

SUMMARY
Hypopigmentation is a feature of copper deficiency in humans, as caused by mutation of the copper (Cu2+) transporter ATP7A in Menkes disease, or an inability to absorb copper after gastric surgery. However, many causes of copper deficiency are unknown, and genetic polymorphisms might underlie sensitivity to suboptimal environmental copper conditions. Here, we combined phenotypic screens in zebrafish for compounds that affect copper metabolism with yeast chemical-genetic profiles to identify pathways that are sensitive to copper depletion. Yeast chemical-genetic interactions revealed that defects in intracellular trafficking pathways cause sensitivity to low-copper conditions; partial knockdown of the analogous Ap3s1 and Ap1s1 trafficking components in zebrafish sensitized developing melanocytes to hypopigmentation in low-copper environmental conditions. Because trafficking pathways are essential for copper loading into cuproproteins, our results suggest that hypomorphic alleles of trafficking components might underlie sensitivity to reduced-copper nutrient conditions. In addition, we used zebrafish-yeast screening to identify a novel target pathway in copper metabolism for the small-molecule MEK kinase inhibitor U0126. The zebrafish-yeast screening method combines the power of zebrafish as a disease model with facile genome-scale identification of chemical-genetic interactions in yeast to enable the discovery and dissection of complex multigenic interactions in disease-gene networks.
INTRODUCTION
Melanins are pigment granules that provide the color for human hair, skin and eyes, and serve to protect surrounding cells from the DNA-damaging effects of ultraviolet (UV) light. Melanins are synthesized and stored in the melanosomes, specialized lysosome-related organelles of the melanocytes that are transferred to surrounding skin or hair cells or concentrated within the retinal pigmented epithelium (Raposo and Marks, 2007). The restriction of melanosome biogenesis to melanocytes results from the cell-specific expression and production of pigment enzymes coupled with their trafficking through the Golgi and early endosomal pathways to the maturing melanosomal organelles. For example, the melanocyte regulator Mitf promotes expression of the rate-limiting enzyme tyrosinase, which is directed through the Golgi and then to the endosome-to-melanosome pathway by the AP1 and AP3 adaptor complexes. Once at the melanosome, tyrosinase promotes melanin synthesis and deposition along the Pmel fibrillar matrix (Chin et al., 2006; Raposo and Marks, 2007).
Copper (Cu2+) is essential for melanin production, and consequently hypopigmentation is a feature of copper deficiency. Genetic mutations of the copper transporters ATP7A or ATP7B lead to Menkes disease or Wilson’s disease, respectively; these childhood syndromes cause acute clinical symptoms in the bone, skin, hair, blood and nervous system (Madsen and Gitlin, 2008). The transporters ATP7A and ATP7B reside in the trans-Golgi network and shuttle copper from the cytoplasm to copper-dependent enzymes in the Golgi. Environmental conditions can also lead to copper deficiency. For example, lambs born to ewes feeding on copper-deficient grass share many clinical features with children diagnosed with Menkes disease; this observation established copper-metabolism deficiencies as an underlying cause of the pathology of Menkes disease (Danks et al., 1972). Copper deficiency can also occur in patients undergoing gastric surgery or after excessive consumption of zinc or iron, all of which can inhibit absorption of copper through the intestine (Kumar, 2006). However, as the causes of many cases of copper deficiency remain unknown (Madsen and Gitlin, 2007b), a more systematic approach to understanding copper-metabolism deficiency in humans is required, including the development of robust cell and animal models (Thiele and Gitlin, 2008).
Melanocytes have a cell-type-specific requirement for copper in pigmentation because tyrosinase is a copper-dependent enzyme, and its activity is dependent on ATP7A (Petris et al., 2000). Transport of ATP7A into melanosomes spatially restricts melanin synthesis to melanocytes; this process is dependent on the biogenesis of the lysosome-related organelle complex (BLOC)-1, which is required for exit of endosomal cargo (Setty et al., 2008). Excess exogenous copper can restore pigmentation to BLOC-1-deficient melanocytes in vitro (Setty et al., 2008). Components of BLOC-1 are mutated in Hermansky-Pudlak syndrome (HPS), a disorder characterized by defects in lysosome-related organelles, particularly melanosomes and platelet-dense granules (Wei, 2006). Aberrant transport of ATP7A might be responsible for the HPS pathology of melanocytes from BLOC-1-deficient individuals (Setty et al., 2008). In zebrafish, mutation of atp7a or treatment with the copper-chelator neocuproine causes a phenotype with features of Menkes disease, including hypopigmentation, neurological disorders, and loss of lysyl oxidase cuproenzyme activity in the developing notochord (Mendelsohn et al., 2006). In zebrafish, hypomorphic alleles of atp7a and the vacuolar ATPase (v-ATPase) cause hypopigmentation at low copper concentrations, demonstrating that genetic mutations can result in copper-deficiency symptoms in suboptimal nutrient conditions (Madsen and Gitlin, 2008).
We have developed a coupled zebrafish phenotypic and yeast chemical-genetic screening approach to identify novel genetic pathways that underlie copper-nutrient sensitivity in the developing vertebrate melanocyte (Fig. 1A). Phenotypic chemical screens in zebrafish enable the identification of novel biological pathways by virtue of chemically induced phenotypes (Zon and Peterson, 2005). Such zebrafish screens are typically carried out by treating developing embryos with libraries of small molecules to identify compounds that elicit a specific phenotype. The small size and transparency of the embryo, which can be arrayed in 96-well plates, coupled with tractable genetic tools for target validation and disease modelling make the zebrafish a highly effective system for phenotype-driven small-molecule screens. However, despite its tractability, identification of the target pathways in zebrafish is usually arduous (Peterson, 2008). Chemical-genetic approaches in the budding yeast Saccharomyces cerevisiae can provide rich functional information on the mechanism of action of compounds (Boone et al., 2007; Hillenmeyer et al., 2008). Mutations that render yeast sensitive to a particular compound – that is, chemical-genetic interactions – often establish links to the cellular target pathways (Hartwell et al., 1997; Parsons et al., 2004; Parsons et al., 2006). The yeast genome-wide gene-deletion collection, in which each of the ∼6000 genes has been individually replaced with a deletion cassette marked with unique sequence identifiers (barcodes) that can be quantified in parallel by microarray hybridization, is a powerful resource for chemical genetics (Winzeler et al., 1999; Giaever et al., 2002). Chemical-genetic profiling, whereby growth of the pooled deletion collection is scored for hypersensitivity to a compound, enables the identification of target and off-target pathways. This approach has been used to interrogate the mechanism of action of both novel and clinically approved drugs, including chemotherapeutics and neuroleptics (Lum et al., 2004; Parsons et al., 2004; Parsons et al., 2006; Ericson et al., 2008; Hillenmeyer et al., 2008). Compounds with similar chemical-genetic profiles can be grouped to reveal similar modes of action and similar targets (Parsons et al., 2006; Hillenmeyer et al., 2008). Often the gene networks that confer sensitivity or resistance to a given compound are conserved from yeast to mammalian cells (Ericson et al., 2008; Yu et al., 2008).
Fig. 1.

A combined zebrafish and yeast approach to probe chemical-genetic interactions in melanocyte pigmentation. (A) Overview of work flow. Phenotypic screening in zebrafish informs as to the developmental and physiological target pathway. Small molecules that promote similar phenotypes in zebrafish are then tested in genome-wide chemical-genetic screens in yeast to reveal potential target genetic pathways. Conserved target pathways are then directly re-tested for chemical-genetic interactions in the zebrafish system using genetic mutant lines, morpholino oligonucleotides or small molecules known to impinge on the same target. (B) Phenotypic screening of a >1500-compound yeast-bioactive library (S.D., J.W., M. Spitzer and M.T., unpublished data) and the Sigma LOPAC library for altered pigmentation in zebrafish embryos revealed a copper-metabolism phenotype characterized by hypopigmentation, an expanded hindbrain and an undulating notochord (Mendelsohn et al., 2006). Representative phenotype caused by 10 μM JFD00692 was rescued by treatment with exogenous copper chloride (5 μM or 15 μM) during development. (C) Examples of the copper-metabolism phenotype induced by some of the compounds identified in the screen.
We identified a novel panel of compounds that affect pathways of copper metabolism and pigmentation in zebrafish and then interrogated the mode of action of several of these compounds. Based on the identification of target genes in yeast encoding intracellular transport proteins, we show that reduced expression of the orthologous target genes, ap1s1 and ap3s2, in zebrafish cause hypopigmentation specifically under conditions of copper limitation. We also demonstrate that the commonly used MEK inhibitor U0126 affects copper metabolism in zebrafish, and use chemical-genetic profiles in yeast to show that U0126 has at least two target pathways in vivo. This combined zebrafish-yeast chemical-genetic approach should be applicable to the study of many other processes associated with disease and development.
