

Abstract
Aims
Endothelial activation, macrophage infiltration, and foam cell formation are pivotal steps in atherogenesis. Our aim in this study was to analyse the role of SIRT1, a class III deacetylase with important metabolic functions, in plaque macrophages and atherogenesis.
Methods and results
Using partial SIRT1 deletion in atherosclerotic mice, we demonstrate that SIRT1 protects against atherosclerosis by reducing macrophage foam cell formation. Peritoneal macrophages from heterozygous SIRT1 mice accumulate more oxidized low-density lipoprotein (oxLDL), thereby promoting foam cell formation. Bone marrow-restricted SIRT1 deletion confirmed that SIRT1 function in macrophages is sufficient to decrease atherogenesis. Moreover, we show that SIRT1 reduces the uptake of oxLDL by diminishing the expression of lectin-like oxLDL receptor-1 (Lox-1) via suppression of the NF-κB signalling pathway.
Conclusion
Our findings demonstrate protective effects of SIRT1 in atherogenesis and suggest pharmacological SIRT1 activation as a novel anti-atherosclerotic strategy by reducing macrophage foam cell formation.
Keywords: SIRT1, Macrophage foam cell, Atherogenesis
Introduction
Atherosclerosis is a chronic inflammatory disease that results from interaction between oxidized low-density lipoprotein (oxLDL), activated endothelial cells, monocyte-derived macrophages, T cells, and the arterial wall. Activated endothelial cells express adhesion molecules, e.g. vascular cell adhesion molecule 1 (VCAM-1) and intercellular adhesion molecule-1 (ICAM-1), which attract and recruit blood monocytes to the vessel wall. These monocytes differentiate into macrophages and infiltrate to the sub-endothelial space where they release and respond to inflammatory mediators such as tumour necrosis factor-α (TNFα), VCAM-1, and interleukins (IL). Eventually, these inflammatory macrophages ingest oxLDL via scavenger receptors, such as scavenger receptor-A (SR-A), CD36 or lectin-like oxLDL receptor 1 (Lox-1), becoming foam cells, and thereby promoting plaque formation.1
Sir2 is an NAD-dependent class III deacetylase that was found to increase lifespan in yeast.2 Its mammalian orthologue SIRT1 senses caloric restriction, improves insulin secretion in pancreatic beta cells, and reduces accumulation of fatty acids in white adipose tissue (WAT).3–5 Various targets of SIRT1 have been characterized, including PPARγ coactivator 1α (PGC-1α), nuclear factor κB (NF-κB), p53, FOXO transcription factors, and endothelial nitric oxide synthase (eNOS).6–13 Interestingly, many of these targets that are critically involved in regulating metabolism have also been shown to play a role in atherogenesis.14–18 However, little is known about the relevance of SIRT1 in the latter.
In atherogenesis, chronic endothelial dysfunction is a trigger of plaque formation,19 and endothelial SIRT1 overexpression has been shown to prevent atherosclerosis by improving vascular function.20 Nevertheless, the relevance of SIRT1 on the cellular and molecular events governing atherogenesis is unknown. As multiple targets of SIRT1 may play a role in plaque formation, it is likely that eNOS is not the only mechanism by which SIRT1 prevents atherogenesis. In particular, the role of SIRT1 in monocyte adhesion, macrophage infiltration, lipid uptake, and foam cell formation remains to be determined.
To investigate the role of SIRT1 in these cellular and molecular processes, we compared hypercholesterolaemic ApoE−/− SIRT1+/+ with ApoE−/− SIRT1+/− mice. These mice have an SIRT1 haploinsufficiency, but do not display the autoimmune and dysmorphic phenotype of SIRT1−/− mice.21,22
Methods
Animals
SIRT1 knockout mice on a 129 background22 were crossed into ApoE−/− C57BL/6 mice23 to generate ApoE−/− SIRT1+/− mice and ApoE−/− SIRT1+/+ littermates. Of those, male mice were fed a high-cholesterol diet (1.25% total cholesterol, Research Diets) for 12 weeks starting at the age of 8 weeks. Because the few ApoE−/− SIRT1−/− mice showed a similar dysmorphic phenotype as SIRT1−/− mice,21,22 we did not use them in this study. All animal procedures were approved by the local animal committee and performed in accordance with our institutional guidelines.
Bone marrow transplantation
Bone marrow donor mice were ApoE−/− SIRT1+/− (n = 3; pooled) and ApoE−/− SIRT1+/+ (n = 3; pooled) mice, and recipients pure ApoE−/− mice (from Jackson Laboratories). Donor mice were split—dose-irradiated under SPF conditions in filter cages with a total irradiation of 1100 rad.24 Recipient mice were injected intravenously with 106 bone marrow cells (ApoE−/− SIRT1+/−→7 ApoE−/− mice; ApoE−/− SIRT1+/+→6 ApoE−/− mice). Transplanted mice recovered for 5 weeks and were then fed a high-cholesterol diet for 11 weeks.
Lipoprotein uptake
RAW 264.7 and thioglycolate-elicited peritoneal macrophages were starved for 48 h and then incubated with 10 µg/mL oxLDL for 2 h at 37°C/5% CO2. After washing away unspecifically bound LDL, cells were fixed and stained with Oil red O (ORO). Experiments were done twice with six independent pools isolated from six mice of each genotype, ORO staining analysed using a light microscope and quantified using analySIS. Low-density lipoprotein uptake was quantified as the ratio of the percentage of the ORO-positive area divided by the percentage of the total cell area in at least 150 cells per genotype. For CD36 blocking studies, RAW 264.7 macrophages were first pre-stimulated for 5 h with 10 ng/mL murine TNFα, and then pre-incubated with 2 µg/mL mouse anti-CD36 (Cascade Bioscience) before adding 10 µg/mL oxLDL over night.
