

Abstract
Nonhomologous end joining (NHEJ) plays a major role in double-strand break DNA repair, which involves a series of steps mediated by multiprotein complexes. A ring-shaped Ku70/Ku80 heterodimer forms first at broken DNA ends, DNA-dependent protein kinase catalytic subunit (DNA-PKcs) binds to mediate synapsis and nucleases process DNA overhangs. DNA ligase IV (LigIV) is recruited as a complex with XRCC4 for ligation, with XLF/Cernunnos, playing a role in enhancing activity of LigIV. We describe how a combination of methods—X-ray crystallography, electron microscopy and small angle X-ray scattering—can give insights into the transient multicomponent complexes that mediate NHEJ. We first consider the organisation of DNA-PKcs/Ku70/Ku80/DNA complex (DNA-PK) and then discuss emerging evidence concerning LigIV/XRCC4/XLF/DNA and higher-order complexes. We conclude by discussing roles of multiprotein systems in maintaining high signal-to-noise and the value of structural studies in developing new therapies in oncology and elsewhere.
1. Introduction
Nonhomologous End Joining (NHEJ) and Homologous Recombination (HR) comprise the two major modes of DNA double-strand break (DSB) repair in human cells. Although HR is dominant in late S/G2 phases when a sister chromatid is available [1], NHEJ, which does not require a template [2], plays a major role in G1/early S phase [1]. It is predicted that in humans about 50 endogenous DSBs per cell during each cell cycle may occur [3]. These are mainly generated by ionizing radiation, reactive oxygen species, and DNA replication across a nick [2]. Unrepaired DSBs can cause catastrophic gene loss during cell division, leading to chromosomal translocations, increased mutation rates, and carcinogenesis [4]. The NHEJ system is also responsible for programmed DSBs in V(D)J recombination [5] and class switch recombination [6] during development of immune diversity. NHEJ has an alternative end-joining pathway, which is mostly microhomology-mediated end joining [7] and is independent of NHEJ components [8]. Here NHEJ implies the main NHEJ pathway.
The NHEJ pathway comprises three major steps: synapsis, end processing and ligation [9]. Synapsis is carried out by DNA-dependent protein kinase (DNA-PK) consisting of Ku70, Ku80, DNA-PK catalytic subunit (DNA-PKcs), and DNA. Ku70 and Ku80 form a ring-shaped heterodimer around the broken DNA ends and maintain them in proximity [10, 11]. DNA-PKcs, a very large protein belonging to the phosphatidylinositol-3-OH kinase (PI3K)-related kinase (PIKKs) family [12], is recruited through interaction with the C-terminus of Ku80 [13, 14], and causes the Ku70/80 heterodimer to move about one helical turn inward from the end [15] to make space for DNA-PKcs to bind DNA. Two DNA-PK assemblies are probably required to hold the two DNA ends close together [16]. Activated DNA-PKcs phosphorylates itself and various proteins, including the other NHEJ components [17, 18]. Synapsis induces the autophosphorylation of DNA-PKcs and allows other NHEJ proteins access to DNA ends [19, 20].
The end processing involves nucleases such as Artemis [21], which is capable of cutting an array of DNA overhangs and is thought to be sufficient as a nuclease, although other nucleases in particular PNK, aprataxin (APTX), and PNK-APTX-like factor (PALF), a 3′ exonuclease, cannot be ruled out [22]. Artemis interacts with DNA-PKcs and opens DNA hairpins in the V(D)J recombination process [23]. Mutations in Artemis gene cause Radiosensitive Severe Combined Immunodeficiency (RS-SCID) [21]. Polymerases Polμ and Polλ use their BRCT domains to bind to Ku/DNA complexes and terminal deoxynucleotidyl transferase (TdT) is exclusively expressed in initial lymphoid cells to engage in NHEJ of the V(D)J recombination process [24–26]. Furthermore, as recently shown Ku in its role as a lyase also participates in end processing cutting DNA 3′ at abasic sites, indicating that this protein, like its partner DNA-PKcs, has enzymatic properties and thus fulfils a number of roles in the NHEJ pathway [27].
The final ligation step of rejoining is mediated by DNA ligase IV (LigIV), which is associated with dimeric X-ray cross-complementation group 4 (XRCC4) [28]. These proteins form a very stable complex, which is maintained at 2 M NaCl or 7 M urea [29]. XRCC4 stimulates adenylation and ligase activity [30–32]. Knockouts of these genes in mice result in the late embryonic lethality in the p53-dependent manner [33–36] while mutations in lig4 gene result in LIG4 syndrome characterized by radiosensitivity, unusual facial features, microcephaly, developmental and growth delay, pancytopenia, and skin abnormality [37]. XRCC4-Like Factor (XLF)/Cernunnos (XLF), mutations of which in humans cause Severe Combined Immunodeficiency, also interacts with XRCC4, and enhances the ligation by LigIV [38, 39].
Here we review what is known of the architectures of the transient multicomponent complexes that mediate Nonhomologous End Joining. Figure 1 is an attempt to construct an interaction diagram that summarises our current understanding of NHEJ protein interactions and phosphorylation by DNA-PKcs, indicating where structural information is available. Although the existence of DNA-PK—the complex between DNA-PKcs, heterodimeric Ku and DNA—is clearly defined, as is the tight complex between XRCC4 and LigIV, the temporal and spatial organisation of higher-order complexes is unclear. Do subcomplexes exist that allow the Ku to get off the DNA before ligation, or is there one supercomplex in which DNA-PK, LigIV/XRCC4, and XLF coexist to achieve ligation? In this case, how does Ku leave when ends are ligated? In this paper, we first consider what is known about the structure of the huge single chain DNA-PKcs and how this might lead to a better understanding of the target for use in structure-guided drug discovery. We then discuss the organisation of DNA-PKcs/Ku70/Ku80/DNA complex (known as DNA-PK), in order to shed light on the initial events that take place in the NHEJ pathway. We discuss the emerging evidence concerning 3D structures of LigIV/XRCC4/XLF/DNA complexes, which should give clues about the binding and functional mechanism of LigIV/XRCC4 and XLF in NHEJ. Finally we consider the spatial arrangement of higher-order complexes in order to give a picture of the NHEJ repair system as a whole.
Figure 1.
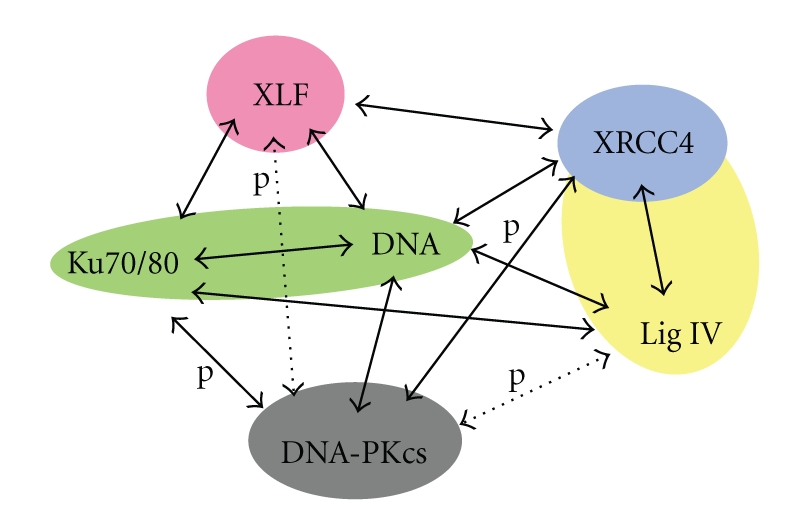
Schematic diagram of interactions of the NHEJ machinery. Colour-filled shapes indicate the proteins and complexes with known 3D structures. Solid arrows indicate confirmed whereas dashed arrows are plausible interactions. Phosphorylation events are indicated by letter “p”.
2. Structural Biology of Individual Components
Considerable advances have been made in the structural biology of individual components and complexes of the NHEJ repair machinery, but further work is required to understand the spatial organisation of this complicated and dynamic process. Here we discuss what is known about each component before discussing the multiprotein complexes that mediate their functions in NHEJ.
2.1. Ku70/80
The double-stranded (ds) DNA end-binding activity of Ku70 and Ku80 requires their association to form a heterodimer [40]. The crystal structure of the Ku70/Ku80 heterodimer reveals a similar topology and domain organisation, comprising an amino-terminal α/β domain, a central β-barrel domain, and a helical C-terminal arm [10]. These proteins, when associated, form a pseudosymmetrical structure, in which residues that contribute to the dimer interface show a low level of sequence identity (approximately 15%; Figure 2), favouring heterodimer formation over Ku70-Ku70 or Ku80-Ku80 homodimerisation.
Figure 2.

Ku70 and Ku80 aligned sequences on the basis of their structures. They show a similar domain organisation despite the low sequence identity. The alignment was created with ClustalW2 [41] and visualized with ESPript [42]. Many residues are conserved (black box) or semiconserved/similar (gray box).
The crystal structure of the Ku70/80 heterodimer in complex with one 55-nucleotide long Y-shaped DNA fragment shows that the Ku70/80 heterodimer adopts the shape of a ring that encircles duplex DNA (Figure 3). No large conformational changes occur on binding DNA to heterodimeric Ku except for the C-terminal domains of Ku70 and Ku80. Indeed, no contacts with DNA bases and only a few interactions with the sugar-phosphate backbone are made. The DNA duplex is embraced through the Ku70/80 preformed ring in such a way that one DNA face is relatively accessible to the solvent and therefore exposed to processing enzymes that remove damaged nucleotides and fill gaps prior to ligation. These features can provide structural support to broken DNA ends and bring the DNA helix into phase across the junction during end processing and ligation. Although Ku70/80 heterodimer shows low affinity for circularized DNA [43] and does not bind any DNA substrates shorter than 14 bp, it does bind dsDNA fragments of similar length and structure in a DNA sequence-independent fashion and irrespective of whether the DNA ends are blunt, with hairpin loops, or 5′ or 3′ overhangs.
Figure 3.

Heterodimerisation of Ku70/80 defines a ring shape that binds DNA. Crystal structures of Ku70/80 heterodimer in the absence of DNA (a) and in DNA-bound form (b). Adapted from [10].
2.2. DNA-PKcs
The structure of DNA-PKcs has proved quite elusive. Some beautiful work performed using cryo-electron microscopy single particle reconstruction of DNA-PKcs [44–47] has given a good impression of the overall structure (see Figure 4(a)). This has now been complemented by work in our laboratory. We have shown that DNA-PKcs crystals can be grown and diffract to about 8.5 Å resolution but the diffraction is better for the complexes with C-terminal fragments of Ku80, presumably due to a stabilization of the DNA-PKcs in the complex leading to better ordering of the crystal packing. We have recently used multiwavelength anomalous dispersion with the Ta6Br12 2+ heavy-metal cluster [48] to solve the structure of DNA-PKcs in complex with C-terminal domain of Ku80 at 6.6 Å resolution (Figure 4(b)).
Figure 4.

