow star; (Das-Bradoo et al., 2006). PCNA is recruited to the lagging strand, and a polymerase switch occurs between pol-α and pol-δ, after which pol-α is released from ubiquitinated Mcm10 and recognized by Ubc4 and Not4. The question mark denotes the fact that it is currently unclear whether the interaction is direct or indirect. Accumulation of Cdc17 and Mcm10 interferes with the switch to pol-δ, allowing pol-α to synthesize more DNA than in wild-type cells.
Inhibition of replicative pol activity induces chromosomal breaks, even at drug doses that are low enough to not affect the proliferation rate of cells (Glover et al., 1984). In addition, a mutant of Cdc17, pol1–1, increases mutation frequency and microsatellite instability in budding yeast (Gutierrez and Wang, 2003). Furthermore, low levels of pol-α cause chromosomal translocations in budding yeast, even in the presence of a functional S phase checkpoint (Lemoine et al., 2005). All of these investigations demonstrate the effects of a decline in pol-α function. In contrast, we have shown here that increasing steady-state levels of wild-type pol-α causes genomic instability as well. In a plasmid-based in vivo assay, cooverexpression of Cdc17 and Mcm10 resulted in a significant increase in microsatellite-mediated recombination (Figure 10, C–E). These events almost certainly require DNA breakage, and we speculate that breaks occur via improper progression of lagging strand synthesis. In this context it is interesting to note that a recent study in Arabidopsis suggests that the failure to properly switch from pol-α to pol-δ might trigger recombination events at replication forks (Schuermann et al., 2009).
DNA rearrangements have been linked to many different types of cancer (Frohling and Dohner, 2008; Nambiar et al., 2008). We hypothesize that microsatellite-induced instability could arise through increased levels of pol-α and its binding partner, Mcm10. In support of this hypothesis, p180, the catalytic subunit of pol-α in humans, and Mcm10 are up-regulated in glioma cells (Sun et al., 2006). Intriguingly, UbcH5, the human holomog of yeast Ubc4/5, is down-regulated in these cells. Furthermore, p180 and Mcm10 levels are increased, whereas CNOT4 levels, the human homolog of yeast Not4, are decreased in human male germ cell tumors (Korkola et al., 2006). These data indicate that the degradation pathway identified here may also be important in human cells. More specifically, because the levels of p180, Mcm10, UbcH5, and CNOT4 are misregulated in cancer cells, this pathway may be important for cancer prevention and perhaps serve as a novel target for cancer therapy.
ACKNOWLEDGMENTS
We thank Drs. T. S. F. Wang, M. Peter, and P. Burgers for strains and plasmids; J. Wood for critical reading of the manuscript; P. Jauert from the Kirkpatrick laboratory and members of the Bielinsky laboratory for helpful discussions. We acknowledge Paul Enberg for the construction of key yeast strains. We thank Dr. D. Clarke for help with the fluorescence microscope. This work was supported by a National Institutes of Health Cancer Biology Training Grant (CA009138), and a grant from the National Institutes of Health (GM074917). A.-K. B. is a scholar of the Leukemia and Lymphoma Society.
Abbreviations used:
- Mcm
Minichromosome maintenance
- pol-α
DNA polymerase-α
- PCNA
proliferating cell nuclear antigen
- Ubc
ubiquitin conjugating enzyme.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E09-06-0452) on July 21, 2010.