RESULTS
A chemical screen identifies small molecules that elicit a copper-deficiency phenotype in zebrafish
Using phenotypic screening in zebrafish, we identified a panel of 45 small molecules that caused a phenotype characteristic of copper depletion (Fig. 1; Table 1). We screened the 1280-member Library of Pharmacologically Active Compounds (LOPAC) and a collection of 1576 bioactive compounds (S.D., J.W., M. Spitzer and M.T., unpublished data) for effects on zebrafish pigmentation. Two embryos at 4 hours post-fertilization (hpf) were placed in each well of a 96-well plate and treated with 10 μM of compound and observed at multiple time points over 48 hours for a copper-metabolism phenotype as exemplified by the atp7a-mutant zebrafish or treatment with neocuproine. These phenotypic manifestations include hypopigmentation, an expanded hindbrain and an undulating notochord in a 2- to 3-day-old fish (Fig. 1B). Active compounds included known copper-binding molecules, such as 1-phenyl-3-(2-thiazolyl)-2-thiourea (PTT) (Mendelsohn et al., 2006), as well as the metal-binding thiosemicarbazones, thiourea derivates, phenanthrolines and pyridin-pyrimidinones (Fig. 1B). Unexpectedly, the commonly used MEK inhibitor U0126 was also identified as causing a phenotype consistent with defective copper metabolism (Table 1); this phenotypic effect of U0126 has been recently described elsewhere (Hawkins et al., 2008).
Table 1.
Small molecules and copper-metabolism phenotypes

Further phenotypic characterization showed the copper-metabolism phenotype caused by many of the small molecules could be prevented with the addition of exogenous copper, indicating that the characteristic zebrafish Menkes phenotypes in pigmentation, notochord, blood and hindbrain development are dependent upon the loss of environmental copper availability (Fig. 1B; Table 1). Interestingly, three compounds caused hypersensitivity to exogenous copper addition (Table 1; supplementary material Fig. S1). One possibility is that these three compounds interfere with detoxification of excess copper ions in the cell; elevated copper levels are highly toxic, and intracellular levels are finely balanced by copper chaperones and metallothionein levels (Rae et al., 1999).
Genome-wide genetic-sensitivity profiles reveal potential copper-metabolism pathways
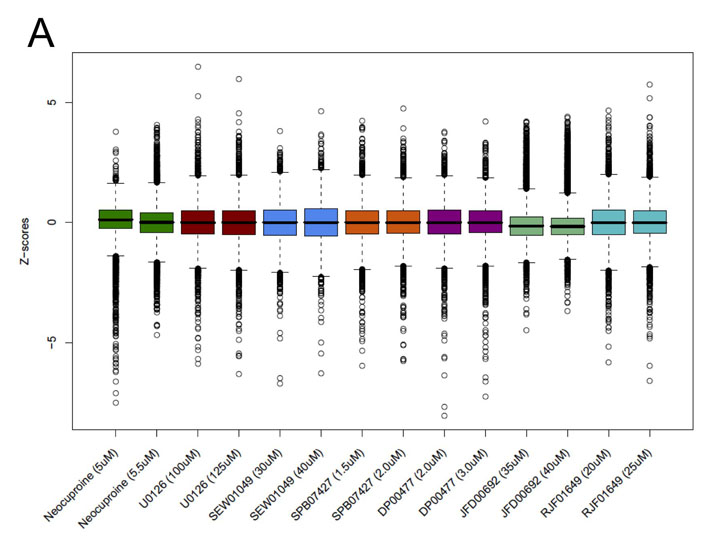
We then used yeast chemical-genetic profiles to identify gene deletions that resulted in sensitivity to a selected subset of compounds that affected copper metabolism in our zebrafish screen: this copper-metabolism subset (CM subset) comprised the compounds neocuproine, U0126, DP00477, SEW01049, SPB07427, JFD00692 and RJF01649. Evolutionarily conserved genetic pathways that are sensitive to copper nutrition in yeast could then be directly tested in the zebrafish vertebrate system (Fig. 1A). We screened pools of ∼5000 haploid yeast deletion mutants and ∼1000 heterozygous diploid mutants for growth after treatment with the CM subset. Compound concentrations were titrated such that overall growth of the yeast pool was diminished by 20–30% (Parsons et al., 2006). Aliquots of the deletion pools were grown for 20 generations in the presence of the different compounds as specified in supplementary material Table S1 or in the presence of 0.4% DMSO (control samples). Genomic DNA was isolated from each pool, the barcodes were amplified with fluorescently labeled primers, and the PCR products were hybridized to spotted oligonucleotide microarrays (Cook et al., 2008). Scatter plots demonstrated that for most chemical-genetic screens, the majority of deletion mutants were not affected by treatment with compound (supplementary material Fig. S2). Boxplots and quantile statistics of z-scores for each microarray allowed identification of sensitive and resistant mutants (see Methods).
Although all the chemicals in the CM subset produced a similar developmental atp7a-mutant phenotype in zebrafish, we found both distinct and common genetic pathways in yeast to be sensitive or resistant to individual compounds in the CM subset, reflecting the unique action of each chemical (Fig. 2). First, we examined the sensitivity of deletions in genes known to be involved in copper uptake and metabolism. Deletion strains defective in copper metabolism are in fact rarely affected by chemical treatments in large-scale surveys (Hillenmeyer et al., 2008), probably because yeast cultures in these studies are typically grown in rich medium that contains adequate amounts of available copper. We found the fitness profiles of strains deleted for known copper-uptake and -metabolism genes to be unique to each compound (Fig. 2A). Deletion of genes such as the copper transporter CTR1, the copper chaperones ATX1 and ATX2, the copper-transporting ATPase CCC2, the copper-zinc superoxide dismutase SOD1 and the copper-dependent transcription factor CUP2 all caused sensitivity to chemically induced copper deficiency. Copper transport into the cell by high-affinity transporters requires copper to be in the reduced Cu(I) state, which is dependent on the ferric and cupric (ferric/cupric) reductases Fre1 and Fre2 (De Freitas et al., 2003). In yeast, and in human disease, copper and iron homeostasis are intimately linked (De Freitas et al., 2003; van Bakel et al., 2005; Madsen and Gitlin, 2007a). We observed that both fre1Δ and fre2Δ strains were highly sensitive to copper starvation caused by different compounds. Finally, the iron transporters Fet3 and Fet5 are known copper-dependent enzymes, and we found that a fet3Δ strain was also highly sensitive to copper depletion. The effects of the CM subset on specific gene-deletion strains thus reflected action of compounds on copper homeostasis, as well as iron homeostasis (van Bakel et al., 2005; Rustici et al., 2007).
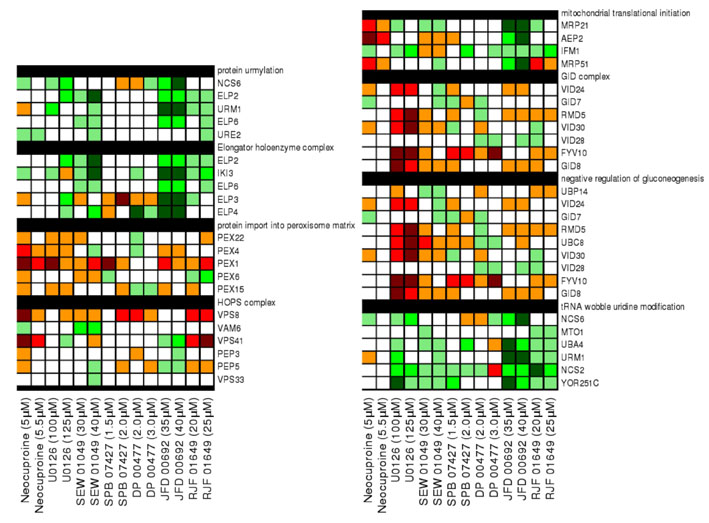
Fig. 2.