Equivalent views of DNA-PKcs as defined by (a) Cryo-electron microscopy and (b) X-ray protein crystallography. References to the publications and resolutions of the models are given above. Colour coding of these EM structures are as given in their respective publications [44–47]. The X-ray crystallographic experimental electron density map is as defined by Sibanda et al. [48].
Much of the DNA-PKcs polypeptide chain is constructed from HEAT repeat units (Figure 5) to form several separate domains. The DNA-PKcs tertiary structure measures 160 Å high and 120 Å across as viewed in Figure 6(a). From the N-terminus, HEAT-repeat motifs comprising about 66 α-helices fold into a hollow circular structure, which when viewed from the side resembles a cradle (Figure 6(b)). The chain changes direction before the circle is complete, thus leaving a gap (Figure 6(a)). Within this circular structure the regularity of the HEAT repeats breaks down at certain points, as indicated in Figure 6(a) with blue arrows. These points of irregularity may play a part in conformational changes that have been implicated in the function of this molecule [16]. It is possible that these conformational changes could have a bearing on the size of the gap (Figure 6(a)), which may have a role in the release of DNA-PKcs from DNA ends when NHEJ is complete. The ring structure most likely acts as a platform for proteins that engage in repair of broken DNA and together with Ku holds in place the DNA while it is being repaired.
Figure 5.
An example of HEAT-repeat motifs. Two consecutive α-helical hairpins are shown.
Figure 6.

Crystal structure of DNA-PKcs. Molecular surface of the DNA-PKcs structure showing (a) front and (b) side views. Also shown in (a), is the overall size of DNA-PKcs with the potential flexible sites indicated in blue arrows. The molecule is colour coded as follows: the ring structure that is predominantly HEAT repeats is green; the forehead that is part of the ring structure is light green; the putative DNA binding domain is magenta; the larger C-terminal part that includes the FAT and FATC domains is red, and the kinase domain is yellow.
In the second part of the structure the polypeptide chain exploits HEAT repeats to fold into a small, globular, putative DNA-binding domain within the circular structure. It is known that DNA-PKcs binds both double-stranded and single-stranded DNA. Williams et al. (2008) have proposed that “the protrusion” in their cryo-EM structure binds DNA [47], and this protrusion is equivalent to the small globular domain located within the circular region of the crystal structure. This remains the best candidate for both single- and double-stranded DNA recognition, but further work on DNA-PK (DNA-PKcs, Ku, DNA complex) crystals at a higher resolution structure of DNA-PKcs will be needed to confirm this. Thirdly, the C-terminal region folds into the Head/Crown that is perched right at the top of the cradle shaped circular structure and extends further back. This part contains the FAT, kinase domain, FATC, and various parts where other proteins, as indicated by biochemical studies, may bind to form complexes with DNA-PKcs (Figure 7).
Figure 7.

Schematic diagram of DNA-PKcs sequence-function implications. Shown are four highly conserved regions (HCR) [49]; Kinase, FAT, and FATC domains; putative sites for nuclear localization signals (NLSs) [49]; Autophosphorylation sites PQR [50], ABCDE [19], S3205 [51], S3821, S4026, T4102 [52], and T3950 [53]; cleavage sites for apoptotic protease caspase-3 [49, 54–56]; binding sites for Lyn tyrosine kinase [57], C1D protein that interacts at the leucine-zipper motif of DNA-PKcs [58], protein phosphatase-5 (PP5) that binds, using its tetratricopeptide repeat (TPR), to dephosphorylate DNA-PKcs at S2056 and T2609 [59], Ku70/80 that binds to present the damaged DNA double strand ends to DNA-PKcs [60], the kinase interacting protein (KIP) [61], and c-Abl that binds using its SH3 a binding that is triggered by DNA damage [60].
The core of the kinase structure from PI(3)Kγ, one of the family members, was superposed onto this Head/Crown region resulting in a plausible fit to the N-lobe β-strands and the C-lobe α-helices (Figure 8). In this location the kinase is exposed and easily accessible to substrates (Figure 8). From the location of the kinase domain the positions of the FAT and FATC regions can be inferred (Figure 7) as the kinase domain likely “snuggles” in between these two regions [62].
Figure 8.

DNA-PKcs kinase domain. The figure shows a cartoon of the overall structure of DNA-PKcs depicting the position of the kinase domain. The colour coding is as shown in Figure 6. The modelling of DNA-PKcs catalytic domain was based on the crystal structure of one of its family members, the PI(3)Kγ kinase (PDB code: 1E8X). Also shown is a closeup of DNA-PKcs catalytic domain. Helices of DNA-PKcs labelled (a) and (b) could be occupied by helices from the N-lobe of PI(3)Kγ. The N-lobe helices displayed in light blue were omitted from the final structure of DNA-PKcs due to unclear electron density in this region.
The size of the monomer of DNA-PKcs is predicted by small angle X-ray scattering (SAXS) to be about 155 Å [63], broadly in agreement with that of the crystallographic structure. DNA-PKcs dimerizes without DNA in a concentration-dependent manner. SAXS data indicate a large conformational change between autophosphorylated and unphosphorylated DNA-PKcs; the dimension and radius of gyration of phosphorylated DNA-PKcs increased 25 and 2 Å, respectively, compared to mock DNA-PKcs. Also, shape reconstruction of phosphorylated DNA-PKcs shows a wider cleft between head and palm domains than in the unphosphorylated enzyme.
2.3. DNA Ligase IV
Human LigIV has also proved difficult to study in isolation due to instability and flexibility but it is stabilised by interaction with XRCC4 [28]. In human, LigIV is one of three ATP-dependent DNA ligases, I, III, and IV, and plays a central role in eukaryotic NHEJ. LigIV can be divided into the catalytic and interaction regions. There are excellent reviews of the comparison of structures of DNA and RNA ligases, and RNA capping enzymes elsewhere [64–67].
LigIV belongs to the nucleotidyltransferase superfamily and carries out a three-step nucleotidyl transfer reaction: the formation of covalent enzyme-nucleotide monophosphate (NMP) intermediate (step 1), the transformation of the NMP to a 5′-phosphate of polynucleotide (step 2), and the joining of the 5′-phosphate with 3′-hydroxyl to seal two polynucleotides (step 3) [68]. These enzymes have four common motifs (I, III, IV, V) and in addition two more motifs (IIIα and VI), which are conserved among the cellular and ASF virus capping enzymes and eukaryotic ATP-dependent DNA ligases [69]. A recently found motif, Va, is well conserved among human DNA ligases [70]. The motif I KX(D/N)G has the catalytic lysine which forms the NMP-covalent intermediate. Motifs I-V are located in the nucleotidyltransferase domain (NTase) (Figure 9), the core of which comprises three mainly antiparallel β-sheets flanked by six α-helices [64]. Motifs Va and VI belong to the oligonucleotide/oligosaccharide-binding domain (OBD) (Figure 9), which has the five stranded Greek-key β-barrel capped by an α-helix [64, 71]. NTase and OBD are conserved among capping enzymes and DNA ligases [64]. Most LigIV syndrome mutations are found in the NTase and OBD [72] (Figure 9).
Figure 9.

Schematic diagram of human DNA ligase IV. Domain boundaries shown in the figure are based on the crystallographic structure of human DNA ligase I [73] and BRCT domains of LigIV [94]. Conserved motifs (I, III, IIIa, IV, V, Va, and VI) are shown in orange rectangles [69, 70]. The conserved catalytic lysine is indicated in green line. Reported phosphorylation sites are indicated by red lines [81, 99]. LigIV syndrome mutations are shown under domains [72, 100]. Ku and XRCC4 interaction regions are indicated with black brackets [29, 84, 94].
Many enzymes in the nucleotidyltrasferase superfamily have extra domains in addition to conserved catalytic core domains. N-terminal to the NTase, for instance, the human DNA ligases have DNA-binding domain (DBD), the three-dimensional structure of which was first uncovered by Pascal et al. (2004) with the catalytic domains of human DNA ligase I (LigI) complexed with an unligatable, nicked DNA fragment [73]. This domain is also found in archaeal DNA ligases [74–76] and possibly other eukaryotic, Poxvirus, and archaeal DNA ligases [77]. Pascal et al. (2004) showed that DBD is essential for LigI to bind DNA and to carry out ligation of DNA nicks [73]. However, this does not seem to be the case for DNA ligase III [78], although most DNA-binding affinity of LigIV seems to come from its DBD (T Ochi and TL Blundell, unpublished results). These results suggest that DBD of each human DNA ligase has different DNA-binding properties, although they are likely to have similar structures [79]. Two LigIV syndrome mutations are severe only when they are combined with R278H, and they seem to have little impact on LigIV activity [37]. On the basis of structural similarities of the catalytic regions of LigI and LigIV, they are likely to bind DNA in a similar manner.
In addition to the catalytic region, human DNA ligases have extra domains [79]. LigIV has a tandem BRCT domain with a linker predicted to be mostly disordered. This linker seems to be important for the catalytic activity of LigIV [80] and has a phosphorylation site at T650 by DNA-PKcs, the phosphorylation of which stabilizes LigIV [81]. The BRCT domain, which typically has four parallel β-strands surrounded by three α-helices [82], is common in cell cycle checkpoint proteins that respond to DNA damage [83]. LigIV interacts with XRCC4 mainly through the linker between the two BRCT [80]. As noted above, in addition to the interaction with XRCC4, the first BRCT domain (BRCT1) has been shown to interact with Ku70/80 [84].
Structures of tandem BRCT domains of human BRCA1 and MDC1, yeast Crb2, Nbs1, and Brc1, have been solved with different phosphopeptides. Four key residues that form the phospho-serine binding pocket—the (S/T)G motif at the end of the first β-strand (β1) and the (S/T)XK motif at the beginning of the second helix (α2) [85–93] have been identified; the residues are conserved in the tandem BRCT domain of LigIV except that the second motif is replaced by NXR. Thus BRCT1 might bind to phosphoserines [94], although the interactions with the two proximal BRCT domains found in BRCA1, MDC1, Crb2, Nbs1, and Brc1 are unlikely to occur in LigIV as the tandem BRCT domains are probably positioned apart [94, 95]. Indeed, in vitro phosphopeptide binding experiments showed that BRCT domains of LigIV bound phosphopeptides [96, 97]. However, the precise sequence of a phosphopeptide that binds to the BRCT domains has not yet been determined.
Since the tandem BRCT domains have a common globular arrangement of the BRCT domains, LigIV domains may interact when LigIV is in the free form. The main dimerization interface of the tandem BRCT domain is α2 in BRCT1, and the first and third helices α′1 and α′3 in the second BRCT domain (BRCT2) [98]. Interestingly, the interaction surface of those BRCT domains and XRCC4 is similar to that of other tandem BRCT domains (as discussed below). It is possible that a BRCT domain from another protein interacts with α2 of BRCT1, which is exposed to the solvent. Thus, although the tandem BRCT domains of LigIV have a long linker, they have features in common with other tandem BRCT domains.
2.4. XRCC4
In solution, XRCC4 exists as a salt-dependent equilibrium of dimers and tetramers [102] see Figure 10. Tail-to-tail tetramerisation was observed in XRCC4 protein crystals [103]. Binding of LigIV with the XRCC4 C-terminal 〈 α-helical coiled-coil stabilizes the XRCC4 dimer formation. The binding region between XRCC4 and LigIV overlaps with the XRCC4 tetramerisation region, which may explain why LigIV functions to shift XRCC4 to dimer form in solution [102]. Whether tetrameric XRCC4 has a function during NHEJ repair pathway is still not known.
Figure 10.