REFERENCES
- Aves S. J., Tongue N., Foster A. J., Hart E. A. The essential Schizosaccharomyces pombe cdc23 DNA replication gene shares structural and functional homology with the Saccharomyces cerevisiae DNA43 (MCM10) gene. Curr. Genetics. 1998;34:164–171. doi: 10.1007/s002940050382. [DOI] [PubMed] [Google Scholar]
- Bell S. P., Dutta A. DNA replication in eukaryotic cells. Annual Rev. Biochem. 2002;71:333–374. doi: 10.1146/annurev.biochem.71.110601.135425. [DOI] [PubMed] [Google Scholar]
- Blake D., Luke B., Kanellis P., Jorgensen P., Goh T., Penfold S., Breitkreutz B. J., Durocher D., Peter M., Tyers M. The F-box protein Dia2 overcomes replication impedance to promote genome stability in Saccharomyces cerevisiae. Genetics. 2006;174:1709–1727. doi: 10.1534/genetics.106.057836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bochman M. L., Schwacha A. The Mcm2–7 complex has in vitro helicase activity. Molecular Cell. 2008;31:287–293. doi: 10.1016/j.molcel.2008.05.020. [DOI] [PubMed] [Google Scholar]
- Boeke J. D., LaCroute F., Fink G. R. A positive selection for mutants lacking orotidine-5′-phosphate decarboxylase activity in yeast: 5-fluoro-orotic acid resistance. Mol. Gen. Genet. 1984;197:345–346. doi: 10.1007/BF00330984. [DOI] [PubMed] [Google Scholar]
- Brachmann C. B., Davies A., Cost G. J., Caputo E., Li J., Hieter P., Boeke J. D. Designer deletion strains derived from Saccharomyces cerevisiae S288C: a useful set of strains and plasmids for PCR-mediated gene disruption and other applications. Yeast (Chichester, England) 1998;14:115–132. doi: 10.1002/(SICI)1097-0061(19980130)14:2<115::AID-YEA204>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Burgers P. M. Polymerase dynamics at the eukaryotic DNA replication fork. J. Biol. Chem. 2009;284:4041–4045. doi: 10.1074/jbc.R800062200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chattopadhyay S., Bielinsky A. K. Human Mcm10 regulates the catalytic subunit of DNA polymerase-alpha and prevents DNA damage during replication. Mol. Biol. Cell. 2007;18:4085–4095. doi: 10.1091/mbc.E06-12-1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J., Rappsilber J., Chiang Y. C., Russell P., Mann M., Denis C. L. Purification and characterization of the 1.0 MDa CCR4-NOT complex identifies two novel components of the complex. J. Mol. Biol. 2001;314:683–694. doi: 10.1006/jmbi.2001.5162. [DOI] [PubMed] [Google Scholar]
- Chuang S. M., Madura K. Saccharomyces cerevisiae Ub-conjugating enzyme Ubc4 binds the proteasome in the presence of translationally damaged proteins. Genetics. 2005;171:1477–1484. doi: 10.1534/genetics.105.046888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collart M. A., Struhl K. NOT1(CDC39), NOT2(CDC36), NOT3, and NOT4 encode a global-negative regulator of transcription that differentially affects TATA-element utilization. Genes Dev. 1994;8:525–537. doi: 10.1101/gad.8.5.525. [DOI] [PubMed] [Google Scholar]
- Das-Bradoo S., Ricke R. M., Bielinsky A. K. Interaction between PCNA and diubiquitinated Mcm10 is essential for cell growth in budding yeast. Mol. Cell. Biol. 2006;26:4806–4817. doi: 10.1128/MCB.02062-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das-Bradoo S., Nguyen H. D., Wood J. L., Ricke R. M., Haworth J. C., Bielinsky A. K. Defects in DNA ligase I trigger PCNA ubiquitylation at Lys 107. Nat. Cell Biol. 2010;12:74–79. doi: 10.1038/ncb2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diffley J. F., Cocker J. H., Dowell S. J., Rowley A. Two steps in the assembly of complexes at yeast replication origins in vivo. Cell. 1994;78:303–316. doi: 10.1016/0092-8674(94)90299-2. [DOI] [PubMed] [Google Scholar]