Cell culture
Murine RAW 264.7 cells (Mouse leukaemic monocyte macrophage cell line) were treated with 200 µM splitomicin (Sigma-Aldrich) to perform cholesterol efflux and oxLDL uptake studies. RAW 264.7 cells were stimulated for with 10 ng/mL murine TNFα for 5 h. SIRT1−/− mouse embryonic fibroblasts (MEF)25 were kindly provided by Frederick W. Alt (Harvard University, Boston, MA, USA), and RelA/p65−/− MEF with reconstituted wt-RelA/p65 or non-acetylatable RelA/p65 were described previously.26
Plasmid and siRNA transfection
Transient transfection of pcDNA3.1::SIRT1 or siRNA into RAW 264.7 or MEF were done with lipofectamin 2000 or lipofectamin RNAi MAX (both Invitrogen). The oligos used for SIRT1-siRNA have been described previously.5
Immunohistochemistry and immunocytochemistry
Serial cryosections from the aortic sinus were stained with ORO, rat anti-CD68, rat anti-CD3 (Abcam), rat anti-CD31, rabbit anti-SIRT1 (Upstate/Millipore). Means were taken from n = 6 different mice evaluating six serial cryosections/tissue from each mouse. Thoraco-abdominal aortae were fixed and plaques stained with ORO. Collagen, fibrous cap thickness, and necrotic core size were analysed by Elastica van Gieson and Massons trichrome stainings. Cell death was assessed with the terminal deoxyribonucleotidyl transferase-mediated dUTP nick-end labelling kit (Roche).
RNA and protein analysis
Total RNA isolated from proximal aortae was extracted with TRIZOL (Invitrogen), reverse transcribed, and the cDNA quantified by SYBR green qPCR using specific primers. For protein analysis, aortic tissue lysates were blotted and incubated with rabbit anti-SIRT1 (Upstate/Millipore), rabbit anti-eNOS, rabbit anti-phospho-eNOS (Ser1177).
Cholesterol efflux
For cholesterol efflux experiments, RAW 264.7 cells were labelled with 2 µCi/mL [1,2-3H]cholesterol (Perkin Elmer) for 24 h. Following the labelling period, cells were washed and allowed to equilibrate overnight in DMEM containing 0.2% BSA supplemented with cholesterol in the presence or absence of 0.1 mM splitomicin together with 0.3 mM 8-Br-cAMP or 22(R)-HC and 9-cisRA (Sigma). After 24 h stimulation, cells were washed and incubated for 6 h in DMEM containing 0.2% BSA in the presence or absence of 28 µg/mL lipid-free apoA-I. The radioactivity recovered in the culture medium and cell lysates was measured. The apoA-I-mediated cholesterol efflux was calculated as the percentage of total [1,2-3H]cholesterol released into the medium after subtracting the values obtained in the absence of apoA-I. The cholesterol efflux assays were performed in duplicates with three pairs per treatment group.
Plasma lipids and cytokines
Total plasma cholesterol, triglycerides, and free fatty acids were analysed using TR13421, TR22421 (Thermo Electron, Inc.), and 994–75409 (Wako Chemicals). The lipid distribution in plasma lipoprotein fractions was assessed by fast-performance liquid chromatography gel filtration with a Superose 6 HR 10/30 column (Pharmacia).27 Plasma values of VCAM-1 (MVC00) and ICAM-1 (MIC100; R&D) were determined using ELISA, and TGF-β, IFNγ, IL-6, IL-10, and mKC using multiplex array systems (Becton, Dickinson and Company).
Flow cytometry
For blood and spleen FACS analyses, single cell suspensions were incubated with antibodies against CD4, CD8, B220, CD11c, CD11b, CD62L, CD44, and CD25 (BD Pharmingen) and then cells were analysed with a FACSCantoII (BD Pharmingen) and FACSDiva software. Post-acquisition analysis was done with FACSDiva (BD Pharmingen) or FlowJo7 software (Tree Star).
Statistical analysis
Data are presented as mean ± SEM. The en face ORO quantifications were analysed with a non-parametric Mann–Whitney U-test. Statistical significance of differences was calculated using an ANOVA with post hoc Tukey's test or Student unpaired t-test. Significance was accepted at the level of P < 0.05.