XRCC4 dimer, tetramer, and binding with DNA ligase IV peptide. The equilibrium is shifted to dimer when DNA ligase IV peptide binds to XRCC4.
The protein sequence of XRCC4 after residue 213 is not included in XRCC4 crystal structures due to the expected highly disordered and flexible structure of XRCC4 C-terminal domain [103]. However, EM studies have revealed that mouse XRCC4 C-terminal structure is a globular domain [104]. This domain includes putative nuclear localization sequences [105]. These authors also suggested that a cluster of acidic amino acids 229–238 is important for the auto-transcription activity. Furthermore, the XRCC4 C-terminal domain is the target for NHEJ regulatory proteins. DNA-PKcs phosphorylates XRCC4 and regulates its binding with DNA [31]. Residues S260 and S318 in the XRCC4 C-terminal region were identified to be the main phosphorylation sites by DNA-PKcs [106]. XRCC4 is also phosphorylated by CK2, residue T233 and the phosphorylation by CK2 recruits PNK, which is likely to participate in NHEJ [107]. Indeed the structure of the ForkHead-Associated (FHA) Domain of PNK with a XRCC4-derived phosphopeptide has been solved [108]. XRCC4 residue K210 was also reported to be important for small ubiquitin-like modifier (SUMO) modification, which regulates XRCC4 cellular localization [109]. The XRCC4 C-terminal region, together with the N-terminal region (residues 1–28) and central region (residues 168–200), may facilitate cooperative DNA binding [31]. Thus, definition of the structure of the C-terminal region structure will contribute to understanding how XRCC4 binds to LigIV and DNA in order to carry out its function.
2.5. XLF
XLF was identified through a cDNA functional complementation cloning study of patient 2BN following discovery of a group of NHEJ deficiency patients (2BN) [38, 110]. It was also independently identified through yeast two-hybrid screening for XRCC4 interactors [39]. XLF is evolutionarily conserved throughout a wide range of eukaryotes such as vertebrates, insects, and even in filamentous fungi [111]. Full-length human XLF contains 299 residues. At its extreme C-terminus, a small conserved basic cluster constitutes the nuclear localization sequence. Using immunofluorescence staining, XLF was observed localizing in nucleus of human cells [39].
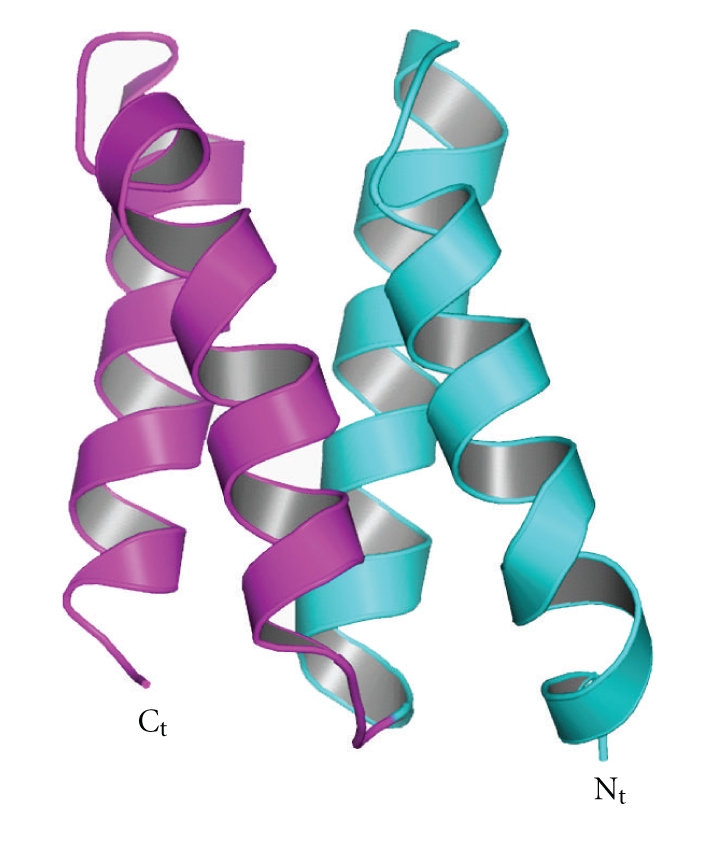
The crystal structure of XLF with a C-terminal truncation, solved independently at 2.3 Å resolution by Andres et al. [112] and in our laboratory [101], exists as a homodimer containing a globular N-terminal head domain and extended coiled-coil helical tail, which is folded back around the coiled-coil (Figure 11). The N-terminal head domain starts with a single helix α1, which is followed by a seven-stranded antiparallel β structure sandwiching a helix-turn-helix motif between β4 and β5. The tail structure contains three helices α4, α5, and α6. While α4 extends away from N-terminal head domain around 60 Å, α5 and α6 fold back and make contact with the head domain. The α4 helices from the two protomers interact as a coiled-coil structure burying highly conserved and hydrophobic residues at the interface. This dimerization of XLF is further enhanced through the folding back of the α5 and α6 helices to encircle the α4 helices of the other protomer to form a clamp, leading to burying of a surface area of ~6500 Å2. Gel filtration, protein crosslinking and analytical ultracentrifugation are also consistent with a stable homodimer form of XLF in solution [101]. XLF was found to have concentration-dependent higher-order complex formation during gel filtration experiments [112]. The homodimer of XLF, however, is the smallest stable functional unit.
Figure 11.

Crystal structure of XLF/Cernunnos. Ribbon diagram of the XLF/Cernunnos dimer. One protomer is rainbow colour going from N-terminus (blue) to C-terminus (red). Adapted from Li et al. [101].
Due to the predicted disordered structure for the XLF C-terminal region after residue 245, around 70 residues were removed from the XLF C-terminus in the crystal structure analyses [101, 112]. The approximate location of the XLF C-terminal region, however, can be predicted to be near the N-terminal head domain region according to the helix α6 direction.
Although XLF and XRCC4 have similar architectures, large structural differences from head to tail occur between these two proteins. For the head domain, both proteins contain the same seven-stranded antiparallel β-structure sandwiching a helix-turn-helix motif, but XLF contains an extra helix at the N-terminus. As we have seen, the tail structure of XLF contains distinct helices folding back, while the extended coiled-coil tail structure of XRCC4 contains the LigIV binding region near the C-terminus. The differences in sequence and structure between XLF and XRCC4 tails explain why LigIV does not bind to XLF in the same way as XRCC4.
The functions and mechanisms of action of XLF in NHEJ are still not fully understood. XLF not only stabilizes LigIV/XRCC4 at broken DNA ends, but also enhances the LigIV/XRCC4 end-joining process. XLF has also been found to be essential for repairing mismatched 3′ overhangs and the gap-filling process together with DNA polymerase polλ and polμ [113, 114]. Understanding how XLF functions in NHEJ through studying its interaction with other NHEJ proteins structurally will help unravel the exact role of XLF. It will contribute towards our current understanding of DNA repair in NHEJ and may also potentially lead to future therapeutic application for NHEJ defects patients.
3. Structural Biology of Complexes
3.1. DNA-PKcs/Ku70/Ku80/DNA Ternary Complex (DNA-PK)
The crystal structure of the Ku70/80 heterodimer does not include the C-terminal DNA-PKcs interaction domain of Ku80 (Ku80CTD), which is dispensable for the binding of Ku70/80 to DNA but is required for DNA-PK recruitment to the sites of damaged DNA [13, 14]. Nuclear magnetic resonance analysis of 19 kDa Ku80CTD (residues 545–732) defines an α-helical structure [115, 116]. Further structural studies of full-length Ku70/80 with and without DNA have been conducted using single-particle electron microscopy (EM) [117] and SAXS combined with live cell imaging [63]. The position of Ku80CTD was proposed to be under the α/β domain of Ku70 by EM, but the domain was found to be flexible in the SAXS study. Molecular dynamic simulations of Ku80CTD produced an ensemble of conformations, supporting the idea of Ku80CTD being a region of high flexibility [63]. Taken together, the studies show that association of Ku70 and Ku80 to form a heterodimer is required for binding dsDNA ends, that Ku-dependent DNA binding drives the recruitment of DNA-PKcs and that the latter interaction involves the helical domain located at the C-terminus of Ku80. Although Ku80CTD was included in the crystal structure of DNA-PKcs, its position could not be unequivocally defined, presumably due to the dominance of similar alpha helical structures in the DNA-PKcs itself [48].
Insights into the DNA-PKcs/Ku70/Ku80 holoenzyme structures and possible synaptic complexes have been obtained using cryo-electron microscopy and SAXS. Boskovic et al. (2003) used electron microscopy at low resolution (~30 Å) to demonstrate large conformational changes in human DNA-PKcs when double-stranded DNA binds, and suggested that this may correlate with the activation of the kinase [118]. Subsequently, Spagnolo et al. (2006) have used single-particle electron microscopy at ~25 Å resolution to study human DNA-PKcs/Ku70/Ku80 holoenzyme assembled on DNA [16]. They again found evidence for conformational changes on binding of Ku and DNA to DNA-PKcs. They identified dimeric particles comprising two DNA-PKcs/Ku70/Ku80 holoenzymes, which they consider are likely to be synaptic complexes, maintaining broken ends and providing a platform for other components required for end processing and ligation. A SAXS study of DNA-PK revealed that it had two different modes of dimerization as was observed previously with DNA-PKcs [63]. Depending on the presence of either 40 bp hairpin DNA or 40 bp Y-shaped DNA, DNA-PK formed the head-to-head or palm-to-palm dimer. Very recently Perry et al. (2010) have taken study of the DNA-PKcs/Ku70/Ku80 holoenzyme further by analyzing their earlier SAXS studies in the light of the crystal structure of DNA-PKcs [119]. They have impressively demonstrated that DNA-PK phosphorylation causes a large conformational change, sufficient to open the gap in the ring and provide access to or release from DNA. Ku80CTD has been shown to be flexible and to extend in solution to the benefit of recruitment of DNA-PKcs. It is possible that Ku80 interacts with DNA-PKcs on both sides of BSB [63].
3.2. DNA Ligase IV/XRCC4 Complexes
LigIV is stabilized by forming a tight complex with XRCC4 [28]. About 99% of LigIV is preadenylated when purified together with XRCC4 and it is difficult to readenylate after single-nick ligation [120], implying that the LigIV/XRCC4 complex is ready to ligate DNA. Unlike other human DNA ligases LigIV/XRCC4 can efficiently ligate one of the nicks of a DSB, although the other is unligatable [26], and it can ligate DNA strands across gaps and fully incompatible ends [121]. Furthermore, it has been shown that LigIV can ligate single-stranded poly-T DNA [122]. Interestingly, the ligation efficiency is higher with long DNA substrates ≥157 bp than short ones ≤53 bp [123]. This might be related to the observation that a single LigIV/XRCC4 bridges two DNA ends [124].
The crystal structures of the XRCC4 dimer complexed with the tandem BRCT domain of LigIV shows that the linker between the two BRCT domains is well ordered and forms a helix-loop-helix (HLH) clamp around the coiled-coil [29, 94] (Figures 12(a) and 12(b)). The same interaction mode and secondary structure arrangement are observed in the orthologous yeast complex between XRCC4 (Lif1p) and LigIV (Lig4p) [95] (Figure 12(c)). The two BRCT domains in the human and yeast complexes extend the clamp, encircling the coiled-coil domain. The 310-helix in BRCT1 is located close to the conserved XRCC4 interaction region of the linker (XIR: residue 748–784) between two BRCT domains. The corresponding 310-helix in BRCT1 of 53BP1 participates in the interaction surface of p53 [125, 126]. The interaction of XRCC4 with LigIV produces a kink in one helix of the coiled-coil of XRCC4 dimer and switches the left-handed heptad repeat into a right-handed undecad coiled-coil; as a result, the LigIV interaction surface becomes flat [29, 94]. The kink bends in the opposite direction in the complex between XRCC4 with XIR and with the tandem BRCT domain [94]. The former structure might be an intermediate state of LigIV/XRCC4 interaction. If so, this dynamical conformational change might have a biological role in vivo. This kink does not appear in Lif1p/Lig4p even though the refinement of the structure against a new 3.5 Å diffraction data set was carried out (see [127], T Ochi and TL Blundell, unpublished results). Thus, the kink may be unique to human and some other higher organisms.
Figure 12.