- Ellison M. J., Hochstrasser M. Epitope-tagged ubiquitin. A new probe for analyzing ubiquitin function. J. Biol. Chem. 1991;266:21150–21157. [PubMed] [Google Scholar]
- Fien K., Cho Y. S., Lee J. K., Raychaudhuri S., Tappin I., Hurwitz J. Primer utilization by DNA polymerase alpha-primase is influenced by its interaction with Mcm10p. J. Biol. Chem. 2004;279:16144–16153. doi: 10.1074/jbc.M400142200. [DOI] [PubMed] [Google Scholar]
- Fleming J. A., Lightcap E. S., Sadis S., Thoroddsen V., Bulawa C. E., Blackman R. K. Complementary whole-genome technologies reveal the cellular response to proteasome inhibition by PS-341. Proc. Natl. Acad. Sci. USA. 2002;99:1461–1466. doi: 10.1073/pnas.032516399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frohling S., Dohner H. Chromosomal abnormalities in cancer. New Engl. J. Med. 2008;359:722–734. doi: 10.1056/NEJMra0803109. [DOI] [PubMed] [Google Scholar]
- Garg P., Stith C. M., Sabouri N., Johansson E., Burgers P. M. Idling by DNA polymerase delta maintains a ligatable nick during lagging-strand DNA replication. Genes Dev. 2004;18:2764–2773. doi: 10.1101/gad.1252304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glover T. W., Berger C., Coyle J., Echo B. DNA polymerase alpha inhibition by aphidicolin induces gaps and breaks at common fragile sites in human chromosomes. Human Genetics. 1984;67:136–142. doi: 10.1007/BF00272988. [DOI] [PubMed] [Google Scholar]
- Goldsby R. E., Lawrence N. A., Hays L. E., Olmsted E. A., Chen X., Singh M., Preston B. D. Defective DNA polymerase-delta proofreading causes cancer susceptibility in mice. Nat. Medicine. 2001;7:638–639. doi: 10.1038/88963. [DOI] [PubMed] [Google Scholar]
- Gutierrez P. J., Wang T. S. Genomic instability induced by mutations in Saccharomyces cerevisiae POL1. Genetics. 2003;165:65–81. doi: 10.1093/genetics/165.1.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson S. T., Petes T. D. Instability of simple sequence DNA in Saccharomyces cerevisiae. Mol. Cell. Biol. 1992;12:2749–2757. doi: 10.1128/mcb.12.6.2749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hershko A., Heller H., Elias S., Ciechanover A. Components of ubiquitin-protein ligase system. Resolution, affinity purification, and role in protein breakdown. J. Biol. Chem. 1983;258:8206–8214. [PubMed] [Google Scholar]
- Hochstrasser M. Ubiquitin-dependent protein degradation. Annual Rev. Genetics. 1996;30:405–439. doi: 10.1146/annurev.genet.30.1.405. [DOI] [PubMed] [Google Scholar]
- Hofmann R. M., Pickart C. M. Noncanonical MMS2-encoded ubiquitin-conjugating enzyme functions in assembly of novel polyubiquitin chains for DNA repair. Cell. 1999;96:645–653. doi: 10.1016/s0092-8674(00)80575-9. [DOI] [PubMed] [Google Scholar]
- Homesley L., Lei M., Kawasaki Y., Sawyer S., Christensen T., Tye B. K. Mcm10 and the MCM2–7 complex interact to initiate DNA synthesis and to release replication factors from origins. Genes Dev. 2000;14:913–926. [PMC free article] [PubMed] [Google Scholar]
- Izumi M., Yanagi K., Mizuno T., Yokoi M., Kawasaki Y., Moon K. Y., Hurwitz J., Yatagai F., Hanaoka F. The human homolog of Saccharomyces cerevisiae Mcm10 interacts with replication factors and dissociates from nuclease-resistant nuclear structures in G(2) phase. Nucleic Acids Res. 2000;28:4769–4777. doi: 10.1093/nar/28.23.4769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koepp D. M., Kile A. C., Swaminathan S., Rodriguez-Rivera V. The F-box protein Dia2 regulates DNA replication. Mol. Biol. Cell. 2006;17:1540–1548. doi: 10.1091/mbc.E05-09-0884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korkola J. E., Houldsworth J., Chadalavada R. S., Olshen A. B., Dobrzynski D., Reuter V. E., Bosl G. J., Chaganti R. S. Down-regulation of stem cell genes, including those in a 200-kb gene cluster at 12p13.31, is associated with in vivo differentiation of human male germ cell tumors. Cancer Res. 2006;66:820–827. doi: 10.1158/0008-5472.CAN-05-2445. [DOI] [PubMed] [Google Scholar]