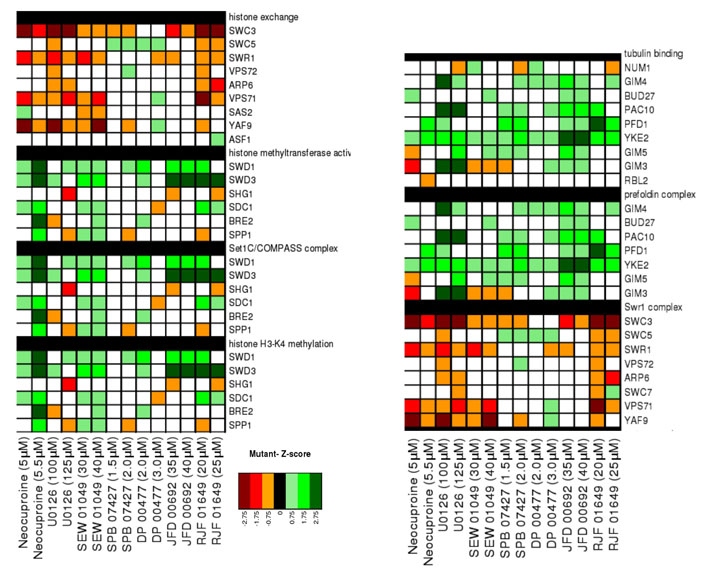
Yeast chemical-genetic profiles of small molecules that affect copper metabolism. (A) Copper-pathway mutant strains that were specifically sensitive or resistant to U0126, neocuproine or the CM subset compounds. At least two concentrations for each compound were used for analysis. Genes with copper-dependent functions were compiled from Saccharomyces Genome Database (SGD, http://www.yeastgenome.org); this list includes copper-containing proteins and known factors in copper and/or iron transport and homeostasis. Color scale indicates degrees of sensitivity (red) or resistance (green); white indicates no data available in a given experiment. (B) Yeast chemical-genetic profiles reveal shared and distinct modes of action for CM subset compounds. A heatmap of GO biological process categories that correlated with sensitivity (red) or resistance (green) to different compounds is shown. Two different concentrations of each compound were screened. Scores for each GO category that contained at least four genes were calculated as the average z-score of all mutants in the category. GO categories contained in the heatmap represent the top 2% of affected categories. GO categories with overlapping gene sets are clustered (see also Table 2).
To understand the biological processes affected by the CM subset, we next assessed the Gene Ontology (GO) biological processes affected by each compound (Fig. 2B; Table 2; supplementary material Fig. S3; see Methods). Although each chemical caused a unique fingerprint of genetic sensitivity, several core pathways were shared by the different compounds. These shared processes included mitochondrial translation initiation, the Swr1 histone-remodeling complex, tubulin-complex assembly, vacuolar organization and intracellular transport, and peroxisome formation. Many of these pathways are linked to copper homeostasis. For example, copper plays a vital role in mitochondrial function, such as in cytochrome c oxidase activity (Madsen and Gitlin, 2007b). Expression of the copper metallothionein gene in yeast is regulated by chromatin modification (Kuo et al., 2005), such that sensitivity of the Swr1 histone-exchange complex (Zilberman et al., 2008) to low-copper conditions might reflect inappropriate control of metallothionein (Kuo et al., 2005). Interestingly, loss of genes involved in threonine biosynthesis resulted in sensitivity to some CM subset compounds. Depletion of genes involved in the biosynthesis of certain amino acids causes sensitivity to multiple treatment conditions, including high-copper concentrations (Ericson et al., 2008; Hillenmeyer et al., 2008; Jo et al., 2008). However, genes involved in the threonine-biosynthesis pathway are not among this known set of pleiotropic genes (Ericson et al., 2008; Hillenmeyer et al., 2008), suggesting a previously unknown relationship between threonine biosynthesis and copper metabolism in yeast.
Table 2.
Genes within Gene Ontology (GO) category biological processes in Fig. 2

The identification of strains defective in tubulin binding and assembly (Geissler et al., 1998) as resistant to copper starvation revealed an unexpected link between tubulin and copper homeostasis. Tubulins require post-translation folding, assembly into heterodimers, and then assembly into microtubules. In vitro and cell-based studies have shown that microtubules disassemble in excess copper (Liliom et al., 1999; Liliom et al., 2000; Nawaz et al., 2005; Pribyl et al., 2008; Liu et al., 2009). Copper might bind the sulfhydral groups of microtubules and thereby block microtubule assembly and/or induce disassembly (Wallin et al., 1977). The GIM (genes involved in microtubule biogenesis) proteins are highly conserved and form a complex that promotes the formation of functional tubulins (Geissler et al., 1998). The gimΔ strains all have a slow-growth phenotype in rich medium, such that the resistance of gimΔ strains to copper limitation might reflect reduced activity of a copper-dependent process that normally inhibits tubulin biogenesis. This effect seems to be specific to the gimΔ strains because all other strains with a slow-growth phenotype did not suppress growth defects caused by copper depletion.
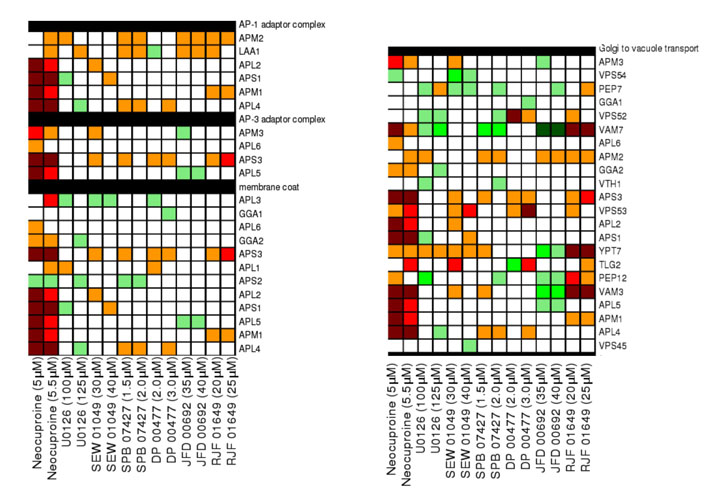
Copper transport is highly regulated in the cell and involves transport into the endoplasmic reticulum, as well as through secretory and vacuolar pathways for delivery to copper-dependent enzymes, copper excretion and copper detoxification (Yuan et al., 1997; Howell et al., 2006; Gonzalez et al., 2008; Jo et al., 2008; Jo et al., 2009). GO biological process category enrichment for cell growth in low-copper conditions revealed the importance of the AP1 adaptor complex, the AP3 adaptor complex, the HOPS complex, the clathrin adaptor complex and the Golgi-to-vacuole transport pathway. AP1, AP3 and HOPS complex subunits are highly conserved and mutated in some human genetic disorders (Dell’Angelica, 2009). Although disruption of the AP1, AP3 and HOPS complexes has not previously been shown to cause sensitivity to low-copper conditions, intracellular transport and vacuolar pathways share some overlap with sensitivity to iron homeostasis. For example, disruption of the AP3 adaptor complex in aps3Δ and apm3Δ strains causes sensitivity to both low-iron and low-copper conditions (Jo et al., 2009) (supplementary material Fig. S3). The requirement for these pathways in the presence of low levels of copper might reflect the inability of copper to reach essential copper-dependent proteins, including the activity of copper-dependent proteins in iron homeostasis, such as Fet3.
Finally, yeast gene-deletion profiles uncovered an important role for the peroxisome in coping with copper depletion. We speculate this dependency arises because the copper-zinc SOD is a constituent enzyme of the peroxisome matrix, which is essential for its dismutase and peroxidase function (Titorenko and Mullen, 2006; Islinger et al., 2009). Deletions in other peroxisomal genes have also recently been shown to sensitize to low-iron conditions, possibly because of an elevated requirement for peroxisomal β-oxidation in energy production when iron-dependent mitochondrial energy production is impaired (Jo et al., 2009). A similar effect might occur upon copper deficiency because copper is also essential for mitochondrial functions and iron homeostasis. Notably, human disorders of peroxisome biogenesis such as Zellweger syndrome are characterized in part by high levels of copper and iron in blood and tissues (Wanders and Waterham, 2005).
The MEK inhibitor U0126 elicits an unexpected copper-metabolism phenotype
The identification of U0126 in our zebrafish phenotypic screen is consistent with a recently suggested role for this MEK inhibitor in copper metabolism (Hawkins et al., 2008). Although U0126 partially phenocopies the atp7a-mutant zebrafish, the speculative link between MEK signaling and copper pathways has not been investigated (Hawkins et al., 2008). We thus set out to determine whether the copper-metabolism phenotype induced by U0126 in zebrafish development and in yeast chemical-genetic profiles was directly due to inhibition of MEK activity or whether U0126 might have an additional target in copper homeostasis independent of its activity as a MEK inhibitor.