Crystal structures of LigIV/XRCC4 and Lif1p-Lig4p. Conserved XRCC4 or Lif1 interaction region of LigIV (XIR) or Lig4p (LIR) are indicated with blue arrows. (a) XRCC4(residues 1–213)(blue) and XIR (violet) (PDB code: 1IK9). (b) XRCC(residues 1–203)(blue) and BRCT domains of LigIV(residues 654–911)(violet) (PDB code: 3II6) (c) Lif1p(residues 1–246)(blue) and BRCT domains of Lig4p(residues 680–944)(violet).
The second helix in HLH mediates a hydrophobic interaction with the opposite side of the flat surface of the XRCC4 to where XIR interacts [94] and a similar extensive hydrophobic interaction is observed in Lif1p/Lig4 (residues 827–839) [95]. LigIV additionally interacts with the coiled-coil of XRCC4 via α′1 and α′3 of BRCT2, in a manner that resembles the interaction between BRCT1 and BRCT2 of other tandem BRCT domains. Superposition of LigIV/XRCC4 and Lif1p/Lig4p based on XIR and the corresponding region of Lig4p (LIR) shows that, apart from the kink described above, a further change occurs in the position of BRCT1 (Figure 13). This may be a crystallographic artefact because BRCT1 is closely packed with BRCT2 belonging to another molecule in both human and yeast structures. However, the NMR structure of BRCT1 (PDB code: 2E2W) has the same conformation as the crystallographic one, suggesting at least human BRCT1 and the following linker is likely to have the same conformation in solution.
Figure 13.

Comparison of positions of BRCT1 between LigIV/XRCC4 and Lif1p/Lig4p. Structures of LigIV/XRCC4 (violet/silver) and Lifp1/Lig4p (green/gold) were superimposed based on XIR and LIR. The figure is a view of the C- to N-terminal direction of the coiled-coils of XRCC4 and Lif1p. The N-terminals and helices of BRCT1 of LigIV and Li4p are labeled with violet and green, respectively.
A recently published EM structure of the LigIV/XRCC4 complex shows the N-terminal of LigIV in proximity to the head domain of XRCC4 [128]. The authors compared two LigIV/XRCC4 constructs, one with the full-length sequences, and the other with a full-length LigIV and a truncated XRCC4 (residues 1–213). From the differences of the two EM images, they determined the position of the C-terminal of XRCC4 and by labelling the hexahistidine tag with gold, they identified the N-terminus of LigIV. Although the authors reconstructed 2D averaged images of LigIV/XRCC4, the 3D reconstruction failed partially because of heterogeneity of the LigIV/XRCC4 conformation. Thus, they proposed that the catalytic region of LigIV is connected to the C-terminal region by a flexible linker and this may have functional importance (see also Perry et al. (2010) [119]).
We have carried out SAXS studies of the tandem BRCT domain of LigIV/mutated XRCC4 (BmX4) and LigIV/mutated XRCC4 (LmX4) in order to investigate the conformation of the catalytic region in solution (Figure 14(a)) [129]. Here, mutated XRCC4 is identical to the one used for solving the structure of XIR/XRCC4 [29]. The linearity of the respective Guinier plots confirmed that the protein solutions were homogeneous and monodisperse (Figure 14(b)). The deduced radius of gyration and the maximum molecular dimension of LmX4 are 9 Å and 43 Å larger, respectively, than those of BmX4. The simulated scattering profile using the crystallographic structure of BmX4 (PDB code: 3II6) fitted the measured SAXS curve well (χ 2 = 3.687, data not shown). Moreover, the ab initio 3D shape restoration of BmX4 reproduced an overall conformation consistent with the crystal structure (Figure 14(c)). The ab initio shape reconstruction of LmX4 revealed that the catalytic region may contribute additional density to the head domain of XRCC4 or the tandem BRCT domain of LigIV when compared with the conformation of BmX4 (Figure 14(c)). Since the extended, open conformation of the catalytic region in solution has also been observed in an archaeal DNA ligase [74], the extra density may correspond to a similar conformation of the catalytic region of LigIV to the closed conformation observed in other archaea DNA ligases [75, 76]. As the shape restorations of LmX4 yielded a reproducible conformation (also indicated by the normalized spatial discrepancy (NSD) value after shape averaging), this finding might imply interactions between the catalytic region and BmX4. However, electrophoretic mobility shift assay and protease analysis (data not shown) indicate that the catalytic region is unlikely to have strong interactions with BmX4. Thus, although the majority of LmX4 in solution may have the extended open conformation, the catalytic region is flexibly attached to BmX4. Our observations agree with the EM study [128].
Figure 14.

SAXS study of LmX4 and Bm4. (a) SAXS data of LmX4 (green) and BmX4 (gold) where q = (4πsin θ)/λ. (b) Guinier plots of LmX4 and BmX4 with liner fits (red line) q < 1.3/Rg. (c) Shape reconstructions of BmX4 (gold) and LmX4 (green) created by using Dammin [130]. Ten individual models were calculated and averaged by Damaver [131]. NSD of BmX4 and LmX4 are 0.780 and 0.814, respectively. Molecular envelopes were produced using the UCSF Chimera package from the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco (supported by NIH P41 RR-01081) [132]. The crystallographic model of BmX4 (PDB code: 3II6) was superimposed into its molecular envelope by using Chimera.
3.3. XLF/XRCC4 Complexes
The interaction between XLF and XRCC4 is salt sensitive, it does not depend on DNA [39, 133] and interactions occur through the head regions as shown by yeast two-hybrid study of various mutants [134]. XLF bound to beads at its C-terminal was still able to pull down LigIV/XRCC4, implying that the C-terminal of XLF is not important for interaction with LigIV/XRCC4 [135].
Mutagenesis studies indicate that the structurally exposed XLF residue L115 (Figure 16 shown in green) located in the β6-β7 loop is important for XLF/XRCC4 interaction [112]. Residues K63, K65, and K99 (Figure 15 shown in green) of XRCC4 are essential for interaction and are located on the region of the head domain close to the helical tail [112]. Nonessential interaction residues of XLF are mainly located outside the head domain region whereas the nonessential XLF/XRCC4 binding residues in XRCC4 are mainly located on the topside of N-terminal head domain and on the helical tail structure before the LigIV binding region (Figures 15 and 16 shown in grey) [112]. These studies are consistent with a linear side-by-side interaction model, in which XLF head domains slide into the space created by XRCC4 head domains and N-terminal part of the tail structure [112] (Figure 17). However, we cannot exclude a model for XLF/XRCC4, involving XLF and XRCC4 binding together in a side-by-side manner but with a degree of twist introducing curvature and possibly a circular complex. This would have the advantage of forming a finite and discrete complex. Further X-ray small angle scattering experiments may be the best approach to resolving this, especially if the complexes are dynamic as gel filtration experiments suggest. However, some encouragement that well-defined complexes can be identified is found in the observation that XLF/XRCC4 complexes have been crystallized and X-ray data collected, albeit to low resolution (Q Wu, TL Blundell unpublished data).
Figure 16.

Surface views of XLF1–233 (PDB code: 2QM4). Residue L115, which is important for XLF-XRCC4 binding is labeled in green. Nonessential residues for XLF-XRCC4 interaction I105, E111, E169, L174, R178, L179, E185, and I195 and residues after 224 are labeled in grey [112].
Figure 15.

Surface views of XRCC41–213-DNA ligase IV peptide748–784 structure (PDB code:1IK9). Residues K63, K65, and K99, which are important for XLF-XRCC4 binding are labeled in green. Nonessential residues for XLF-XRCC4 interaction K26, R71, K72, K90, R107, K115, K146, R150, R153, R161, and R164 are labeled in grey [112].
Figure 17.