Results
SIRT1 protects against atherosclerosis
To address the role of SIRT1 in atherogenesis, we compared SIRT1 expression in aortic lysates obtained from atherosclerotic ApoE−/− and normal wild-type (WT) mice. Aortic SIRT1 protein expression was lower in ApoE−/− than in WT mice (see Supplementary material online, Figure S1A), suggesting a protective role of SIRT1 in atherogenesis. In order to establish a cause–effect relationship between SIRT1 expression and atherosclerosis, we applied genetic deletion of SIRT1 in atherosclerotic mice. For this purpose, we compared 20-week-old male ApoE−/− SIRT1+/+ and ApoE−/− SIRT1+/− mice that were kept on a high-cholesterol diet for 12 weeks (see Supplementary material online, Figure S2A). Of note, SIRT1 expression is only slightly reduced in WT mice treated with a high-cholesterol diet (see Supplementary material online, Figure S1B). To examine any SIRT1 compensation for the missing SIRT1 allele, we assessed SIRT1 expression in aortic lysates of the two genotypes. Aortic SIRT1 protein in ApoE−/− SIRT1+/− mice amounted to about 60% of protein in ApoE−/− SIRT1+/+ mice (see Supplementary material online, Figure S1C). Importantly, en face plaque quantifications in thoraco-abdominal aortae and in serial cross sections of aortic roots revealed fewer atherosclerotic plaques in ApoE−/− SIRT1+/+ compared with ApoE−/− SIRT1+/− mice (Figures 1A and 2). Massons trichrome and van Gieson stainings revealed that total collagen content, necrotic core size, and fibrous cap thickness did not differ between the two groups (Figure 1B, see Supplementary material online, Figure S1D–F). No difference was observed in the amount of apoptotic cells (see Supplementary material online, Figure S1F). These findings indicate that endogeneous SIRT1 protects against atherosclerosis.
Figure 1.

SIRT1 protects mice against atherosclerosis. (A) En face Oil red O (ORO) staining of thoraco-abdominal aortae and quantifications of plaque area. n = 9, ApoE−/− SIRT1+/+(▪); n = 11, ApoE−/− SIRT1+/−(□). (B) Representative images for Massons trichrome and Elastica van Gieson stainings from animals with comparable plaque sizes. Magnification: X 40. *P < 0.05.
Figure 2.

SIRT1 deletion increases macrophage and T-cell accumulation in plaques. Immunohistochemistry with quantifications of Oil red O (ORO), CD68, and CD3 (arrows) on plaques from aortic sinus. Magnifications: Oil red O, CD68: ×40; CD3: ×400. n = 6 per genotype. *P < 0.05.
Accumulation of plaque macrophages and T cells is reduced by SIRT1
Accumulation of macrophages and T cells in the subintimal space plays a central role in atherogenesis.1 Our analyses revealed increased accumulation of macrophages and T cells in atherosclerotic plaques of ApoE−/− SIRT1+/− compared with ApoE−/− SIRT1+/+ mice (Figure 2). SIRT1 staining in aortae of healthy WT mice or ApoE−/− mice under normal diet showed that SIRT1 is expressed in smooth muscle and endothelial cells (see Supplementary material online, Figure S3A). In diseased aortae from ApoE−/− mice under high-cholesterol diet, SIRT1 is also expressed in cells within the plaques (see Supplementary material online, Figure S3A). Double stainings revealed that SIRT1 is expressed in both plaque endothelial cells and macrophages in diseased ApoE−/− aortae (see Supplementary material online, Figure S3B).
SIRT1 in macrophages is sufficient to reduce foam cell formation and atherogenesis
We observed no difference neither in fasted nor fed plasma glucose or insulin levels, total body or epididymal fat weight (see Supplementary material online, Figure S4) and found no difference in total cholesterol and its subfractions between ApoE−/− SIRT1+/+ and ApoE−/− SIRT1+/− mice (see Supplementary material online, Figure S5 and Table S1). Further, plasma cytokine levels were similar in the two genotypes (see Supplementary material online, Table S2). Since inflammatory factors from WAT may enhance atherogenesis,28,29 we examined the expression of several adipokines and adipose-derived hormones in epididymal WAT. Expression of Adiponectin (Adipoq), Leptin, Visfatin (Nampt), Chemerin (Rarres2), Resistin (Retn) were equivalent, whereas expression of Plasminogen activator inhibitor 1 (PAI-1 or Serpine1) was slightly, but not significantly elevated in ApoE−/− SIRT1+/− compared with ApoE−/− SIRT1+/+ epididymal WAT (see Supplementary material online, Figure S6). These data do not exclude a contribution of systemic inflammatory factors such as WAT, but suggest that the damaging effects of partial SIRT1 deficiency are mainly mediated via inflammatory cells localized in plaques.
Therefore, we focused on the role of SIRT1 in macrophages. Foam cell formation is a crucial step in atherogenesis.1 We observed no difference in basal LDL uptake in peritoneal-elicited macrophages from ApoE−/− SIRT1+/− compared with ApoE−/− SIRT1+/+ mice, but found enhanced uptake upon stimulation with oxLDL (Figure 3A). Pharmacological inhibition of SIRT1 with splitomicin in RAW 264.7 macrophages showed a trend towards increasing oxLDL uptake (see Supplementary material online, Figure S7). Consistently, siRNA-induced SIRT1 knockdown increased uptake of oxLDL in RAW 264.7 macrophages compared with control cells (Figure 3B and C). To study a potential role of CD36 in oxLDL uptake, we blocked CD36-mediated oxLDL uptake using an anti-CD36 antibody in scrambled or SIRT1-siRNA-treated macrophages. Neutralization of CD36 decreased uptake of oxLDL by ∼50% compared with non-neutralized cells (Figure 3C). A higher elevation of oxLDL uptake was observed in SIRT1-siRNA-treated macrophages with additional CD36 neutralization, but did not reach significance difference compared with any other group (Figure 3C). These data suggest that SIRT1 exerts CD36-dependent and -independent effects in oxLDL uptake.
Figure 3.