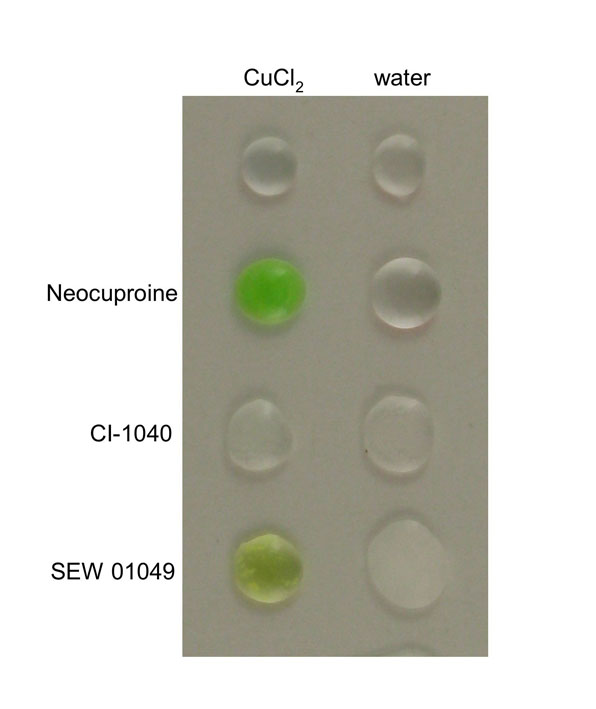
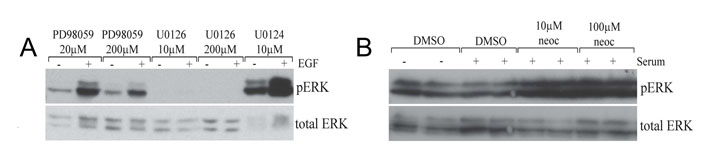
We compared the U0126-induced phenotype with those induced by the selective MEK inhibitor PD0325901 and the known copper chelator neocuproine (Fig. 3A). In vitro kinase-inhibition profiles suggest that the MEK inhibitors CI-1040 and PD0325901 are the most highly selective inhibitors of MEK activity, as compared with U0126 and other commercially available compounds (Davies et al., 2000; Sebolt-Leopold, 2008). Despite the fact that the RAS-RAF-MEK-ERK signaling pathway plays an important role in melanoma development in humans, mice and zebrafish, we found that neither CI-1040 nor PD0325901 affects melanocyte pigmentation in zebrafish development (Fig. 3A) (see also Grzmil et al., 2007; Anastasaki et al., 2009). This result suggests that selective inhibition of MEK activity does not affect pigmentation in zebrafish melanocytes, and that the similarity between the U0126-induced phenotype and the atp7a-mutant phenotype most likely reflects a previously undetected additional target pathway for U0126 in copper homeostasis. Like neocuproine, U0126 also causes a loss of blood development in the embryo and an undulating notochord (Fig. 3A,B). In support of the hypothesis that U0126 can directly affect copper metabolism, we found that U0126, but not CI-1040, avidly binds copper in aqueous solution (Fig. 3C; supplementary material Fig. S4). Thus, despite its frequent use as a specific inhibitor of MEK kinase activity, our analysis based on pigment formation in zebrafish has identified a novel action for U0126 in vivo.
Fig. 3.

Phenotype elicited by U0126 treatment in zebrafish embryos. (A) U0126 phenocopy of neocuproine (neo) in 2-day-old zebrafish embryos. As a control, the more selective MEK inhibitor PD0325901 did not affect pigmentation but instead prevented development of posterior features. (B) U0126 caused blood loss as detected by o-dianisidine (red) staining. (C) U0126 and neocuproine complex with copper, as indicated by color change upon addition to aqueous copper chloride solution. (D) Inhibition of MEK activity in vivo. Lysates from 12-hpf zebrafish embryos were assessed for phospho-ERK and phospho-MEK levels after treatment with PD0325901 (PD) and U0126 (U0). Note inhibitor treatment is known to increase phospho-MEK (Pratilas et al., 2009). A short (2-hour) treatment (asterisk) with PD0325901 also reduced phospho-ERK levels in zebrafish embryos, demonstrating the elevated potency of PD0325901. (E) U0126 (10 μM) treatments prevent pigmentation when administered before 21 hpf. When administered at 21 hpf or later, the embryos develop pigment at about 283 hpf, similar to untreated zebrafish embryos. U0126 treatments are each represented by a barline, and the color of the barline represents the color of the zebrafish melanocytes. The effects on pigmentation are reversible, and pigmentation can rapidly recover from U0126 treatment. For example, U0126-treated unpigmented 33 hpf zebrafish embryos recover pigmentation 4 hours after shifting to fresh embryo medium (asterisk). These phenotypes are consistent with a role for U0126 in blocking the copper-dependent pigmentation enzyme, tyrosinase (Rawls and Johnson, 2000). (F) Electron microscopy of 2-day-old untreated and U0126-treated (5 μM) zebrafish embryos reveals smaller and less densely pigmented melanosomes in the latter.
Western blotting of extracts from treated zebrafish embryos with an anti-phospho-ERK antibody confirmed that PD0325901 and U0126 both effectively inhibit MEK activity during development, whereas neocuproine had no obvious inhibitory effect on phospho-ERK levels in zebrafish (Fig. 3D) or in human cells (supplementary material Fig. S7). PD0325901 was a substantially more potent inhibitor than U0126 as a 2-hour treatment was sufficient to strongly inhibit ERK activity (Fig. 3D). To study the U0126-induced pigmentation phenotype in more detail, we mapped the timing of action of U0126 on developing melanocytes (Fig. 3E). In zebrafish development, melanocytes develop from the neural crest and become pigmented at about 28 hpf. Treatment with U0126 after 21 hpf did not affect normal melanocyte pigmentation, whereas treatment before 21 hpf prevented pigmentation; this effect was not a permanent developmental defect as melanocyte pigmentation rapidly recovered after U0126 wash-out (Fig. 3E). By electron microscopy, we also found evidence of fewer and less-dense melanosomes in melanocytes of embryos treated with 5 μM of U0126 (Fig. 3F). The timing of action of U0126 on melanocyte pigmentation is consistent with the known kinetics of appearance of the rate-limiting copper-dependent enzyme tyrosinase in development (Hultman and Johnson, 2010).
We compared the genome-wide chemical-genetic profiles of U0126, CI-1040 and neocuproine to determine the potential target pathways of U0126. These profiles revealed that 13 deletion strains were sensitive to both U0126 and neocuproine, and that 12 deletion strains exhibited shared sensitivities to U0126 and CI-1040 (Fig. 4A). By contrast, only the pex10Δ strain was sensitive to both CI-1040 and neocuproine, and only the yafΔ9 strain was sensitive to all three compounds. Therefore, although U0126 has a partially overlapping chemical-genetic fingerprint with CI-1040 and neocuproine, CI-1040 and neocuproine have overtly different activities. These profiles are fully consistent with the shared molecular phenotype between U0126 and CI-1040 of inhibition of ERK phosphorylation in zebrafish lysates (Fig. 3D), and the shared developmental and chemical phenotype between U0126 and neocuproine (Fig. 3A).
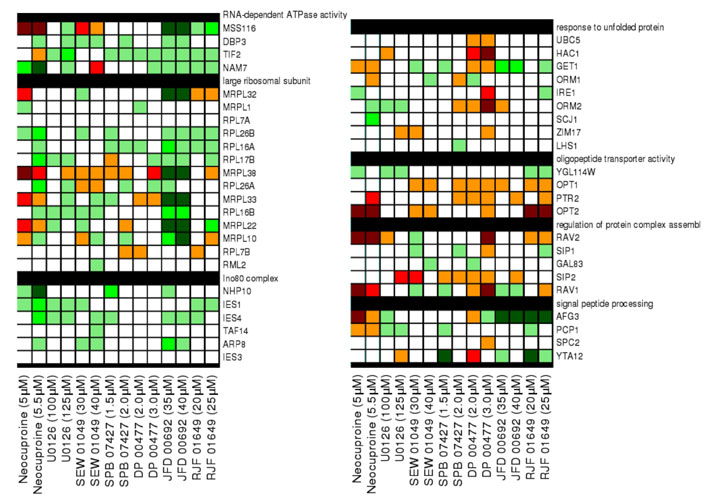
Fig. 4.

Novel target pathways for the MEK inhibtior U0126. (A) Heat map of deletion strains that are sensitive (red) or resistant (green) to U0126, neocuproine and CI-1040. U0126 shares a chemical profile with neocuproine and with CI-1040, whereas neocuproine and CI-1040 do not share a common profile. (B) Neocuproine (yellow), U0126 (green) and CI-1040 (blue) treatment affect common and distinct biological processes. GO categories were examined for each of the deletion mutants that were affected by each compound (see Table 2). The genes in a specific GO category were compared with the number of strains actually affected by each compound and the P values determined. Only GO categories with more than four genes were examined and only P values <0.05 were considered.