XLF/XRCC4 linear side-by-side interaction model. Adapted from [112].
3.4. Spatial Arrangement of Higher-Order Complexes
In order to give a picture of the spatial and temporal organisation of the NHEJ repair system as a whole, an understanding of the order of interactions during the assembly of the DNA-PKcs/Ku70/Ku80/DNA ternary complex and the LigIV/XRCC4/XLF/DNA quaternary complex will be essential.
Ku70/80 and DNA-PKcs, which have higher DNA-binding affinity compared to LigIV/XRCC4/XLF, most likely form the DNA-PKcs/Ku70/Ku80/DNA ternary complex first. For the following LigIV/XRCC4/XLF/DNA complex formation, the order and dynamics of protein assembly are still to be determined. The interaction between XRCC4 and XLF is relatively weak compared to the strong binding between XRCC4 and LigIV. It is not clear whether the XLF-dimer interactions with XRCC4-dimer are maintained when the ligase is recruited. Protein interaction assays have confirmed the XRCC4 independent, XLF recruitment to DSBs ends through interaction with Ku70/80 only in the present of DNA. This may imply that XLF can act independently without XRCC4.
Live cell imaging techniques have identified the immediate recruitment of XLF to laser-induced DSBs with Ku70/80 protein bound [136]. XRCC4 is dispensable for XLF recruitment to DNA ends, but its presence can stabilize the XLF/DNA interaction [136]. Protein interaction assays have confirmed the interaction between Ku70/80 and XLF, and this interaction only occurs in the presence of DNA [136].
Both XRCC4 and XLF require a long piece of DNA for binding. How DNA is structurally involved in all the higher-order protein complexes is of fundamental interest. The phosphorylation of LigIV, XRCC4, and XLF by DNA-PKcs does not interfere greatly with the core functions of these proteins, but could alter the relative binding affinities of various protein-protein or protein-DNA interactions, which are important for correct spatial arrangement of the higher-order complexes. All of this uncertainty underlines the need for further studies to characterise complexes temporally as well as spatially.
4. Discussion
The challenges of structural characterisation of dynamic multiprotein systems clearly demand a combination of SAXS, EM, X-ray crystallography, and other approaches. All will be advantaged by methods for stabilization and fixation of the complexes. Modified constructs, for example, phospho-mimicking mutation and truncation as well as postmodification, for example, phosphorylation and methylation, need to be explored in order to identify stable complexes. For single particle cryo-EM studies, GraFix has been successfully introduced to stabilize macromolecules [137]. This exploits glycerol gradient centrifugation into increasing concentrations of chemical fixation reagent to stabilize individual macromolecules and to prevent opportunistic aggregation. A similar approach might be used for other structural studies including X-ray crystallography, although here molecular surface modification of the complexes may prevent the formation of ordered crystals.
Crystals of large multiprotein assemblies suitable for high-resolution X-ray diffraction remain a challenge. So the development of methods to analyse low-resolution X-ray diffraction data is essential. In this respect free-electron laser (FEL) light sources may allow single particle X-ray FEL (XFEL) imaging. X-ray crystallography with nanocrystals is also a promising method.
X-ray crystallography is still the only technique to give atomic resolution of large structures and high resolution is essential for studying the binding of small molecules. Indeed chemical tools that allow specific intervention in NHEJ should allow dissection of the roles of the various components. These tools would also likely contribute to the discovery of lead compounds and preclinical candidates for therapeutic intervention at allosteric and other regulatory interaction sites in oncology and for patients with defects in the NHEJ pathway.
The immediate interest, developing from the emerging structure of DNA-PKcs, is the improvement of design of inhibitors that bind at the ATP site of the protein kinase moiety. Such inhibitors would not only inform the development of useful therapeutic agents but should also be of immediate value in investigating the possibility of improving stability of the kinase domain, and the quality and resolution of crystals.
Eventually, we would hope to pursue a structure-guided approach to optimize the design of such inhibitors. Similar approaches could be taken with the ligase active site. In our view a more exciting and adventurous approach would be to design new chemical entities that bind at allosteric sites, templates or adaptor binding sites—so called allo-targeting—that are critical to the activation, colocalisation and/or specificity of the regulation of NHEJ. The use of fragment-based methods [138–140] in this context is attractive. Likely targets would be the head-to-head interactions of XRCC4 and XLF, the interactions of BRCT domains, and the interaction of Ku70/80 and the DNA-PKcs.
In conclusion, a spatial and temporal understanding of NHEJ should provide insights into the mechanism of this critical cellular process and also suggest approaches to designing useful chemical tools. Indeed the design of small chemical agents that noncovalently modulate interactions would also likely contribute to the discovery of lead compounds that allow therapeutic intervention in oncology and treatment of patients with defects in the NHEJ pathway.
Acknowledgments
The authors would like to thank Dr. J. Günter Grossmann for useful discussions and comments on the SAXS data. B. L. Sibanda and D. Y. Chirgadze were supported by Wellcome Trust Programme Grant: 079281/Z/06/Z. T. Ochi was supported by Overseas Research Studentship (ORS). Molecular graphics except for Figures 4 and 14 were prepared by using PyMOL [141].
References
- 1.Takata M, Sasaki MS, Sonoda E, et al. Homologous recombination and non-homologous end-joining pathways of DNA double-strand break repair have overlapping roles in the maintenance of chromosomal integrity in vertebrate cells. The EMBO Journal. 1998;17(18):5497–5508. doi: 10.1093/emboj/17.18.5497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.O’Driscoll M, Jeggo PA. The role of double-strand break repair—insights from human genetics. Nature Reviews Genetics. 2006;7(1):45–54. doi: 10.1038/nrg1746. [DOI] [PubMed] [Google Scholar]
- 3.Vilenchik MM, Knudson AG. Endogenous DNA double-strand breaks: production, fidelity of repair, and induction of cancer. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(22):12871–12876. doi: 10.1073/pnas.2135498100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Khanna KK, Jackson SP. DNA double-strand breaks: signaling, repair and the cancer connection. Nature Genetics. 2001;27(3):247–254. doi: 10.1038/85798. [DOI] [PubMed] [Google Scholar]
- 5.Gellert M. V(D)J recombination: RAG proteins, repair factors, and regulation. Annual Review of Biochemistry. 2002;71:101–132. doi: 10.1146/annurev.biochem.71.090501.150203. [DOI] [PubMed] [Google Scholar]
- 6.Chaudhuri J, Alt FW. Class-switch recombination: interplay of transcription, DNA deamination and DNA repair. Nature Reviews Immunology. 2004;4(7):541–552. doi: 10.1038/nri1395. [DOI] [PubMed] [Google Scholar]
- 7.Yan CT, Boboila C, Souza EK, et al. IgH class switching and translocations use a robust non-classical end-joining pathway. Nature. 2007;449(7161):478–482. doi: 10.1038/nature06020. [DOI] [PubMed] [Google Scholar]
- 8.Nussenzweig A, Nussenzweig MC. A backup DNA repair pathway moves to the forefront. Cell. 2007;131(2):223–225. doi: 10.1016/j.cell.2007.10.005. [DOI] [PubMed] [Google Scholar]
- 9.Lieber MR, Ma Y, Pannicke U, Schwarz K. The mechanism of vertebrate nonhomologous DNA end joining and its role in V(D)J recombination. DNA Repair. 2004;3(8-9):817–826. doi: 10.1016/j.dnarep.2004.03.015. [DOI] [PubMed] [Google Scholar]
- 10.Walker JR, Corpina RA, Goldberg J. Structure of the Ku heterodimer bound to dna and its implications for double-strand break repair. Nature. 2001;412(6847):607–614. doi: 10.1038/35088000. [DOI] [PubMed] [Google Scholar]
- 11.Downs JA, Jackson SP. A means to a DNA end: the many roles of Ku. Nature Reviews Molecular Cell Biology. 2004;5(5):367–378. doi: 10.1038/nrm1367. [DOI] [PubMed] [Google Scholar]
- 12.Hartley KO, Gell D, Smith GCM, et al. DNA-dependent protein Kinase catalytic subunit: a relative of phosphatidylinositol 3-Kinase and the ataxia telangiectasia gene product. Cell. 1995;82(5):849–856. doi: 10.1016/0092-8674(95)90482-4. [DOI] [PubMed] [Google Scholar]
- 13.Singleton BK, Torres-Arzayus MI, Rottinghaus ST, Taccioli GE, Jeggo PA. The C terminus of Ku80 activates the DNA-dependent protein Kinase catalytic subunit. Molecular and Cellular Biology. 1999;19(5):3267–3277. doi: 10.1128/mcb.19.5.3267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gell D, Jackson SP. Mapping of protein-protein interactions within the DNA-dependent protein Kinase complex. Nucleic Acids Research. 1999;27(17):3494–3502. doi: 10.1093/nar/27.17.3494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yoo S, Dynan WS. Geometry of a complex formed by double strand break repair proteins at a single DNA end: recruitment of DNA-PKcs induces inward translocation of Ku protein. Nucleic Acids Research. 1999;27(24):4679–4686. doi: 10.1093/nar/27.24.4679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Spagnolo L, Rivera-Calzada A, Pearl LH, Llorca O. Three-dimensional structure of the human DNA-PKcs/Ku70/Ku80 complex assembled on DNA and its implications for DNA DSB repair. Molecular Cell. 2006;22(4):511–519. doi: 10.1016/j.molcel.2006.04.013. [DOI] [PubMed] [Google Scholar]
- 17.Smith GCM, Jackson SP. The DNA-dependent protein Kinase. Genes and Development. 1999;13(8):916–934. doi: 10.1101/gad.13.8.916. [DOI] [PubMed] [Google Scholar]
- 18.Weterings E, Chen DJ. The endless tale of non-homologous end-joining. Cell Research. 2008;18(1):114–124. doi: 10.1038/cr.2008.3. [DOI] [PubMed] [Google Scholar]