SIRT1 reduces foam cell formation. (A) Increased uptake of oxLDL in peritoneal thioglycolate-elicited macrophages from ApoE−/− SIRT1+/− compared with ApoE−/− SIRT1+/+ mice. oxLDL uptake is given as the ratio of the percentage of ORO-positive area divided by the percentage of total cell area in at least 150 cells per genotype. (B) siRNA-induced SIRT1 knockdown increases uptake of oxLDL in RAW 264.7 macrophages compared with scr-siRNA-treated cells. (C) siRNA-mediated SIRT1 silencing in 5 h TNFα-pretreated RAW 264.7 macrophages: left panel, expression of Lox-1; right panel, uptake of oxLDL in neutralizing anti-CD36 antibody-treated cells. *P < 0.05. ***P < 0.001. Magnifications: ×400.
To examine whether an SIRT1-dependent mechanism in macrophages accounts for decreased atherosclerosis in vivo, we performed bone marrow transplantation experiments. Bone marrow from ApoE−/− SIRT1+/− or ApoE−/− SIRT1+/+ mice was transplanted into irradiated 6-week-old ApoE−/− mice (see Supplementary material online, Figure S2B), respectively. Flow cytometry analyses of blood and spleen samples from transplanted mice revealed an increased number of blood monocytes of ApoE−/− recipient mice receiving ApoE−/− SIRT1+/− bone marrow cells, but no proportional difference in T-cell subtypes, MHCII+ cells, B cells or macrophages (see Supplementary material online, Figure S8). Chimeric ApoE−/− recipient mice receiving ApoE−/− SIRT1+/− bone marrow showed more atherosclerotic lesions compared with mice transplanted with ApoE−/− SIRT1+/+ bone marrow (Figure 4). These findings support the notion that SIRT1 function in macrophages is sufficient to decrease atherogenesis.
Figure 4.
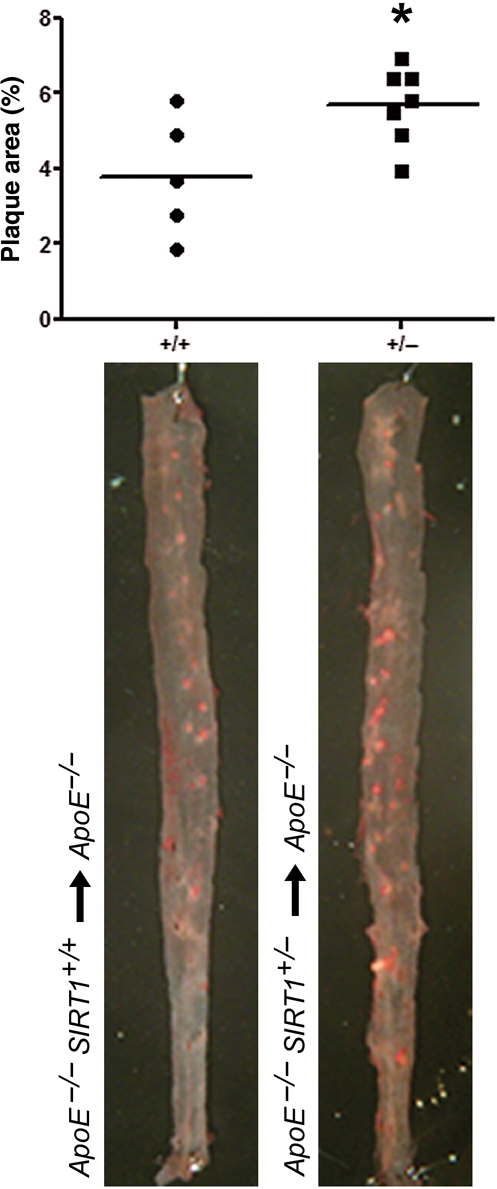
SIRT1 function in macrophages is sufficient to decrease atherogenesis. Chimeric ApoE−/− mice that received ApoE−/− SIRT1+/− (n = 7) bone marrow cells develop more atherosclerosis than those which received ApoE−/− SIRT1+/+ (n = 5) bone marrow cells. *P < 0.05.
Deacetylation of RelA/p65 by SIRT1 diminishes Lox-1 expression
Active uptake of modified cholesterol in macrophages is mainly regulated by SR-A, SR-B, CD36, and Lox-1,30 whereas cholesterol efflux is driven by ATP-binding cassette transporters.31 Expression of CD36, SR-A, and SR-B was not altered (see Supplementary material online, Figure S9), whereas Lox-1 expression was higher in ApoE−/− SIRT1+/− aortic lysates (Figure 5A). Because expression of Lox-1 in SIRT1-siRNA-treated macrophages was increased (Figure 3C), we planned to study the SIRT1–Lox-1 pathway more in detail. Deletion of Lox-1 has been shown to reduce atherosclerosis in mice.32 Since Lox-1 is an NF-κB target33 and SIRT1 deacetylates RelA/p65 in murine macrophages,34 we compared Lox-1 RNA levels in TNFα-stimulated RAW 264.7 cells pretreated with splitomicin. We observed an increase in Lox-1 expression in splitomicin-treated macrophages compared with untreated control cells (Figure 5B). These data suggest that SIRT1 suppresses NF-κB signalling in macrophages, thereby reducing Lox-1 expression and oxLDL uptake.
Figure 5.