Comparison of GO biological processes (Table 2) affected by the different inhibitors revealed both shared and distinct processes. Disruption of the peroxisome and the Swr1 histone-exchange complexes caused sensitivity to both U0126 and neocuproine (Fig. 4B). Impaired intracellular transport and the function of the proteasome 19S regulatory particle compromised growth only in the presence of neocuproine. Loss of the GID ubiquitin ligase complex reduced growth in the presence of U0126, whereas disruption of tubulin-binding processes actually led to resistance in the presence of U0126 (Fig. 4B). A comparison of GO processes affected by the CM subset compounds and CI-1040 allowed us to determine whether the U0126 target pathways were due to copper-dependent pathways, MEK pathways or additional target pathways. Whereas CI-1040 and neocuproine had no effect on tubulin binding and tubulin-complex assembly, treatment with SPB07427, DP00477, JFD00692 and RJF01649 caused resistance of strains defective in tubulin binding and tubulin-complex assembly. Given the shared zebrafish and yeast phenotypes between U0126 and these compounds, it seems that U0126 acts on tubulin-associated processes via alterations in copper metabolism. Notably, in contrast to the shared chemical profiles between U0126, neocuproine and CI-1040, mutations in all six components of the GID complex resulted in high sensitivity to U0126 (Fig. 3B, Fig. 4B). The GID complex is an E3 ubiquitin ligase that targets a key enzyme in gluconeogenesis, fructose 1,6-bisphosphatase (FBPase), for proteasomal degradation (Santt et al., 2008). With the exception of neocuproine, the GID complex also showed sensitivity to CM subset compounds, albeit most strongly with U0126 (Fig. 2; supplementary material Fig. S3). The GID complex might thus represent a new target of copper metabolism in yeast. Collectively, these data reveal new copper-dependent target pathways for U0126, which have remained cryptic despite the extensive use of U0126 in biochemical and cell biological assays in many different signaling contexts.
Knockdown of trafficking components sensitizes zebrafish to copper-dependent hypopigmentation
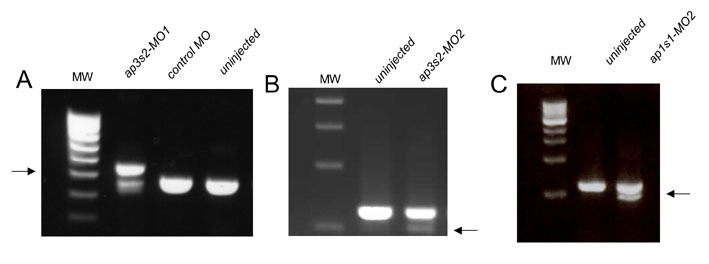
Intracellular transport pathways, although essential for all cells, have a specific role in melanosome biogenesis. Pigmentation enzymes and the copper transporter ATP7A are selectively transported from the trans-Golgi network to the maturing melanosomes (Raposo and Marks, 2007; Setty et al., 2008). To test whether the genes that affect copper sensitivity in yeast might inform about relevant copper pathways in melanocytes, we selected two genes that had orthologs in zebrafish for knockdown by morpholino oligonucleotides (MOs) in the developing embryo. For this purpose we designed MOs against genes that encode a component of the AP1 transport complex (APS1 in yeast, ap1s1 in zebrafish) and a component of the AP3 transport complex (APS3 in yeast, ap3s2 in zebrafish) (supplementary material Fig. S5). We separately injected a splice-site MO designed against ap3s2 (ap3s2-MO1) and a translation-blocking MO designed against ap1s1 (ap1s2-MO1) (Montpetit et al., 2008) into single-celled zebrafish embryos and monitored development in low-neocuproine conditions. Splice-site MOs were verified by PCR analysis (supplementary material Fig. S6). At low MO concentrations (1–3 pg), the ap1s1 and ap3s2 morphants developed normally to at least 2 days pf (Fig. 5A). Next, we treated 4–6 hpf morphants with a range of neocuproine concentrations (1μM, 2 μM and 5 μM) and followed the development of the treated animals over 2 days. Without neocuproine, neither the uninjected controls nor the morphant animals displayed any of the principal features of developmental copper deficiency, including reduced pigmentation, a buckling notochord or an expanded hindbrain (Fig. 5A). This result suggests that under optimal nutrition conditions, partial reduction of ap1s1 or ap3s2 function does not affect development of the zebrafish embryo. At 5 μM neocuproine, developing wild-type and morphant embryos exhibited features of developmental copper-metabolism deficiency, which could be prevented with the addition of exogenous copper chloride at a concentration of 5 μM (Fig. 5A). At intermediate neocuproine concentrations (1 μM, 2 μM), we often observed severe hypopigmentation in the ap3s2 morphants (n=37/71 embryos), whereas control animals exhibited only a mild (at 1 μM) to moderate (at 2 μM) reduction in melanocyte pigmentation (n>100). The ap1s1 morphants also displayed melanocyte pigmentation sensitivity at 1 μM neocuproine (n=13/39) compared with the control-treated animals. A similar effect was seen using the splice-site MOs ap3s2 (ap3s2-MO2) and ap1s1 (ap1s1-MO2) (see Methods). Thus, at least two of the yeast intracellular transport pathways that affected sensitivity to the CM subset compounds proved to be physiologically relevant in the developing vertebrate.
Fig. 5.

Identification of two new gene-nutrient interactions in melanocyte pigmentation. Morpholino oligonucleotides (1–3 pg) directed against ap1s1 and ap3s2 result in morphants with normal development and pigmentation. Higher concentrations of ap1s1-MO have been reported to cause pigmentation phenotypes (Montpetit et al., 2008). Treatment with 5 μM neocuproine caused a copper-deficiency phenotype in all animals that could be rescued with the addition of 15 μM copper chloride. At a concentration of 1.0 μM neocuproine, copper limitation caused a loss of pigmentation in the ap3s2 morphants compared with control-treated embryos, and a severe loss of pigmentation at 2 μM neocuproine, compared with mild (1 μM) to moderate (2 μM) loss of pigmentation in control animals. A milder pigmentation phenotype was also visible in the ap1s1 morphants treated with 1 μM neocuproine. Experiments were repeated multiple times (more than three times for each morpholino; n>20 per treatment), and similar results could be obtained with an additional splice-site MO designed to target each gene (see Methods). (B) Simplified schematic of gene-copper interactions in a zebrafish melanocyte. Partial reduction of AP3 complex activity (such as in the ap3s2 morphant; represented by a dotted line) retains sufficient AP3 activity for melanosome pigmentation in optimal-copper nutrient conditions, and the melanocyte is darkly pigmented. Likewise, melanocytes with normal AP3 activity in suboptimal-copper nutrient conditions retain the ability to generate pigment. By contrast, in low-copper nutrient conditions, reduced AP3 function is no longer sufficient for promoting proper melanosome maturation, and the melanocytes are hypopigmented. Cu, copper; E, endosomal bodies; G, trans-Golgi network; M, melanosomes.
DISCUSSION
Identification of the allelic variants that underlie gene-nutrient interactions is important for understanding the genetic differences between individuals that contribute to disease susceptibility (Thiele and Gitlin, 2008). We used the zebrafish and yeast model systems to identify small molecules that affect copper metabolism, to uncover conserved cognate genetic pathways that sensitize to copper deficiency and to test physiological relevance of these pathways in zebrafish (Fig. 1A). The identification of the dual targets of the commonly used MEK inhibitor U0126 underscores the power of a combined zebrafish and yeast platform to elucidate multiple target pathways of small molecules in vivo.
The conservation of copper-metabolism pathways in yeast, zebrafish and humans suggests that our analysis of genetic sensitivities to copper limitation can provide insight into the molecular pathogenesis of hypocupremia. Comparison of the chemical profiles of the five copper chelators identified by phenotypic screening, as well as neocuproine and U0126, revealed a substantial overlap in target pathways, including intracellular transport pathways. Intracellular trafficking is crucial for transport of copper to copper-dependent enzymes, recycling of the copper transporter ATP7a, and also for copper detoxification in environments of high copper concentrations (Jo et al., 2008). In melanocytes, ATP7A itself is transported to maturing melanosomes in order to restrict the activation of copper-dependent pigmentation enzymes, such as tyrosinase, to this specialized structure (Setty et al., 2008). Tyrosinase, as well as other pigmentation enzymes, is selectively transported to the maturing melanosome by components of the AP1 and AP3 complex; another transporter, the BLOC-1 complex, is required for the selective transport of ATP7A (Wei, 2006; Raposo and Marks, 2007). Known but rare alleles of the AP3 gene give rise to the disorder HPS, and AP1s1 components are mutated in a neurocutaneous syndrome that is characterized by mental retardation, enteropathy, deafness, peripheral neuropathy, ichthyosis and keratodermia, termed MEDNIK. Our yeast and zebrafish data suggest that reduced but otherwise functional levels of AP1 and AP3 that have no effect on pigmentation in optimal nutritional conditions can cause dramatic effects on pigmentation under reduced-copper nutrition conditions (Fig. 5B). The combination of zebrafish and yeast screens might be particularly suited to detection of gene-environment interactions in vertebrates that are characterized by allelic variants of modest quantitative effect (Manolio et al., 2009).