- 19.Ding Q, Reddy YVR, Wang W, et al. Autophosphorylation of the catalytic subunit of the DNA-dependent protein Kinase is required for efficient end processing during DNA double-strand break repair. Molecular and Cellular Biology. 2003;23(16):5836–5848. doi: 10.1128/MCB.23.16.5836-5848.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Block WD, Yu Y, Merkle D, et al. Autophosphorylation-dependent remodeling of the DNA-dependent protein Kinase catalytic subunit regulates ligation of DNA ends. Nucleic Acids Research. 2004;32(14):4351–4357. doi: 10.1093/nar/gkh761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Moshous D, Callebaut I, de Chasseval R, et al. Artemis, a novel DNA double-strand break repair/V(D)J recombination protein, is mutated in human severe combined immune deficiency. Cell. 2001;105(2):177–186. doi: 10.1016/s0092-8674(01)00309-9. [DOI] [PubMed] [Google Scholar]
- 22.Lieber MR. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annual Review of Biochemistry. 2010;79:181–211. doi: 10.1146/annurev.biochem.052308.093131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ma Y, Pannicke U, Schwarz K, Lieber MR. Hairpin opening and overhang processing by an Artemis/DNA-dependent protein Kinase complex in nonhomologous end joining and V(D)J recombination. Cell. 2002;108(6):781–794. doi: 10.1016/s0092-8674(02)00671-2. [DOI] [PubMed] [Google Scholar]
- 24.Mahajan KN, Nick McElhinny SA, Mitchell BS, Ramsden DA. Association of DNA polymerase μ (pol μ) with Ku and ligase IV: role for pol μ in end-joining double-strand break repair. Molecular and Cellular Biology. 2002;22(14):5194–5202. doi: 10.1128/MCB.22.14.5194-5202.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee JW, Blanco L, Zhou T, et al. Implication of DNA Polymerase λ in alignment-based gap filling for nonhomologous DNA end hoining in human nuclear extracts. The Journal of Biological Chemistry. 2004;279(1):805–811. doi: 10.1074/jbc.M307913200. [DOI] [PubMed] [Google Scholar]
- 26.Ma Y, Lu H, Tippin B, et al. A biochemically defined system for mammalian nonhomologous DNA end joining. Molecular Cell. 2004;16(5):701–713. doi: 10.1016/j.molcel.2004.11.017. [DOI] [PubMed] [Google Scholar]
- 27.Roberts SA, Strande N, Burkhalter MD, et al. Ku is a 5′-dRP/AP lyase that excises nucleotide damage near broken ends. Nature. 2010;464(7292):1214–1217. doi: 10.1038/nature08926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bryans M, Valenzano MC, Stamato TD. Absence of DNA ligase IV protein in XR-1 cells: evidence for stabilization by XRCC4. Mutation Research/DNA Repair. 1999;433(1):53–58. doi: 10.1016/s0921-8777(98)00063-9. [DOI] [PubMed] [Google Scholar]
- 29.Sibanda BL, Critchlow SE, Begun J, et al. Crystal structure of an Xrcc4-DNA ligase IV complex. Nature Structural Biology. 2001;8(12):1015–1019. doi: 10.1038/nsb725. [DOI] [PubMed] [Google Scholar]
- 30.Grawunder U, Wilm M, Wu X, et al. Activity of DNA ligase IV stimulated by complex formation with XRCC4 protein in mammalian cells. Nature. 1997;388(6641):492–495. doi: 10.1038/41358. [DOI] [PubMed] [Google Scholar]
- 31.Modesti M, Hesse JE, Gellert M. DNA binding of Xrcc4 protein is associated with V(D)J recombination but not with stimulation of DNA ligase IV activity. The EMBO Journal. 1999;18(7):2008–2018. doi: 10.1093/emboj/18.7.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang Y, Lamarche BJ, Tsai M-D. Human DNA ligase IV and the ligase IV/XRCC4 complex: analysis of nick ligation fidelity. Biochemistry. 2007;46(17):4962–4976. doi: 10.1021/bi0621516. [DOI] [PubMed] [Google Scholar]
- 33.Gao Y, Sun Y, Frank KM, et al. A critical role for DNA end-joining proteins in both lymphogenesis and neurogenesis. Cell. 1998;95(7):891–902. doi: 10.1016/s0092-8674(00)81714-6. [DOI] [PubMed] [Google Scholar]
- 34.Frank KM, Sekiguchi JM, Seidl KJ, et al. Late embryonic lethality and impaired V(D)J recombination in mice lacking DNA ligase IV. Nature. 1998;396(6707):173–177. doi: 10.1038/24172. [DOI] [PubMed] [Google Scholar]
- 35.Gao Y, Ferguson DO, Xie W, et al. Interplay of p53 and DNA-repair protein XRCC4 in tumorigenesis, genomic stability and development. Nature. 2000;404(6780):897–900. doi: 10.1038/35009138. [DOI] [PubMed] [Google Scholar]
- 36.Frank KM, Sharpless NE, Gao Y, et al. DNA ligase IV deficiency in mice leads to defective neurogenesis and embryonic lethality via the p53 pathway. Molecular Cell. 2000;5(6):993–1002. doi: 10.1016/s1097-2765(00)80264-6. [DOI] [PubMed] [Google Scholar]
- 37.O’Driscoll M, Cerosaletti KM, Girard P-M, et al. DNA ligase IV mutations identified in patients exhibiting developmental delay and immunodeficiency. Molecular Cell. 2001;8(6):1175–1185. doi: 10.1016/s1097-2765(01)00408-7. [DOI] [PubMed] [Google Scholar]
- 38.Buck D, Malivert L, de Chasseval R, et al. Cernunnos, a novel nonhomologous end-joining factor, is mutated in human immunodeficiency with microcephaly. Cell. 2006;124(2):287–299. doi: 10.1016/j.cell.2005.12.030. [DOI] [PubMed] [Google Scholar]
- 39.Ahnesorg P, Smith P, Jackson SP. XLF interacts with the XRCC4-DNA Ligase IV complex to promote DNA nonhomologous end-joining. Cell. 2006;124(2):301–313. doi: 10.1016/j.cell.2005.12.031. [DOI] [PubMed] [Google Scholar]
- 40.Feldmann E, Schmiemann V, Goedecke W, Reichenberger S, Pfeiffer P. DNA double-strand break repair in cell-free extracts from Ku80-deficient cells: implications for Ku serving as an alignment factor in non-homologous DNA end joining. Nucleic Acids Research. 2000;28(13):2585–2596. doi: 10.1093/nar/28.13.2585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Larkin MA, Blackshields G, Brown NP, et al. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23(21):2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 42.Gouet P, Courcelle E, Stuart DI, Métoz F. ESPript: analysis of multiple sequence alignments in PostScript. Bioinformatics. 1999;15(4):305–308. doi: 10.1093/bioinformatics/15.4.305. [DOI] [PubMed] [Google Scholar]
- 43.Dynan WS, Yoo S. Interaction of Ku protein and DNA-dependent protein Kinase catalytic subunit with nucleic acids. Nucleic Acids Research. 1998;26(7):1551–1559. doi: 10.1093/nar/26.7.1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chiu CY, Cary RB, Chen DJ, Peterson SR, Stewart PL. Cryo-EM imaging of the catalytic subunit of the DNA-dependent protein Kinase. Journal of Molecular Biology. 1998;284(4):1075–1081. doi: 10.1006/jmbi.1998.2212. [DOI] [PubMed] [Google Scholar]
- 45.Leuther KK, Hammarsten O, Kornberg RD, Chu G. Structure of DNA-dependent protein Kinase: implications for its regulation by DNA. The EMBO Journal. 1999;18(5):1114–1123. doi: 10.1093/emboj/18.5.1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rivera-Calzada A, Maman JD, Spagnolo L, Pearl LH, Llorca O. Three-dimensional structure and regulation of the DNA-dependent protein Kinase catalytic subunit (DNA-PKcs) Structure. 2005;13(243):p. 495. doi: 10.1016/j.str.2004.12.006. [DOI] [PubMed] [Google Scholar]
- 47.Williams DR, Lee KJ, Chen DJ, Shi J, Stewart PL. Cryo-EM structure of the DNA-dependent protein Kinase catalytic subunit at subnanometer resolution reveals alpha helices and insight into DNA binding. Structure. 2008;16:468–477. doi: 10.1016/j.str.2007.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sibanda BL, Chirgadze DY, Blundell TL. Crystal structure of DNA-PKcs reveals a large open-ring cradle comprised of HEAT repeats. Nature. 2010;463(7277):118–121. doi: 10.1038/nature08648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fujimori A, Araki R, Fukumura R, et al. Identification of four highly conserved regions in DNA-PKcs. Immunogenetics. 2000;51(11):965–973. doi: 10.1007/s002510000227. [DOI] [PubMed] [Google Scholar]
- 50.Xiaoping C, Yaping Y, Gupta S, Cho Y-M, Lees-Miller SP, Meek K. Autophosphorylation of DNA-dependent protein Kinase regulates DNA end processing and may also alter double-strand break repair pathway choice. Molecular and Cellular Biology. 2005;25(24):10842–10852. doi: 10.1128/MCB.25.24.10842-10852.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Douglas P, Sapkota GP, Morrice N, et al. Identification of in vitro and in vivo phosphorylation sites in the catalytic subunit of the DNA-dependent protein Kinase. The Biochemical Journal. 2002;368(1):243–251. doi: 10.1042/BJ20020973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ma Y, Pannicke U, Lu H, Niewolik D, Schwarz K, Lieber MR. The DNA-dependent protein Kinase catalytic subunit phosphorylation sites in human artemis. The Journal of Biological Chemistry. 2005;280(40):33839–33846. doi: 10.1074/jbc.M507113200. [DOI] [PubMed] [Google Scholar]
- 53.Douglas P, Cui X, Block WD, et al. The DNA-dependent protein Kinase catalytic subunit is phosphorylated in vivo on threonine 3950, a highly conserved amino acid in the protein Kinase domain. Molecular and Cellular Biology. 2007;27(5):1581–1591. doi: 10.1128/MCB.01962-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Le Romancer M, Cosulich SC, Jackson SP, Clarke PR. Cleavage and inactivation of DNA-dependent protein Kinase catalytic subunit during apoptosis in Xenopus egg extracts. Journal of Cell Science. 1996;109(13):3121–3127. doi: 10.1242/jcs.109.13.3121. [DOI] [PubMed] [Google Scholar]
- 55.Song Q, Lees-Miller SP, Kumar S, et al. DNA-dependent protein Kinase catalytic subunit: a target for an ICE-like protease in apoptosis. The EMBO Journal. 1996;15(13):3238–3246. [PMC free article] [PubMed] [Google Scholar]
- 56.Teraoka H, Yumoto Y, Watanabe F, et al. CPP32/Yama/apopain cleaves the catalytic component of DNA-dependent protein Kinase in the holoenzyme. The FEBS Letters. 1996;393(1):1–6. doi: 10.1016/0014-5793(96)00842-3. [DOI] [PubMed] [Google Scholar]
- 57.Kumar S, Pandey P, Bharti A, et al. Regulation of DNA-dependent protein Kinase by the Lyn tyrosine Kinase. The Journal of Biological Chemistry. 1998;273(40):25654–25658. doi: 10.1074/jbc.273.40.25654. [DOI] [PubMed] [Google Scholar]