- Kostova Z., Tsai Y. C., Weissman A. M. Ubiquitin ligases, critical mediators of endoplasmic reticulum-associated degradation. Sem. Cell Dev. Biol. 2007;18:770–779. doi: 10.1016/j.semcdb.2007.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laribee R. N., Shibata Y., Mersman D. P., Collins S. R., Kemmeren P., Roguev A., Weissman J. S., Briggs S. D., Krogan N. J., Strahl B. D. CCR4/NOT complex associates with the proteasome and regulates histone methylation. Proc. Natl. Acad. Sci. USA. 2007;104:5836–5841. doi: 10.1073/pnas.0607996104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C., Liachko I., Bouten R., Kelman Z., Tye B. K. Alternative mechanisms for coordinating polymerase alpha and MCM helicase. Mol. Cell. Biol. 2010;30:423–435. doi: 10.1128/MCB.01240-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemoine F. J., Degtyareva N. P., Lobachev K., Petes T. D. Chromosomal translocations in yeast induced by low levels of DNA polymerase a model for chromosome fragile sites. Cell. 2005;120:587–598. doi: 10.1016/j.cell.2004.12.039. [DOI] [PubMed] [Google Scholar]
- Makhnevych T., Sydorskyy Y., Xin X., Srikumar T., Vizeacoumar F. J., Jeram S. M., Li Z., Bahr S., Andrews B. J., Boone C., Raught B. Global map of SUMO function revealed by protein-protein interaction and genetic networks. Molecular Cell. 2009;33:124–135. doi: 10.1016/j.molcel.2008.12.025. [DOI] [PubMed] [Google Scholar]
- Merchant A. M., Kawasaki Y., Chen Y., Lei M., Tye B. K. A lesion in the DNA replication initiation factor Mcm10 induces pausing of elongation forks through chromosomal replication origins in Saccharomyces cerevisiae. Mol. Cell Biol. 1997;17:3261–3271. doi: 10.1128/mcb.17.6.3261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mersman D. P., Du H. N., Fingerman I. M., South P. F., Briggs S. D. Polyubiquitination of the demethylase Jhd2 controls histone methylation and gene expression. Genes Dev. 2009;23:951–962. doi: 10.1101/gad.1769209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel J. J., McCarville J. F., Xiong Y. A role for Saccharomyces cerevisiae Cul8 ubiquitin ligase in proper anaphase progression. J. Biol. Chem. 2003;278:22828–22837. doi: 10.1074/jbc.M210358200. [DOI] [PubMed] [Google Scholar]
- Mimura S., Komata M., Kishi T., Shirahige K., Kamura T. SCFDia2 regulates DNA replication forks during S-phase in budding yeast. EMBO J. 2009;28:3693–3705. doi: 10.1038/emboj.2009.320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison A., Bell J. B., Kunkel T. A., Sugino A. Eukaryotic DNA polymerase amino acid sequence required for 3′-5′ exonuclease activity. Proc. Natl. Acad. Sci. USA. 1991;88:9473–9477. doi: 10.1073/pnas.88.21.9473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulder K. W., Inagaki A., Cameroni E., Mousson F., Winkler G. S., De Virgilio C., Collart M. A., Timmers H. T. Modulation of Ubc4p/Ubc5p-mediated stress responses by the RING-finger-dependent ubiquitin-protein ligase Not4p in Saccharomyces cerevisiae. Genetics. 