Phenotypic and genome-scale genetic analysis in model organisms is an effective method for identifying new target pathways for known and novel compounds. A major question in chemical biology is how a chemically induced phenotype relates to the in vivo mechanism of compound action and how this knowledge can be exploited to predict off-target effects. Biochemical, in silico and cell-based methods are usually applied in an effort to identify molecular targets, but these approaches can often fail because of limitations in sensitivity and specificity. Predicted drug-target associations of 3665 US Food and Drug Administration (FDA)-approved drugs and other pharmaceutical agents correctly identified 23 new molecular targets for known drugs (Keiser et al., 2009). However, a molecular target under a specific experimental context does not always reflect the action of the compound within the complexity of the developing animal, as illustrated by the unexpected copper-dependent effects of the MEK inhibitor U0126 in our study. Although not all processes or genes are conserved between yeast, zebrafish and human, we note that complex disease states often converge on highly conserved gene networks (Tan et al., 2009), suggesting that the zebrafish-yeast screening approach might be applicable to many other human genetic diseases. To our knowledge, our study is the first to explicitly couple phenotype-based screens in zebrafish to genome-wide chemical-genetic profiles in yeast in order to identify gene-environment interactions during animal development. This approach holds promise for interogating networks of gene-environment interactions in other complex human genetic diseases.
METHODS
Zebrafish husbandry and phenotypic screening
Zebrafish were raised and maintained in compliance with the Animals (Scientific Procedures) Act 1986 of the UK. Embryos were acquired by pair breedings of wild-type AB, AB* and TL zebrafish lines. For small-molecule screening, two 4-hpf zebrafish embryos were arrayed in 96-well plates, and 300 μl of E3 embryo medium (Westerfield, 2000) with 10 μM of compound in 1% DMSO was added. Compounds were from the LOPAC (1280) Small Scale International Version (Sigma-Aldrich) and a subselection of the Maybridge screening collection (Fisher Scientific International). Embryos were assessed and imaged for phenotypic changes at 28 hpf, 36 hpf, 48 hpf and 56 hpf. Small molecules that prevented pigmentation, and caused an expanded hindbrain and a wavy notochord, were classed as having a copper-metabolism phenotype. The classification was confirmed by treating developing embryos with freshly prepared compound in the presence or absence of excess copper chloride at concentrations of 5 μM and 15 μM. CI-1040 was kindly provided by Richard Marais (London, UK) and PD0325901 was obtained from Hilary McLauchlan (University of Dundee, UK). Embryos were treated with 10 μM or 25 μM neocuproine or U0126 and 1 μM or 2.5 μM PD0325901 at different developmental stages. To inhibit pathway activity during development, 4 hpf embryos were treated with compounds continuously until 12 hpf; acute effects were assessed using a pulse of compound from 10 hpf to 12 hpf.
Western blotting
Embryos (12 hpf) were frozen at −80°C after removal of the embryo buffer. Samples were lysed in RIPA buffer [2 M Tris pH 7.5, 5 M NaCl, 1% NP40, Na-deoxycholate, 10% SDS, 0.5 M NaF, 1 M β-glycosyl phosphate and protease-inhibitor cocktail tablet (Roche)]. Samples were normalized by protein as measured by Bradford assay, separated by SDS-PAGE, transferred to Hybond-C Extra nitrocellulose membrane (Amersham Biosciences), probed with rabbit [p44/42 MAPK (1:2000) and phospho-MEK1/2 (Ser217/221) (1:500) (Cell Signaling Technology)] or mouse [phospho-p44/42 MAPK (E10) (1:2000), alpha-tubulin B-5-1-2 (1:50,000) (Santa Cruz)] antibodies and detected with horseradish-peroxidase-conjugated secondary antibodies (Roche).
Electron microscopy
Embryos were fixed with 2.5% glutaraldehyde, 2% paraformaldehyde in 100 mM cacodylate buffer (pH 7.2) with 2 mM MgCl2 and 0.1% picric acid. Following a wash with 200 mM cacodylate buffer, samples were fixed with 1% osmium tetroxide in 100 mM cacodylate buffer (pH 7.0), washed with distilled water, stained en bloc with 0.5% aqueous uranyl acetate, and dehydrated with ethanol. Samples were embedded in Agar 100 resin and sections were cut, stained with lead citrate and viewed on a FEI Tecnai 12 transmission electron microscope.
Yeast chemical-genetic profiles
The MATa haploid and the essential heterozygous yeast deletion sets were obtained from Research Genetics (Germany). Chemical-genetic profiles were generated essentially as described (Giaever et al., 2002). Briefly, for growth-inhibition screens, 5-ml pool cultures were seeded at an OD600 of 0.025 and compound (or DMSO for control samples) was added after a 2-h pre-growth period. Compound concentrations were selected to inhibit pool growth by 20–30% compared with DMSO controls at 12 hours; cultures were diluted back to an OD600 of 0.025 twice with addition of fresh compound to allow growth for a total of 20 generations. Each compound was screened at two different concentrations (supplementary material Table S1). The final concentration of DMSO was 0.4% for all screens. Genomic DNA was isolated for control and experimental pools, barcodes were amplified with fluorescently labeled primers, and PCR products were hybridized to in-house short-oligonucleotide microarrays (Cook et al., 2008). A GenePix 4200AL was used to scan the slides, and hybridization intensity for each spot was determined using GenePix Pro 6.0 software.
Identification of sensitive and resistant mutant strains
The identification of sensitive and resistant strains in the chemical-genetic screens was performed using R and limma, an R package for the analysis of gene expression microarray data. Spots with low intensity and low quality were excluded from further analysis. Intensities of duplicate spots for each barcode on the array were averaged. z-scores for up and down barcode tags were calculated based on log2 fold-change ratios (compound treated vs DMSO control): z=(x-μ)/σ where μ is the mean log2-fold change for all up or down barcodes on an array and σ is the standard deviation. The two barcode scores were averaged for each deletion strain to give the final z-score. Microarray raw data and z-scores for each mutant were stored in a custom MySQL database. Standard procedures for z-score and quantile-based statistics for microarray data were used to identify sensitive and resistant deletion strains (Quackenbush, 2002; Cheadle et al., 2003). The microarray data are available at ArrayExpress (accession number E-TABM-922).
Assignment of GO biological process terms
Instead of relying on arbitrary cut-offs to define enriched sets of deletion strains that were most affected by treatment with the compounds, we calculated average z-scores for all GO biological process categories in each experiment for GO categories that contained data for at least four genes. Quartile-based statistics were used to identify significantly affected GO biological processes (Saccharomyces Genome Database, http://www.yeastgenome.org, release 02/07/2009).
Morpholino oligonucleotides
The MOs were designed by GeneTools (USA) and prepared as a 1 mM stock solution in water. For Fig. 5A, 1–3 pg was injected into each one-cell stage embryo. MO sequences were designed to block splicing for a zebrafish ortholog of the yeast gene APS3, called ap3s2 (Ensembl(Zv8): ENSDARG00000039882) (ap3s2-MO1: 5′-TGCAAAAGCCTCTCCATCACCTTCC-3′). Confirmation of ap3s2-MO1 activity was determined by PCR with primers AP3S2-F1 [5′-TCAACAACCATGGGAAACCC-3′ (forward primer)] and AP3S2-R1 [5′-TGACTGCAGAAACGGCTCG-3′ (reverse primer)]. Inclusion of intron 2 was determined by sequencing of the longer cDNA product. A translation-block MO for ap1s1 (Ensembl(Zv8): ENSDARG00000056803), an ortholog of APS1, was directly purchased from GeneTools (ap1s1-MO1: 5′-ACAGAAGCATAAAGCGCATCATTTC-3′), and has been previously verified by western blotting morphant embryo extracts with an antibody (Montpetit et al., 2008). Two additional splice-site MOs were designed and ordered from GeneTools: ap3s2-MO2 (5′-TGCAGTTACGTTCACCTGTATAAGA-3′) and ap1s1-MO2 (5′-GACTAGCATACCTACGTAAACACAC-3′) (Montpetit et al., 2008). Confirmation of ap3s2-MO2 activity was determined by PCR with primers AP3S2-F2 [5′-TGCAGCAGCAGATCATCAGGG-3′ (forward primer)] and AP3S2-R2 [5′-GACTGTCAGTAATGGCAAGAGG-3′ (reverse primer)]. Results were consistent with both MOs for each gene, with greater effects seen with increasing concentration of MO (5–10 pg). All MOs were labeled with a florescent tag and assessed for even MO distribution throughout the embryo. Changes in pigmentation were assessed by two or three individuals who scored the phenotypes blinded to the MO genotypes.