Deacetylation of RelA/p65 by SIRT1 diminishes Lox-1 expression. (A) Aortic Lox-1 expression is increased in ApoE−/− SIRT1+/− compared with ApoE−/− SIRT1+/+ mice. (B) Lox-1 expression is higher in 5 h TNFα-stimulated RAW 264.7 macrophages pretreated with 200 µM splitomicin (Splito) compared with untreated cells (nt). (C) Ectopic SIRT1 expression in SIRT1−/− MEF reduces Lox-1 expression. (D) In RelA/p65−/− MEF with reconstituted wt-RelA/p65, siRNA-mediated SIRT1 knockdown enhances Lox-1 expression upon 5 h TNFα stimulation, whereas no effect is observed in RelA/p65−/− MEF with a reconstituted mutated, non-acetylatable K310-RelA/p65. *P < 0.05; ***P < 0.001.
To get more insight into the molecular events underlying SIRT1-dependent NF-κB deacetylation in murine cells, we analysed SIRT1 expression in different MEF cell lines. Ectopic expression of SIRT1 in SIRT1−/− MEF reduced Lox-1 expression upon TNFα treatment (Figure 5C). In RelA/p65−/− MEF with reconstituted wt-RelA/p65 expression,26 siRNA-induced SIRT1 knockdown enhanced Lox-1 expression upon TNFα stimulation. In contrast, SIRT1-siRNA had no effect on Lox-1 expression in TNFα-stimulated RelA/p65−/− MEF with reconstituted non-acetylatable K310R-mutant-RelA/p6526 (Figure 5D). These data show that SIRT1-dependent deacetylation of RelA/p65 at K310 is sufficient to reduce Lox-1 gene expression.
Several reports show a link between Lox-1 and matrix metalloproteinases (MMPs) that are expressed and secreted by human endothelial cells, including the collagenase MMP1, stromelysin-1 (MMP3), the membrane type 1 MMP (MT1-MMP or MMP14), and the tissue inhibitor of metalloproteinase 3 (TIMP3).35–38 To investigate if metalloproteinase expression is affected by SIRT1, we quantified aortic expression of MMP13, MMP3, MMP8, MMP9, MMP14, and TIMP3. No significant change in the expression of these MMPs was observed in ApoE−/− SIRT1+/− compared with ApoE−/− SIRT1+/+ aortae (see Supplementary material online, Figure S10), indicating that the phenotype of ApoE−/− SIRT1+/− mice is not related to SIRT1–Lox-1-mediated expression of MMPs.
Atheroprotective effects of SIRT1 do not affect cholesterol efflux
When examining cholesterol efflux, we found that expression of ABCA1 was not altered in aortic lysates from ApoE−/− SIRT1+/− compared with ApoE−/− SIRT1+/+ mice (see Supplementary material online, Figure S11A). Similarly, expression levels of ABCA1 and ABCG1 were not different in peritoneal macrophages from ApoE−/− SIRT1+/+ and ApoE−/− SIRT1+/− mice (see Supplementary material online, Figure S11B). Cholesterol efflux assays in RAW 264.7 macrophages treated with splitomicin revealed no ApoA-I-dependent changes (see Supplementary material online, Figure S11C). Furthermore, we detected no difference in aortic expression of the ABCA1 regulators LXRα, PPARγ, or its coactivator PGC-1α (PPARγ coactivator 1α) (see Supplementary material online, Figure S11D–F). These data suggest that aortic cholesterol efflux is not affected by SIRT1 in atherosclerotic mice.
Discussion
Recently, SIRT1 has been shown to decrease atherosclerosis by improving endothelium-dependent vascular function in ApoE−/− mice with an endothelial SIRT1 overexpression that were kept on a high-fat diet.20 Our study shows that endogenous SIRT1 prevents macrophage foam cell formation in atherogenesis independently of systemic lipid levels. We demonstrate that loss of a single SIRT1 allele in ApoE−/− SIRT1+/− mice is sufficient to increase plaque formation.
Macrophage-derived foam cell formation is enhanced upon a relative increase in cholesterol uptake or by a defective cholesterol efflux, respectively.39 Our in vitro foam cell assay reveals that SIRT1 activation diminishes oxLDL uptake in peritoneal macrophages. Among the receptors that may account for this increased LDL uptake by macrophages, we identified Lox-1 to be critically involved: SIRT1 inhibits TNFα-induced expression of Lox-1 in macrophages. In fact, the Lox-1 promoter contains NF-κB binding sites and is expressed upon TNFα stimulation.33 Using RelA/p65−/− MEF, we could further show that the deacetylation of RelA/p65 by SIRT1 suppresses Lox-1 expression. We acknowledge that the final proof of a Lox-1-mediated effect on foam cell formation would require additional evidence from genetic loss-of-function or neutralizing antibody experiments. These questions need to be addressed in future studies.
To test whether an SIRT1-dependent mechanism in bone marrow-derived cells accounts for the increase in atherosclerosis in vivo, we performed bone marrow transplantation experiments. Chimeric ApoE−/− mice receiving ApoE−/− SIRT1+/− bone marrow showed increased atherosclerotic plaques compared with mice receiving ApoE−/− SIRT1+/+ bone marrow. These findings demonstrate that partial SIRT1 deletion in bone marrow-derived macrophages is sufficient to enhance atherogenesis.