- 58.Yavuzer U, Smith GCM, Bliss T, Werner D, Jackson SP. DNA end-independent activation of DNA-PK mediated via association with the DNA-binding protein C1D. Genes and Development. 1998;12(14):2188–2199. doi: 10.1101/gad.12.14.2188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wechsler T, Chen BPC, Harper R, et al. DNA-PKcs function regulated specifically by protein phosphatase 5. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(5):1247–1252. doi: 10.1073/pnas.0307765100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jin S, Kharbanda S, Mayer B, Kufe D, Weaver DT. Binding of Ku and c-Abl at the Kinase homology region of DNA-dependent protein Kinase catalytic subunit. The Journal of Biological Chemistry. 1997;272(40):24763–24766. doi: 10.1074/jbc.272.40.24763. [DOI] [PubMed] [Google Scholar]
- 61.Wu X, Lieber MR. Interaction between DNA-dependent protein Kinase and a novel protein, KIP. Mutation Research/DNA Repair. 1997;385(1):13–20. doi: 10.1016/s0921-8777(97)00035-9. [DOI] [PubMed] [Google Scholar]
- 62.Bosotti R, Isacchi A, Sonnhammer ELL. FAT: a novel domain in PIK-related Kinases. Trends in Biochemical Sciences. 2000;25(5):225–227. doi: 10.1016/s0968-0004(00)01563-2. [DOI] [PubMed] [Google Scholar]
- 63.Hammel M, Yu Y, Mahaney BL, et al. Ku and DNA-dependent protein Kinase dynamic conformations and assembly regulate DNA binding and the initial non-homologous end joining complex. The Journal of Biological Chemistry. 2010;285(2):1414–1423. doi: 10.1074/jbc.M109.065615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Doherty AJ, Suh SW. Structural and mechanistic conservation in DNA ligases. Nucleic Acids Research. 2000;28(21):4051–4058. doi: 10.1093/nar/28.21.4051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ellenberger T, Tomkinson AE. Eukaryotic DNA ligases: structural and functional insights. Annual Review of Biochemistry. 2008;77:313–338. doi: 10.1146/annurev.biochem.77.061306.123941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pascal JM. DNA and RNA ligases: structural variations and shared mechanisms. Current Opinion in Structural Biology. 2008;18(1):96–105. doi: 10.1016/j.sbi.2007.12.008. [DOI] [PubMed] [Google Scholar]
- 67.Shuman S. DNA ligases: progress and prospects. The Journal of Biological Chemistry. 2009;284(26):17365–17369. doi: 10.1074/jbc.R900017200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Shuman S, Lima CD. The polynucleotide ligase and RNA capping enzyme superfamily of covalent nucleotidyltransferases. Current Opinion in Structural Biology. 2004;14(6):757–764. doi: 10.1016/j.sbi.2004.10.006. [DOI] [PubMed] [Google Scholar]
- 69.Shuman S, Schwer B. RNA capping enzyme and DNA ligase: a superfamily of covalent nucleotidyl transferases. Molecular Microbiology. 1995;17(3):405–410. doi: 10.1111/j.1365-2958.1995.mmi_17030405.x. [DOI] [PubMed] [Google Scholar]
- 70.Marchetti C, Walker SA, Odreman F, Vindigni A, Doherty AJ, Jeggo P. Identification of a novel motif in DNA ligases exemplified by DNA ligase IV. DNA Repair. 2006;5(7):788–798. doi: 10.1016/j.dnarep.2006.03.011. [DOI] [PubMed] [Google Scholar]
- 71.Murzin AG. OB(oligonucleotide/oligosaccharide binding)-fold: common structural and functional solution for non-homologous sequences. The EMBO Journal. 1993;12(3):861–867. doi: 10.1002/j.1460-2075.1993.tb05726.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chistiakov DA, Voronova NV, Chistiakov AP. Ligase IV syndrome. European Journal of Medical Genetics. 2009;52(6):373–378. doi: 10.1016/j.ejmg.2009.05.009. [DOI] [PubMed] [Google Scholar]
- 73.Pascal JM, O’Brien PJ, Tomkinson AE, Ellenberger T. Human DNA ligase I completely encircles and partially unwinds nicked DNA. Nature. 2004;432(7016):473–478. doi: 10.1038/nature03082. [DOI] [PubMed] [Google Scholar]
- 74.Pascal JM, Tsodikov OV, Hura GL, et al. A flexible interface between DNA Ligase and PCNA supports conformational switching and efficient ligation of DNA. Molecular Cell. 2006;24(2):279–291. doi: 10.1016/j.molcel.2006.08.015. [DOI] [PubMed] [Google Scholar]
- 75.Nishida H, Kiyonari S, Ishino Y, Morikawa K. The closed structure of an archaeal DNA Ligase from pyrococcus furiosus. Journal of Molecular Biology. 2006;360(5):956–967. doi: 10.1016/j.jmb.2006.05.062. [DOI] [PubMed] [Google Scholar]
- 76.Kim DJ, Kim O, Kim H-W, Kim HS, Lee SJ, Suh SW. ATP-dependent DNA ligase from archaeoglobus fulgidus displays a tightly closed conformation. Acta Crystallographica Section F. 2009;65(6):544–550. doi: 10.1107/S1744309109017485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Martin IV, MacNeill SA. ATP-dependent DNA ligases. Genome Biology. 2002;3(4, article 3005) doi: 10.1186/gb-2002-3-4-reviews3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Cotner-Gohara E, Kim I-K, Tomkinson AE, Ellenberger T. Two DNA-binding and nick recognition modules in human DNA ligase III. The Journal of Biological Chemistry. 2008;283(16):10764–10772. doi: 10.1074/jbc.M708175200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Tomkinson AE, Vijayakumar S, Pascal JM, Ellenberger T. DNA ligases: structure, reaction mechanism, and function. Chemical Reviews. 2006;106(2):687–699. doi: 10.1021/cr040498d. [DOI] [PubMed] [Google Scholar]
- 80.Grawunder U, Zimmer D, Lieber MR. DNA ligase IV binds to XRCC4 via a motif located between rather than within its BRCT domains. Current Biology. 1998;8(15):873–876. doi: 10.1016/s0960-9822(07)00349-1. [DOI] [PubMed] [Google Scholar]
- 81.Wang Y-G, Nnakwe C, Lane WS, Modesti M, Frank KM. Phosphorylation and regulation of DNA ligase IV stability by DNA-dependent protein Kinase. The Journal of Biological Chemistry. 2004;279(36):37282–37290. doi: 10.1074/jbc.M401217200. [DOI] [PubMed] [Google Scholar]
- 82.Zhang X, Moréra S, Bates PA, et al. Structure of an XRCC1 BRCT domain: a new protein-protein interaction module. The EMBO Journal. 1998;17(21):6404–6411. doi: 10.1093/emboj/17.21.6404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Bork P, Hofmann K, Bucher P, Neuwald AF, Altschul SF, Koonin EV. A superfamily of conserved domains in DNA damage-responsive cell cycle checkpoint proteins. The FASEB Journal. 1997;11(1):68–76. [PubMed] [Google Scholar]
- 84.Costantini S, Woodbine L, Andreoli L, Jeggo PA, Vindigni A. Interaction of the Ku heterodimer with the DNA ligase IV/Xrcc4 complex and its regulation by DNA-PK. DNA Repair. 2007;6(6):712–722. doi: 10.1016/j.dnarep.2006.12.007. [DOI] [PubMed] [Google Scholar]
- 85.Shiozaki EN, Gu L, Yan N, Shi Y. Structure of the BRCT repeats of BRCA1 bound to a BACH1 phosphopeptide: implications for signaling. Molecular Cell. 2004;14(3):405–412. doi: 10.1016/s1097-2765(04)00238-2. [DOI] [PubMed] [Google Scholar]
- 86.Clapperton JA, Manke IA, Lowery DM, et al. Structure and mechanism of BRCA1 BRCT domain recognition of phosphorylated BACH1 with implications for cancer. Nature Structural and Molecular Biology. 2004;11(6):512–518. doi: 10.1038/nsmb775. [DOI] [PubMed] [Google Scholar]
- 87.Williams RS, Lee MS, Hau DD, Glover JNM. Structural basis of phosphopeptide recognition by the BRCT domain of BRCA1. Nature Structural and Molecular Biology. 2004;11(6):519–525. doi: 10.1038/nsmb776. [DOI] [PubMed] [Google Scholar]
- 88.Stucki M, Clapperton JA, Mohammad D, Yaffe MB, Smerdon SJ, Jackson SP. MDC1 directly binds phosphorylated histone H2AX to regulate cellular responses to DNA double-strand breaks. Cell. 2005;123(7):1213–1226. doi: 10.1016/j.cell.2005.09.038. [DOI] [PubMed] [Google Scholar]
- 89.Varma AK, Brown RS, Birrane G, Ladias JAA. Structural basis for cell cycle checkpoint control by the BRCA1-CtIP complex. Biochemistry. 2005;44(33):10941–10946. doi: 10.1021/bi0509651. [DOI] [PubMed] [Google Scholar]
- 90.Kilkenny ML, Doré AS, Roe SM, et al. Structural and functional analysis of the Crb2-BRCT2 domain reveals distinct roles in checkpoint signaling and DNA damage repair. Genes and Development. 2008;22(15):2034–2047. doi: 10.1101/gad.472808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Williams RS, Dodson GE, Limbo O, et al. Nbs1 flexibly tethers Ctp1 and Mre11-Rad50 to coordinate DNA double-strand break processing and repair. Cell. 2009;139(1):87–99. doi: 10.1016/j.cell.2009.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lloyd J, Chapman JR, Clapperton JA, et al. A Supramodular FHA/BRCT-repeat architecture mediates Nbs1 adaptor function in response to DNA damage. Cell. 2009;139(1):100–111. doi: 10.1016/j.cell.2009.07.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Williams JS, Williams RS, Dovey CL, Guenther G, Tainer JA, Russell P. γh2A binds Brc1 to maintain genome integrity during S-phase. The EMBO Journal. 2010;29(6):1136–1148. doi: 10.1038/emboj.2009.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wu P-Y, Frit P, Meesala S, et al. Structural and functional interaction between the human DNA repair proteins DNA ligase IV and XRCC4. Molecular and Cellular Biology. 2009;29(11):3163–3172. doi: 10.1128/MCB.01895-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Doré AS, Furnham N, Davies OR, et al. Structure of an Xrcc4-DNA ligase IV yeast ortholog complex reveals a novel BRCT interaction mode. DNA Repair. 2006;5(3):362–368. doi: 10.1016/j.dnarep.2005.11.004. [DOI] [PubMed] [Google Scholar]
- 96.Yu X, Chini CCS, He M, Mer G, Chen J. The BRCT domain is a phospho-protein binding domain. Science. 2003;302(5645):639–642. doi: 10.1126/science.1088753. [DOI] [PubMed] [Google Scholar]
- 97.Rodriguez M, Yu X, Chen J, Songyang Z. Phosphopeptide binding specificities of BRCA1 COOH-terminal (BRCT) domains. The Journal of Biological Chemistry. 2003;278(52):52914–52918. doi: 10.1074/jbc.C300407200. [DOI] [PubMed] [Google Scholar]
- 98.Glover JNM, Williams RS, Lee MS. Interactions between BRCT repeats and phosphoproteins: tangled up in two. Trends in Biochemical Sciences. 2004;29(11):579–585. doi: 10.1016/j.tibs.2004.09.010. [DOI] [PubMed] [Google Scholar]
- 99.Imami K, Sugiyama N, Kyono Y, Tomita M, Ishihama Y. Automated phosphoproteome analysis for cultured cancer cells by two-dimensional nanoLC-MS using a calcined titania/C18 biphasic column. Analytical Sciences. 2008;24(1):161–166. doi: 10.2116/analsci.24.161. [DOI] [PubMed] [Google Scholar]
- 100.Unal S, Cerosaletti K, Uckan-Cetinkaya D, Cetin M, Gumruk F. A novel mutation in a family with DNA ligase IV deficiency syndrome. Pediatric Blood and Cancer. 2009;53(3):482–484. doi: 10.1002/pbc.22031. [DOI] [PubMed] [Google Scholar]