2007;176:181–192. doi: 10.1534/genetics.106.060640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulder K. W., Winkler G. S., Timmers H. T. DNA damage and replication stress induced transcription of RNR genes is dependent on the Ccr4-Not complex. Nucleic Acids Res. 2005;33:6384–6392. doi: 10.1093/nar/gki938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muzi Falconi M., Piseri A., Ferrari M., Lucchini G., Plevani P., Foiani M. De novo synthesis of budding yeast DNA polymerase alpha and POL1 transcription at the G1/S boundary are not required for entrance into S phase. Proc. Natl. Acad. Sci. USA. 1993;90:10519–10523. doi: 10.1073/pnas.90.22.10519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nambiar M., Kari V., Raghavan S. C. Chromosomal translocations in cancer. Biochim. Biophys. Acta. 2008;1786:139–152. doi: 10.1016/j.bbcan.2008.07.005. [DOI] [PubMed] [Google Scholar]
- Nethanel T., Kaufmann G. Two DNA polymerases may be required for synthesis of the lagging DNA strand of simian virus 40. J. Vir. 1990;64:5912–5918. doi: 10.1128/jvi.64.12.5912-5918.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nick McElhinny S. A., Gordenin D. A., Stith C. M., Burgers P. M., Kunkel T. A. Division of labor at the eukaryotic replication fork. Mol. Cell. 2008;30:137–144. doi: 10.1016/j.molcel.2008.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panasenko O., Landrieux E., Feuermann M., Finka A., Paquet N., Collart M. A. The yeast Ccr4-Not complex controls ubiquitination of the nascent-associated polypeptide (NAC-EGD) complex. J. Biol. Chem. 2006;281:31389–31398. doi: 10.1074/jbc.M604986200. [DOI] [PubMed] [Google Scholar]
- Panasenko O. O., David F. P., Collart M. A. Ribosome association and stability of the nascent polypeptide-associated complex is dependent upon its own ubiquitination. Genetics. 2009;181:447–460. doi: 10.1534/genetics.108.095422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pursell Z. F., Isoz I., Lundstrom E. B., Johansson E., Kunkel T. A. Yeast DNA polymerase epsilon participates in leading-strand DNA replication. Science. 2007;317:127–130. doi: 10.1126/science.1144067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricke R. M., Bielinsky A. K. Mcm10 regulates the stability and chromatin association of DNA polymerase-alpha. Mol. Cell. 2004;16:173–185. doi: 10.1016/j.molcel.2004.09.017. [DOI] [PubMed] [Google Scholar]
- Ricke R. M., Bielinsky A. K. A conserved Hsp10-like domain in Mcm10 is required to stabilize the catalytic subunit of DNA polymerase-alpha in budding yeast. J. Biol. Chem. 2006;281:18414–18425. doi: 10.1074/jbc.M513551200. [DOI] [PubMed] [Google Scholar]
- Robertson P. D., Warren E. M., Zhang H., Friedman D. B., Lary J. W., Cole J. L., Tutter A. V., Walter J. C., Fanning E., Eichman B. F. Domain architecture and biochemical characterization of vertebrate Mcm10. J. Biol. Chem. 2008;283:3338–3348. doi: 10.1074/jbc.M706267200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rock K. L., Gramm C., Rothstein L., Clark K., Stein R., Dick L., Hwang D., Goldberg A. L. Inhibitors of the proteasome block the degradation of most cell proteins and the generation of peptides presented on MHC class I molecules. Cell. 1994;78:761–771. doi: 10.1016/s0092-8674(94)90462-6. [DOI] [PubMed] [Google Scholar]
- Sawyer S. L., Cheng I. H., Chai W., Tye B. K. Mcm10 and Cdc45 cooperate in origin activation in Saccharomyces cerevisiae. J. Mol. Biol. 2004;340:195–202. doi: 10.1016/j.jmb.2004.04.066. [DOI] [PubMed] [Google Scholar]