The role of NF-κB in plaque macrophages and lipoprotein uptake in atherogenesis is controversial. Disruption of NF-κB signalling by partial IKK2 deletion in macrophages increased atherosclerosis.40 Conversely, studies using macrophage-specific p50 deletion or a dominant-negative IκBα mutant to disrupt NF-κB signalling, resulted in smaller atherosclerotic lesions and diminished uptake of lipoproteins.41,42 Since NF-κB and its transcriptional activity are tightly regulated,43,44 pharmacological modulation of an upstream regulator of NF-κB seems more promising in preventing atherogenesis than direct NF-κB modulation.45 Our study shows that SIRT1 might be an attractive modulator, since its interference with the NF-κB signalling pathway exerts beneficial effects on plaque formation.
Several reports suggest that other SIRT1 targets may also contribute to atherogenesis. For instance, PPARγ, a key transcription factor in adipocyte differentiation, plays a pivotal role in macrophages and modulates the extent of atherosclerosis.14 Both PPARγ and its target LXR are regulated by SIRT1 in adipocytes and macrophages.5,46 Interestingly, Li et al.46 showed reduced cholesterol efflux in primary macrophages from SIRT1−/− compared with SIRT1+/+ mice. We could neither observe a difference in the aortic expression of ABCA1, ABCG1, PPARγ, or LXRα nor an ApoA-I-dependent decrease in cholesterol efflux upon splitomicin treatment in RAW 264.7 macrophages. Possibly, the effect of a single missing SIRT1 allele is not sufficient to affect cholesterol efflux in atherosclerotic mice.
Taken together, our results reveal a novel mechanism by which SIRT1 prevents atherogenesis: SIRT1 suppresses NF-signalling by deacetylating RelA/p65, thereby reducing Lox-1 expression and diminishing uptake of oxLDL and foam cell formation. Given the availability of specific SIRT1-activating drugs that are being tested in clinical trials in patients with type 2 diabetes, pharmacological activation of SIRT1 may also become an attractive anti-atherogenic strategy by preventing macrophage foam cell formation.47
Supplementary material
Supplementary material is available at European Heart Journal online.
Funding
This work was funded by grants from the Swiss National Science Foundation [grant numbers 31-114094/1 to C.M.M. and 3100-068118 to T.F.L.] and the University Research Priority Program ‘Integrative Human Physiology’ at the University of Zurich. Further support was provided by unrestricted grants from the MERCATOR Foundation Switzerland and a strategic alliance with Pfizer, Inc., New York. Funding to pay the Open Access publication charges for this article was provided by the Swiss National Science Foundation.
Conflict of interest: none declared.
Supplementary Material
Acknowledgements
We thank Elin Stenfeldt (University of Gothenburg), Sabine Rütti, and Chad E. Brokopp (University Hospital Zurich) for technical assistance and the Center for Microscopy and Image Analysis at the University of Zurich for using their facilities.
References
- 1.Libby P. Inflammation in atherosclerosis. Nature. 2002;420:868–874. doi: 10.1038/nature01323. doi:10.1038/nature01323. [DOI] [PubMed] [Google Scholar]
- 2.Kaeberlein M, McVey M, Guarente L. The SIR2/3/4 complex and SIR2 alone promote longevity in Saccharomyces cerevisiae by two different mechanisms. Genes Dev. 1999;13:2570–2580. doi: 10.1101/gad.13.19.2570. doi:10.1101/gad.13.19.2570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen D, Steele AD, Lindquist S, Guarente L. Increase in activity during calorie restriction requires Sirt1. Science. 2005;310:1641. doi: 10.1126/science.1118357. doi:10.1126/science.1118357. [DOI] [PubMed] [Google Scholar]
- 4.Bordone L, Motta MC, Picard F, Robinson A, Jhala US, Apfeld J, McDonagh T, Lemieux M, McBurney M, Szilvasi A, Easlon EJ, Lin SJ, Guarente L. Sirt1 regulates insulin secretion by repressing UCP2 in pancreatic beta cells. PLoS Biol. 2006;4:e31. doi: 10.1371/journal.pbio.0040031. doi:10.1371/journal.pbio.0040031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Picard F, Kurtev M, Chung N, Topark-Ngarm A, Senawong T, Machado De Oliveira R, Leid M, McBurney MW, Guarente L. Sirt1 promotes fat mobilization in white adipocytes by repressing PPAR-gamma. Nature. 2004;429:771–776. doi: 10.1038/nature02583. doi:10.1038/nature02583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vaziri H, Dessain SK, Ng Eaton E, Imai SI, Frye RA, Pandita TK, Guarente L, Weinberg RA. hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell. 2001;107:149–159. doi: 10.1016/s0092-8674(01)00527-x. doi:10.1016/S0092-8674(01)00527-X. [DOI] [PubMed] [Google Scholar]