- 101.Li Y, Chirgadze DY, Bolanos-Garcia VM, et al. Crystal structure of human XLF/Cernunnos reveals unexpected differences from XRCC4 with implications for NHEJ. The EMBO Journal. 2008;27(1):290–300. doi: 10.1038/sj.emboj.7601942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Modesti M, Junop MS, Ghirlando R, et al. Tetramerization and DNA ligase IV interaction of the DNA double-strand break repair protein XRCC4 are mutually exclusive. Journal of Molecular Biology. 2003;334(2):215–228. doi: 10.1016/j.jmb.2003.09.031. [DOI] [PubMed] [Google Scholar]
- 103.Junop MS, Modesti M, Guarné A, Ghirlando R, Gellert M, Yang W. Crystal structure of the Xrcc4 DNA repair protein and implications for end joining. The EMBO Journal. 2000;19(22):5962–5970. doi: 10.1093/emboj/19.22.5962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Recuero-Checa MA, Doré AS, Arias-Palomo E, et al. Electron microscopy of Xrcc4 and the DNA ligase IV-Xrcc4 DNA repair complex. DNA Repair. 2009;8(12):1380–1389. doi: 10.1016/j.dnarep.2009.09.007. [DOI] [PubMed] [Google Scholar]
- 105.Mizuta R, Cheng H-L, Gao Y, Alt FW. Molecular genetic characterization of XRCC4 function. International Immunology. 1997;9(10):1607–1613. doi: 10.1093/intimm/9.10.1607. [DOI] [PubMed] [Google Scholar]
- 106.Yu Y, Wang W, Ding Q, et al. DNA-PK phosphorylation sites in XRCC4 are not required for survival after radiation or for V(D)J recombination. DNA Repair. 2003;2(11):1239–1252. doi: 10.1016/s1568-7864(03)00143-5. [DOI] [PubMed] [Google Scholar]
- 107.Koch CA, Agyei R, Galicia S, et al. Xrcc4 physically links DNA end processing by polynucleotide Kinase to DNA ligation by DNA ligase IV. The EMBO Journal. 2004;23(19):3874–3885. doi: 10.1038/sj.emboj.7600375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Bernstein NK, Williams RS, Rakovszky ML, et al. The molecular architecture of the mammalian DNA repair enzyme, polynucleotide Kinase. Molecular Cell. 2005;17(5):657–670. doi: 10.1016/j.molcel.2005.02.012. [DOI] [PubMed] [Google Scholar]
- 109.Yurchenko V, Xue Z, Sadofsky MJ. SUMO modification of human XRCC4 regulates its localization and function in DNA double-strand break repair. Molecular and Cellular Biology. 2006;26(5):1786–1794. doi: 10.1128/MCB.26.5.1786-1794.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Dai Y, Kysela B, Hanakahi LA, et al. Nonhomologous end joining and V(D)J recombination require an additional factor. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(5):2462–2467. doi: 10.1073/pnas.0437964100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Hentges P, Ahnesorg P, Pitcher RS, et al. Evolutionary and functional conservation of the DNA non-homologous end-joining protein, XLF/Cernunnos. The Journal of Biological Chemistry. 2006;281(49):37517–37526. doi: 10.1074/jbc.M608727200. [DOI] [PubMed] [Google Scholar]
- 112.Andres SN, Modesti M, Tsai CJ, Chu G, Junop MS. Crystal structure of human XLF: a twist in nonhomologous DNA end-joining. Molecular Cell. 2007;28(6):1093–1101. doi: 10.1016/j.molcel.2007.10.024. [DOI] [PubMed] [Google Scholar]
- 113.Tsai CJ, Kim SA, Chu G. Cernunnos/XLF promotes the ligation of mismatched and noncohesive DNA ends. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(19):7851–7856. doi: 10.1073/pnas.0702620104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Akopiants K, Zhou R-Z, Mohapatra S, et al. Requirement for XLF/Cernunnos in alignment-based gap filling by DNA polymerases λ and μ for nonhomologous end joining in human whole-cell extracts. Nucleic Acids Research. 2009;37(12):4055–4062. doi: 10.1093/nar/gkp283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Harris R, Esposito D, Sankar A, et al. The 3D solution structure of the C-terminal region of Ku86 (Ku86CTR) Journal of Molecular Biology. 2004;335(2):573–582. doi: 10.1016/j.jmb.2003.10.047. [DOI] [PubMed] [Google Scholar]
- 116.Zhang Z, Hu W, Cano L, Lee TD, Chen DJ, Chen Y. Solution structure of the C-terminal domain of Ku80 suggests important sites for protein-protein interactions. Structure. 2004;12(3):495–502. doi: 10.1016/j.str.2004.02.007. [DOI] [PubMed] [Google Scholar]
- 117.Rivera-Calzada A, Spagnolo L, Pearl LH, Llorca O. Structural model of full-length human Ku70-Ku80 heterodimer and its recognition of DNA and DNA-PKcs. EMBO Reports. 2007;8(1):56–62. doi: 10.1038/sj.embor.7400847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Boskovic J, Rivera-Calzada A, Maman JD, et al. Visualization of DNA-induced conformational changes in the DNA repair Kinase DNA-PKcs. The EMBO Journal. 2003;22(21):5875–5882. doi: 10.1093/emboj/cdg555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Perry JJ, Cotner-gohara E, Ellenberger T, Tainer JA. Structural dynamics in DNA damage signaling and repair. Current Opinion in Structural Biology. 2010;20(3):283–294. doi: 10.1016/j.sbi.2010.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Chen X, Ballin JD, Della-Maria J, et al. Distinct kinetics of human DNA ligases I, IIIα, IIIβ, and IV reveal direct DNA sensing ability and differential physiological functions in DNA repair. DNA Repair. 2009;8(8):961–968. doi: 10.1016/j.dnarep.2009.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Gu J, Lu H, Tippin B, Shimazaki N, Goodman MF, Lieber MR. XRCC4:DNA ligase IV can ligate incompatible DNA ends and can ligate across gaps. The EMBO Journal. 2007;26(4):1010–1023. doi: 10.1038/sj.emboj.7601559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Gu J, Lu H, Tsai AG, Schwarz K, Lieber MR. Single-stranded DNA ligation and XLF-stimulated incompatible DNA end ligation by the XRCC4-DNA ligase IV complex: influence of terminal DNA sequence. Nucleic Acids Research. 2007;35(17):5755–5762. doi: 10.1093/nar/gkm579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Kysela B, Doherty AJ, Chovanec M, et al. Ku stimulation of DNA ligase IV-dependent ligation requires inward movement along the DNA molecule. The Journal of Biological Chemistry. 2003;278(25):22466–22474. doi: 10.1074/jbc.M303273200. [DOI] [PubMed] [Google Scholar]
- 124.Chen L, Trujillo K, Sung P, Tomkinson AE. Interactions of the DNA ligase IV-XRCC4 complex with DNA ends and the DNA-dependent protein Kinase. The Journal of Biological Chemistry. 2000;275(34):26196–26205. doi: 10.1074/jbc.M000491200. [DOI] [PubMed] [Google Scholar]
- 125.Joo WS, Jeffrey PD, Cantor SB, Finnin MS, Livingston DM, Pavletich NP. Structure of the 53BP1 BRCT region bound to p53 and its comparison to the Brca1 BRCT structure. Genes and Development. 2002;16(5):583–593. doi: 10.1101/gad.959202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Derbyshire DJ, Basu BP, Serpell LC, et al. Crystal structure of human 53BP1 BRCT domains bound to p53 tumour suppressor. The EMBO Journal. 2002;21(14):3863–3872. doi: 10.1093/emboj/cdf383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Ochi T, Bolanos-Garcia VM, Stojanoff V, Moreno A. Perspectives on protein crystallisation. Progress in Biophysics and Molecular Biology. 2009;101(1–3):56–63. doi: 10.1016/j.pbiomolbio.2009.12.001. [DOI] [PubMed] [Google Scholar]
- 128.Recuero-Checa MA, Doré AS, Arias-Palomo E, et al. Electron microscopy of Xrcc4 and the DNA ligase IV-Xrcc4 DNA repair complex. DNA Repair. 2009;8(12):1380–1389. doi: 10.1016/j.dnarep.2009.09.007. [DOI] [PubMed] [Google Scholar]
- 129.Ochi T, Grossmann JG, Bolanos-Garcia VM, Blundell TL. To be published. [Google Scholar]
- 130.Svergun DI. Restoring Low Resolution Structure of Biological Macromolecules from Solution Scattering Using Simulated Annealing. Biophysical journal. 1999;76:2879–2886. doi: 10.1016/S0006-3495(99)77443-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Volkov VV, Svergun DI. Uniqueness of ab initio shape determination in small-angle scattering. Journal of Applied Crystallography. 2003;36(3 I):860–864. doi: 10.1107/S0021889809000338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Pettersen EF, Goddard TD, Huang CC, et al. UCSF Chimera—a visualization system for exploratory research and analysis. Journal of Computational Chemistry. 2004;25(13):1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 133.Riballo E, Woodbine L, Stiff T, Walker SA, Goodarzi AA, Jeggo PA. XLF-Cernunnos promotes DNA ligase IV-XRCC4 re-adenylation following ligation. Nucleic Acids Research. 2009;37(2):482–492. doi: 10.1093/nar/gkn957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Deshpande RA, Wilson TE. Modes of interaction among yeast Nej1, Lif1 and Dnl4 proteins and comparison to human XLF, XRCC4 and Lig4. DNA Repair. 2007;6(10):1507–1516. doi: 10.1016/j.dnarep.2007.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Lu H, Pannicke U, Schwarz K, Lieber MR. Length-dependent binding of human XLF to DNA and stimulation of XRCC4·DNA ligase IV activity. The Journal of Biological Chemistry. 2007;282(15):11155–11162. doi: 10.1074/jbc.M609904200. [DOI] [PubMed] [Google Scholar]
- 136.Yano K-I, Chen DJ. Live cell imaging of XLF and XRCC4 reveals a novel view of protein assembly in the non-homologous end-joining pathway. Cell Cycle. 2008;7(10):1321–1325. doi: 10.4161/cc.7.10.5898. [DOI] [PubMed] [Google Scholar]
- 137.Kastner B, Fischer N, Golas MM, et al. GraFix: sample preparation for single-particle electron cryomicroscopy. Nature Methods. 2008;5(1):53–55. doi: 10.1038/nmeth1139. [DOI] [PubMed] [Google Scholar]
- 138.Blundell TL, Jhoti H, Abell C. High-throughput crystallography for lead discovery in drug design. Nature Reviews Drug Discovery. 2002;1(1):45–54. doi: 10.1038/nrd706. [DOI] [PubMed] [Google Scholar]
- 139.Blundell TL, Sibanda BL, Montalvão RW, et al. Structural biology and bioinformatics in drug design: opportunities and challenges for target identification and lead discovery. Philosophical Transactions of the Royal Society B. 2006;361(1467):413–423. doi: 10.1098/rstb.2005.1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Congreve M, Murray CW, Blundell TL. Keynote review: structural biology and drug discovery. Drug Discovery Today. 2005;10(13):895–907. doi: 10.1016/S1359-6446(05)03484-7. [DOI] [PubMed] [Google Scholar]
- 141.DeLano WL. The PyMOL Molecular Graphics System. Palo Alto, Calif, USA: DeLano Scientific LLC; 2008. http://www.pymol.org. [Google Scholar]