- Schrader E. K., Harstad K. G., Matouschek A. Targeting proteins for degradation. Nat. Chem. Biol. 2009;5:815–822. doi: 10.1038/nchembio.250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuermann D., Fritsch O., Lucht J. M., Hohn B. Replication stress leads to genome instabilities in Arabidopsis DNA polymerase delta mutants. Plant Cell. 2009;21:2700–2714. doi: 10.1105/tpc.109.069682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seufert W., Jentsch S. Ubiquitin-conjugating enzymes UBC4 and UBC5 mediate selective degradation of short-lived and abnormal proteins. EMBO J. 1990;9:543–550. doi: 10.1002/j.1460-2075.1990.tb08141.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L., Hui A. M., Su Q., Vortmeyer A., Kotliarov Y., Pastorino S., Passaniti A., Menon J., Walling J., Bailey R., Rosenblum M., Mikkelsen T., Fine H. A. Neuronal and glioma-derived stem cell factor induces angiogenesis within the brain. Cancer Cell. 2006;9:287–300. doi: 10.1016/j.ccr.2006.03.003. [DOI] [PubMed] [Google Scholar]
- Varshavsky A. Regulated protein degradation. Trends Biochem. Sci. 2005;30:283–286. doi: 10.1016/j.tibs.2005.04.005. [DOI] [PubMed] [Google Scholar]
- Vaziri C., Saxena S., Jeon Y., Lee C., Murata K., Machida Y., Wagle N., Hwang D. S., Dutta A. A p53-dependent checkpoint pathway prevents rereplication. Mol. Cell. 2003;11:997–1008. doi: 10.1016/s1097-2765(03)00099-6. [DOI] [PubMed] [Google Scholar]
- Venkatesan R. N., Treuting P. M., Fuller E. D., Goldsby R. E., Norwood T. H., Gooley T. A., Ladiges W. C., Preston B. D., Loeb L. A. Mutation at the polymerase active site of mouse DNA polymerase delta increases genomic instability and accelerates tumorigenesis. Mol. Cell. Biol. 2007;27:7669–7682. doi: 10.1128/MCB.00002-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waga S., Stillman B. Anatomy of a DNA replication fork revealed by reconstitution of SV40 DNA replication in vitro. Nature. 1994;369:207–212. doi: 10.1038/369207a0. [DOI] [PubMed] [Google Scholar]
- Wahl A. F., Geis A. M., Spain B. H., Wong S. W., Korn D., Wang T. S. Gene expression of human DNA polymerase alpha during cell proliferation and the cell cycle. Mol. Cell. Biol. 1988;8:5016–5025. doi: 10.1128/mcb.8.11.5016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X., Ira G., Tercero J. A., Holmes A. M., Diffley J. F., Haber J. E. Role of DNA replication proteins in double-strand break-induced recombination in Saccharomyces cerevisiae. Mol. Cell. Biol. 2004;24:6891–6899. doi: 10.1128/MCB.24.16.6891-6899.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen W., Meinkoth J. L., Tsien R. Y., Taylor S. S. Identification of a signal for rapid export of proteins from the nucleus. Cell. 1995;82:463–473. doi: 10.1016/0092-8674(95)90435-2. [DOI] [PubMed] [Google Scholar]
- Whelan W. L., Gocke E., Manney T. R. The CAN1 locus of Saccharomyces cerevisiae: fine-structure analysis and forward mutation rates. Genetics. 1979;91:35–51. doi: 10.1093/genetics/91.1.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X., Guo D., Yuan F., Wang Z. Accessibility of DNA polymerases to repair synthesis during nucleotide excision repair in yeast cell-free extracts. Nucleic Acids Res. 2001;29:3123–3130. doi: 10.1093/nar/29.14.3123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu W., Ukomadu C., Jha S., Senga T., Dhar S. K., Wohlschlegel J. A., Nutt L. K., Kornbluth S., Dutta A. Mcm10 and And-1/CTF4 recruit DNA polymerase alpha to chromatin for initiation of DNA replication. Genes Dev. 2007;21:2288–2299. doi: 10.1101/gad.1585607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou J., Friesen H., Larson J., Huang D., Cox M., Tatchell K., Andrews B. Regulation of cell polarity through phosphorylation of Bni4 by Pho85 G1 cyclin-dependent kinases in Saccharomyces cerevisiae. Mol. Biol. Cell. 2009;20:3239–3250. doi: 10.1091/mbc.E08-12-1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou L., Stillman B. Formation of a preinitiation complex by S-phase cyclin CDK-dependent loading of Cdc45p onto chromatin. Science. 1998;280:593–596. doi: 10.1126/science.280.5363.593. [DOI] [PubMed] [Google Scholar]