- 7.Brunet A, Sweeney LB, Sturgill JF, Chua KF, Greer PL, Lin Y, Tran H, Ross SE, Mostoslavsky R, Cohen HY, Hu LS, Cheng HL, Jedrychowski MP, Gygi SP, Sinclair DA, Alt FW, Greenberg ME. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science. 2004;303:2011–2015. doi: 10.1126/science.1094637. doi:10.1126/science.1094637. [DOI] [PubMed] [Google Scholar]
- 8.Motta MC, Divecha N, Lemieux M, Kamel C, Chen D, Gu W, Bultsma Y, McBurney M, Guarente L. Mammalian SIRT1 represses forkhead transcription factors. Cell. 2004;116:551–563. doi: 10.1016/s0092-8674(04)00126-6. doi:10.1016/S0092-8674(04)00126-6. [DOI] [PubMed] [Google Scholar]
- 9.Mattagajasingh I, Kim CS, Naqvi A, Yamamori T, Hoffman TA, Jung SB, DeRicco J, Kasuno K, Irani K. SIRT1 promotes endothelium-dependent vascular relaxation by activating endothelial nitric oxide synthase. Proc Natl Acad Sci USA. 2007;104:14855–14860. doi: 10.1073/pnas.0704329104. doi:10.1073/pnas.0704329104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lagouge M, Argmann C, Gerhart-Hines Z, Meziane H, Lerin C, Daussin F, Messadeq N, Milne J, Lambert P, Elliott P, Geny B, Laakso M, Puigserver P, Auwerx J. Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1alpha. Cell. 2006;127:1109–1122. doi: 10.1016/j.cell.2006.11.013. doi:10.1016/j.cell.2006.11.013. [DOI] [PubMed] [Google Scholar]
- 11.Rodgers JT, Lerin C, Haas W, Gygi SP, Spiegelman BM, Puigserver P. Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature. 2005;434:113–118. doi: 10.1038/nature03354. doi:10.1038/nature03354. [DOI] [PubMed] [Google Scholar]
- 12.Yeung F, Hoberg JE, Ramsey CS, Keller MD, Jones DR, Frye RA, Mayo MW. Modulation of NF-kappaB-dependent transcription and cell survival by the SIRT1 deacetylase. EMBO J. 2004;23:2369–2380. doi: 10.1038/sj.emboj.7600244. doi:10.1038/sj.emboj.7600244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Luo J, Nikolaev AY, Imai S, Chen D, Su F, Shiloh A, Guarente L, Gu W. Negative control of p53 by Sir2alpha promotes cell survival under stress. Cell. 2001;107:137–148. doi: 10.1016/s0092-8674(01)00524-4. doi:10.1016/S0092-8674(01)00524-4. [DOI] [PubMed] [Google Scholar]
- 14.Tontonoz P, Spiegelman BM. Fat and beyond: the diverse biology of PPARgamma. Annu Rev Biochem. 2008;77:289–312. doi: 10.1146/annurev.biochem.77.061307.091829. doi:10.1146/annurev.biochem.77.061307.091829. [DOI] [PubMed] [Google Scholar]
- 15.Kim HJ, Park KG, Yoo EK, Kim YH, Kim YN, Kim HS, Kim HT, Park JY, Lee KU, Jang WG, Kim JG, Kim BW, Lee IK. Effects of PGC-1alpha on TNF-alpha-induced MCP-1 and VCAM-1 expression and NF-kappaB activation in human aortic smooth muscle and endothelial cells. Antioxid Redox Signal. 2007;9:301–307. doi: 10.1089/ars.2006.1456. [DOI] [PubMed] [Google Scholar]
- 16.Brand K, Page S, Rogler G, Bartsch A, Brandl R, Knuechel R, Page M, Kaltschmidt C, Baeuerle PA, Neumeier D. Activated transcription factor nuclear factor-kappa B is present in the atherosclerotic lesion. J Clin Invest. 1996;97:1715–1722. doi: 10.1172/JCI118598. doi:10.1172/JCI118598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Knowles JW, Reddick RL, Jennette JC, Shesely EG, Smithies O, Maeda N. Enhanced atherosclerosis and kidney dysfunction in eNOS(−/−)Apoe(−/−) mice are ameliorated by enalapril treatment. J Clin Invest. 2000;105:451–458. doi: 10.1172/JCI8376. doi:10.1172/JCI8376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Guevara NV, Kim HS, Antonova EI, Chan L. The absence of p53 accelerates atherosclerosis by increasing cell proliferation in vivo. Nat Med. 1999;5:335–339. doi: 10.1038/6585. [DOI] [PubMed] [Google Scholar]
- 19.Kawashima S, Yokoyama M. Dysfunction of endothelial nitric oxide synthase and atherosclerosis. Arterioscler Thromb Vasc Biol. 2004;24:998–1005. doi: 10.1161/01.ATV.0000125114.88079.96. doi:10.1161/01.ATV.0000125114.88079.96. [DOI] [PubMed] [Google Scholar]
- 20.Zhang QJ, Wang Z, Chen HZ, Zhou S, Zheng W, Liu G, Wei YS, Cai H, Liu DP, Liang CC. Endothelium-specific overexpression of class III deacetylase SIRT1 decreases atherosclerosis in apolipoprotein E-deficient mice. Cardiovasc Res. 2008;80:191–199. doi: 10.1093/cvr/cvn224. doi:10.1093/cvr/cvn224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sequeira J, Boily G, Bazinet S, Saliba S, He X, Jardine K, Kennedy C, Staines W, Rousseaux C, Mueller R, McBurney MW. Sirt1-null mice develop an autoimmune-like condition. Exp Cell Res. 2008;314:3069–3074. doi: 10.1016/j.yexcr.2008.07.011. doi:10.1016/j.yexcr.2008.07.011. [DOI] [PubMed] [Google Scholar]