Supplementary Material
Acknowledgments
We are grateful to David Harrison for many helpful discussions and on-going support of this work. We thank Karthikeyani Paranthaman for zebrafish husbandry, Nicola Grant for artwork, Phillipe Gautier (for figure S4), and Nick Temperley and John Maule for technical assistance. We are grateful to Ian Jackson, Veronica van Heyningen, Nick Hastie, Corey Nislow, Lea Harrington, Robert Kelsh and Chris Norbury for helpful discussions and critical reading of the manuscript. This work was funded by the Medical Research Scotland (H.I., E.E.P.), National Institutes of Health (J.D.G.), the European Research Council (M.T.), the Canadian Institutes for Health Research Operating Grant MOP-79488 (M.T.), the Scottish Universities Life Sciences Alliance (M.T.), a Royal Society Wolfson Award (M.T.), the Wellcome Trust (E.E.P.), and an MRC Career Development Award (E.E.P.). Deposited in PMC for release after 6 months.
Footnotes
COMPETING INTERESTS
The authors declare no financial, personal or professional associations that interfere with the objectivity of this study.
AUTHOR CONTRIBUTIONS
H.I. and M.S. conceived, designed and performed experiments, and J.W., C.A., Z.Z., M.S., E.M. and E.E.P. performed experiments and analyzed data. S.D., J.G. and R.M. contributed to experimental design and provided reagents. M.T. and E.E.P. conceived and designed experiments, and wrote the paper.
SUPPLEMENTARY MATERIAL
Supplementary material for this article is available at http://dmm.biologists.org/lookup/suppl/doi:10.1242/dmm.005769/-/DC1
REFERENCES
- Anastasaki C, Estep AL, Marais R, Rauen KA, Patton EE. (2009). Kinase-activating and kinase-impaired cardio-facio-cutaneous syndrome alleles have activity during zebrafish development and are sensitive to small molecule inhibitors. Hum Mol Genet. 18, 2543–2554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boone C, Bussey H, Andrews BJ. (2007). Exploring genetic interactions and networks with yeast. Nat Rev Genet. 8, 437–449 [DOI] [PubMed] [Google Scholar]
- Cheadle C, Vawter MP, Freed WJ, Becker KG. (2003). Analysis of microarray data using Z score transformation. J Mol Diagn. 5, 73–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin L, Garraway LA, Fisher DE. (2006). Malignant melanoma: genetics and therapeutics in the genomic era. Genes Dev. 20, 2149–2182 [DOI] [PubMed] [Google Scholar]
- Cook MA, Chan CK, Jorgensen P, Ketela T, So D, Tyers M, Ho CY. (2008). Systematic validation and atomic force microscopy of non-covalent short oligonucleotide barcode microarrays. PLoS One 3, e1546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danks DM, Campbell PE, Walker-Smith J, Stevens BJ, Gillespie JM, Blomfield J, Turner B. (1972). Menkes’ kinky-hair syndrome. Lancet 1, 1100–1102 [DOI] [PubMed] [Google Scholar]
- Davies SP, Reddy H, Caivano M, Cohen P. (2000). Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem J. 351, 95–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Freitas J, Wintz H, Kim JH, Poynton H, Fox T, Vulpe C. (2003). Yeast, a model organism for iron and copper metabolism studies. Biometals 16, 185–197 [DOI] [PubMed] [Google Scholar]
- Dell’Angelica EC. (2009). AP-3-dependent trafficking and disease: the first decade. Curr Opin Cell Biol. 21, 552–559 [DOI] [PubMed] [Google Scholar]
- Ericson E, Gebbia M, Heisler LE, Wildenhain J, Tyers M, Giaever G, Nislow C. (2008). Off-target effects of psychoactive drugs revealed by genome-wide assays in yeast. PLoS Genet. 4, e1000151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geissler S, Siegers K, Schiebel E. (1998). A novel protein complex promoting formation of functional alpha- and gamma-tubulin. EMBO J. 17, 952–966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giaever G, Chu AM, Ni L, Connelly C, Riles L, Veronneau S, Dow S, Lucau-Danila A, Anderson K, Andre B, et al. (2002). Functional profiling of the Saccharomyces cerevisiae genome. Nature 418, 387–391 [DOI] [PubMed] [Google Scholar]
- Gonzalez M, Reyes-Jara A, Suazo M, Jo WJ, Vulpe C. (2008). Expression of copper-related genes in response to copper load. Am J Clin Nutr. 88, 830S–834S [DOI] [PubMed] [Google Scholar]
- Grzmil M, Whiting D, Maule J, Anastasaki C, Amatruda JF, Kelsh RN, Norbury CJ, Patton EE. (2007). The INT6 cancer gene and MEK signaling pathways converge during zebrafish development. PLoS One 2, e959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartwell LH, Szankasi P, Roberts CJ, Murray AW, Friend SH. (1997). Integrating genetic approaches into the discovery of anticancer drugs. Science 278, 1064–1068 [DOI] [PubMed] [Google Scholar]
- Hawkins TA, Cavodeassi F, Erdelyi F, Szabo G, Lele Z. (2008). The small molecule Mek1/2 inhibitor U0126 disrupts the chordamesoderm to notochord transition in zebrafish. BMC Dev Biol. 8, 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillenmeyer ME, Fung E, Wildenhain J, Pierce SE, Hoon S, Lee W, Proctor M, St Onge RP, Tyers M, Koller D, et al. (2008). The chemical genomic portrait of yeast: uncovering a phenotype for all genes. Science 320, 362–365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howell GJ, Holloway ZG, Cobbold C, Monaco AP, Ponnambalam S. (2006). Cell biology of membrane trafficking in human disease. Int Rev Cytol. 252, 1–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hultman KA, Johnson SL. (2010). Differential contribution of direct-developing and stem cell-derived melanocytes to the zebrafish larval pigment pattern. Dev Biol. 337, 425–431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Islinger M, Li KW, Seitz J, Volkl A, Luers GH. (2009). Hitchhiking of Cu/Zn superoxide dismutase to peroxisomes-evidence for a natural piggyback import mechanism in mammals. Traffic 10, 1711–1721 [DOI] [PubMed] [Google Scholar]
- Jo WJ, Loguinov A, Chang M, Wintz H, Nislow C, Arkin AP, Giaever G, Vulpe CD. (2008). Identification of genes involved in the toxic response of Saccharomyces cerevisiae against iron and copper overload by parallel analysis of deletion mutants. Toxicol Sci. 101, 140–151 [DOI] [PubMed] [Google Scholar]
- Jo WJ, Kim JH, Oh E, Jaramillo D, Holman P, Loguinov AV, Arkin AP, Nislow C, Giaever G, Vulpe CD. (2009). Novel insights into iron metabolism by integrating deletome and transcriptome analysis in an iron deficiency model of the yeast Saccharomyces cerevisiae. BMC Genomics 10, 130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keiser MJ, Setola V, Irwin JJ, Laggner C, Abbas AI, Hufeisen SJ, Jensen NH, Kuijer MB, Matos RC, Tran TB, et al. (2009). Predicting new molecular targets for known drugs. Nature 462, 175–181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar N. (2006). Copper deficiency myelopathy (human swayback). Mayo Clin Proc. 81, 1371–1384 [DOI] [PubMed] [Google Scholar]
- Kuo HC, Moore JD, Krebs JE. (2005). Histone H2A and Spt10 cooperate to regulate induction and autoregulation of the CUP1 metallothionein. J Biol Chem. 280, 104–111 [DOI] [PubMed] [Google Scholar]
- Liliom K, Wagner G, Kovacs J, Comin B, Cascante M, Orosz F, Ovadi J. (1999). Combined enhancement of microtubule assembly and glucose metabolism in neuronal systems in vitro: decreased sensitivity to copper toxicity. Biochem Biophys Res Commun. 264, 605–610 [DOI] [PubMed] [Google Scholar]
- Liliom K, Wagner G, Pacz A, Cascante M, Kovacs J, Ovadi J. (2000). Organization-dependent effects of toxic bivalent ions microtubule assembly and glycolysis. Eur J Biochem. 267, 4731–4739 [DOI] [PubMed] [Google Scholar]
- Liu D, Xue P, Meng Q, Zou J, Gu J, Jiang W. (2009). Pb/Cu effects on the organization of microtubule cytoskeleton in interphase and mitotic cells of Allium sativum L. Plant Cell Rep. 28, 695–702 [DOI] [PubMed] [Google Scholar]
- Lum PY, Armour CD, Stepaniants SB, Cavet G, Wolf MK, Butler JS, Hinshaw JC, Garnier P, Prestwich GD, Leonardson A, et al. (2004). Discovering modes of action for therapeutic compounds using a genome-wide screen of yeast heterozygotes. Cell 116, 121–137 [DOI] [PubMed] [Google Scholar]
- Madsen E, Gitlin JD. (2007a). Copper and iron disorders of the brain. Annu Rev Neurosci. 30, 317–337 [DOI] [PubMed] [Google Scholar]
- Madsen E, Gitlin JD. (2007b). Copper deficiency. Curr Opin Gastroenterol. 23, 187–192 [DOI] [PubMed] [Google Scholar]
- Madsen EC, Gitlin JD. (2008). Zebrafish mutants calamity and catastrophe define critical pathways of gene-nutrient interactions in developmental copper metabolism. PLoS Genet. 4, e1000261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manolio TA, Collins FS, Cox NJ, Goldstein DB, Hindorff LA, Hunter DJ, McCarthy MI, Ramos EM, Cardon LR, Chakravarti A, et al. (2009). Finding the missing heritability of complex diseases. Nature 461, 747–753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendelsohn BA, Yin C, Johnson SL, Wilm TP, Solnica-Krezel L, Gitlin JD. (2006). Atp7a determines a hierarchy of copper metabolism essential for notochord development. Cell Metab. 4, 155–162 [DOI] [PubMed] [Google Scholar]
- Montpetit A, Cote S, Brustein E, Drouin CA, Lapointe L, Boudreau M, Meloche C, Drouin R, Hudson TJ, Drapeau P, et al. (2008). Disruption of AP1S1, causing a novel neurocutaneous syndrome, perturbs development of the skin and spinal cord. PLoS Genet. 4, e1000296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nawaz M, Manzl C, Krumschnabel G. (2005). In vitro toxicity of copper, cadmium, and chromium to isolated hepatocytes from carp, Cyprinus carpio L. Bull Environ Contam Toxicol. 75, 652–661 [DOI] [PubMed] [Google Scholar]
- Parsons AB, Brost RL, Ding H, Li Z, Zhang C, Sheikh B, Brown GW, Kane PM, Hughes TR, Boone C. (2004). Integration of chemical-genetic and genetic interaction data links bioactive compounds to cellular target pathways. Nat Biotechnol. 22, 62–69 [DOI] [PubMed] [Google Scholar]
- Parsons AB, Lopez A, Givoni IE, Williams DE, Gray CA, Porter J, Chua G, Sopko R, Brost RL, Ho CH, et al. (2006). Exploring the mode-of-action of bioactive compounds by chemical-genetic profiling in yeast. Cell 126, 611–625 [DOI] [PubMed] [Google Scholar]
- Peterson RT. (2008). Chemical biology and the limits of reductionism. Nat Chem Biol. 4, 635–638 [DOI] [PubMed] [Google Scholar]
- Petris MJ, Strausak D, Mercer JF. (2000). The Menkes copper transporter is required for the activation of tyrosinase. Hum Mol Genet. 9, 2845–2851 [DOI] [PubMed] [Google Scholar]
- Pratilas CA, Taylor BS, Ye Q, Viale A, Sander C, Solit DB, Rosen N. (2009). (V600E)BRAF is associated with disabled feedback inhibition of RAF-MEK signaling and elevated transcriptional output of the pathway. Proc Natl Acad Sci USA 106, 4519–4524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pribyl P, Cepak V, Zachleder V. (2008). Cytoskeletal alterations in interphase cells of the green alga Spirogyra decimina in response to heavy metals exposure: II. The effect of aluminium, nickel and copper. Toxicol Vitro 22, 1160–1168 [DOI] [PubMed] [Google Scholar]
- Quackenbush J. (2002). Microarray data normalization and transformation. Nat Genet. 32, 496–501 [DOI] [PubMed] [Google Scholar]
- Rae TD, Schmidt PJ, Pufahl RA, Culotta VC, O’Halloran TV. (1999). Undetectable intracellular free copper: the requirement of a copper chaperone for superoxide dismutase. Science 284, 805–808 [DOI] [PubMed] [Google Scholar]
- Raposo G, Marks MS. (2007). Melanosomes-dark organelles enlighten endosomal membrane transport. Nat Rev Mol Cell Biol. 8, 786–797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rawls JF, Johnson SL. (2000). Zebrafish kit mutation reveals primary and secondary regulation of melanocyte development during fin stripe regeneration. Development 127, 3715–3724 [DOI] [PubMed] [Google Scholar]
- Rustici G, van Bakel H, Lackner DH, Holstege FC, Wijmenga C, Bahler J, Brazma A. (2007). Global transcriptional responses of fission and budding yeast to changes in copper and iron levels: a comparative study. Genome Biol. 8, R73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santt O, Pfirrmann T, Braun B, Juretschke J, Kimmig P, Scheel H, Hofmann K, Thumm M, Wolf DH. (2008). The yeast GID complex, a novel ubiquitin ligase (E3) involved in the regulation of carbohydrate metabolism. Mol Biol Cell 19, 3323–3333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebolt-Leopold JS. (2008). Advances in the development of cancer therapeutics directed against the RAS-mitogen-activated protein kinase pathway. Clin Cancer Res. 14, 3651–3656 [DOI] [PubMed] [Google Scholar]
- Setty SR, Tenza D, Sviderskaya EV, Bennett DC, Raposo G, Marks MS. (2008). Cell-specific ATP7A transport sustains copper-dependent tyrosinase activity in melanosomes. Nature 454, 1142–1146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan CS, Bodenmiller B, Pasculescu A, Jovanovic M, Hengartner MO, Jorgensen C, Bader GD, Aebersold R, Pawson T, Linding R. (2009). Comparative analysis reveals conserved protein phosphorylation networks implicated in multiple diseases. Sci Signal. 2, ra39. [DOI] [PubMed] [Google Scholar]
- Thiele DJ, Gitlin JD. (2008). Assembling the pieces. Nat Chem Biol. 4, 145–147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Titorenko VI, Mullen RT. (2006). Peroxisome biogenesis: the peroxisomal endomembrane system and the role of the ER. J Cell Biol. 174, 11–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Bakel H, Strengman E, Wijmenga C, Holstege FC. (2005). Gene expression profiling and phenotype analyses of S. cerevisiae in response to changing copper reveals six genes with new roles in copper and iron metabolism. Physiol Genomics 22, 356–367 [DOI] [PubMed] [Google Scholar]
- Wallin M, Larsson H, Edstrom A. (1977). Tubulin sulfhydryl groups and polymerization in vitro. Effects of di- and trivalent cations. Exp Cell Res. 107, 219–225 [DOI] [PubMed] [Google Scholar]
- Wanders RJ, Waterham HR. (2005). Peroxisomal disorders I: biochemistry and genetics of peroxisome biogenesis disorders. Clin Genet. 67, 107–133 [DOI] [PubMed] [Google Scholar]
- Wei ML. (2006). Hermansky-Pudlak syndrome: a disease of protein trafficking and organelle function. Pigment Cell Res. 19, 19–42 [DOI] [PubMed] [Google Scholar]
- Westerfield M. (2000). The Zebrafish Book. A Guide for the Laboratory use of Zebrafish (Danio rerio). Eugene: University of Oregon Press [Google Scholar]
- Winzeler EA, Shoemaker DD, Astromoff A, Liang H, Anderson K, Andre B, Bangham R, Benito R, Boeke JD, Bussey H, et al. (1999). Functional characterization of the S. cerevisiae genome by gene deletion and parallel analysis. Science 285, 901–906 [DOI] [PubMed] [Google Scholar]
- Yu L, Lopez A, Anaflous A, El Bali B, Hamal A, Ericson E, Heisler LE, McQuibban A, Giaever G, Nislow C, et al. (2008). Chemical-genetic profiling of imidazo[1,2-a]pyridines and -pyrimidines reveals target pathways conserved between yeast and human cells. PLoS Genet. 4, e1000284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan DS, Dancis A, Klausner RD. (1997). Restriction of copper export in Saccharomyces cerevisiae to a late Golgi or post-Golgi compartment in the secretory pathway. J Biol Chem. 272, 25787–25793 [DOI] [PubMed] [Google Scholar]
- Zilberman D, Coleman-Derr D, Ballinger T, Henikoff S. (2008). Histone H2A.Z and DNA methylation are mutually antagonistic chromatin marks. Nature 456, 125–129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zon LI, Peterson RT. (2005). In vivo drug discovery in the zebrafish. Nat Rev Drug Discov. 4, 35–44 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}