- 22.McBurney MW, Yang X, Jardine K, Hixon M, Boekelheide K, Webb JR, Lansdorp PM, Lemieux M. The mammalian SIR2alpha protein has a role in embryogenesis and gametogenesis. Mol Cell Biol. 2003;23:38–54. doi: 10.1128/MCB.23.1.38-54.2003. doi:10.1128/MCB.23.1.38-54.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Plump AS, Smith JD, Hayek T, Aalto-Setala K, Walsh A, Verstuyft JG, Rubin EM, Breslow JL. Severe hypercholesterolemia and atherosclerosis in apolipoprotein E-deficient mice created by homologous recombination in ES cells. Cell. 1992;71:343–353. doi: 10.1016/0092-8674(92)90362-g. doi:10.1016/0092-8674(92)90362-G. [DOI] [PubMed] [Google Scholar]
- 24.Becher B, Durell BG, Miga AV, Hickey WF, Noelle RJ. The clinical course of experimental autoimmune encephalomyelitis and inflammation is controlled by the expression of CD40 within the central nervous system. J Exp Med. 2001;193:967–974. doi: 10.1084/jem.193.8.967. doi:10.1084/jem.193.8.967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chua KF, Mostoslavsky R, Lombard DB, Pang WW, Saito S, Franco S, Kaushal D, Cheng HL, Fischer MR, Stokes N, Murphy MM, Appella E, Alt FW. Mammalian SIRT1 limits replicative life span in response to chronic genotoxic stress. Cell Metab. 2005;2:67–76. doi: 10.1016/j.cmet.2005.06.007. doi:10.1016/j.cmet.2005.06.007. [DOI] [PubMed] [Google Scholar]
- 26.Buerki C, Rothgiesser KM, Valovka T, Owen HR, Rehrauer H, Fey M, Lane WS, Hottiger MO. Functional relevance of novel p300-mediated lysine 314 and 315 acetylation of RelA/p65. Nucleic Acids Res. 2008;36:1665–1680. doi: 10.1093/nar/gkn003. doi:10.1093/nar/gkn003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Purcell-Huynh DA, Farese RV, Jr, Johnson DF, Flynn LM, Pierotti V, Newland DL, Linton MF, Sanan DA, Young SG. Transgenic mice expressing high levels of human apolipoprotein B develop severe atherosclerotic lesions in response to a high-fat diet. J Clin Invest. 1995;95:2246–2257. doi: 10.1172/JCI117915. doi:10.1172/JCI117915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Trayhurn P, Wood IS. Adipokines: inflammation and the pleiotropic role of white adipose tissue. Br J Nutr. 2004;92:347–355. doi: 10.1079/bjn20041213. doi:10.1079/BJN20041213. [DOI] [PubMed] [Google Scholar]
- 29.Lyon CJ, Law RE, Hsueh WA. Minireview: adiposity, inflammation, and atherogenesis. Endocrinology. 2003;144:2195–2200. doi: 10.1210/en.2003-0285. doi:10.1210/en.2003-0285. [DOI] [PubMed] [Google Scholar]
- 30.Moore KJ, Freeman MW. Scavenger receptors in atherosclerosis: beyond lipid uptake. Arterioscler Thromb Vasc Biol. 2006;26:1702–1711. doi: 10.1161/01.ATV.0000229218.97976.43. doi:10.1161/01.ATV.0000229218.97976.43. [DOI] [PubMed] [Google Scholar]
- 31.Tiwari RL, Singh V, Barthwal MK. Macrophages: an elusive yet emerging therapeutic target of atherosclerosis. Med Res Rev. 2008;28:483–544. doi: 10.1002/med.20118. doi:10.1002/med.20118. [DOI] [PubMed] [Google Scholar]
- 32.Mehta JL, Sanada N, Hu CP, Chen J, Dandapat A, Sugawara F, Satoh H, Inoue K, Kawase Y, Jishage K, Suzuki H, Takeya M, Schnackenberg L, Beger R, Hermonat PL, Thomas M, Sawamura T. Deletion of LOX-1 reduces atherogenesis in LDLR knockout mice fed high cholesterol diet. Circ Res. 2007;100:1634–1642. doi: 10.1161/CIRCRESAHA.107.149724. doi:10.1161/CIRCRESAHA.107.149724. [DOI] [PubMed] [Google Scholar]
- 33.Nagase M, Abe J, Takahashi K, Ando J, Hirose S, Fujita T. Genomic organization and regulation of expression of the lectin-like oxidized low-density lipoprotein receptor (LOX-1) gene. J Biol Chem. 1998;273:33702–33707. doi: 10.1074/jbc.273.50.33702. doi:10.1074/jbc.273.50.33702. [DOI] [PubMed] [Google Scholar]
- 34.Shen Z, Ajmo JM, Rogers CQ, Liang X, Le L, Murr MM, Peng Y, You M. Role of SIRT1 in regulation of LPS- or two ethanol metabolites-induced TNF{alpha} production in cultured macrophage cell lines. Am J Physiol Gastrointest Liver Physiol. 2009;296:G1047–1053. doi: 10.1152/ajpgi.00016.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sugimoto K, Ishibashi T, Sawamura T, Inoue N, Kamioka M, Uekita H, Ohkawara H, Sakamoto T, Sakamoto N, Okamoto Y, Takuwa Y, Kakino A, Fujita Y, Tanaka T, Teramoto T, Maruyama Y, Takeishi Y. LOX-1-MT1-MMP axis is crucial for RhoA and Rac1 activation induced by oxidized low-density lipoprotein in endothelial cells. Cardiovasc Res. 2009;84:127–136. doi: 10.1093/cvr/cvp177. doi:10.1093/cvr/cvp177. [DOI] [PubMed] [Google Scholar]
- 36.Kakinuma T, Yasuda T, Nakagawa T, Hiramitsu T, Akiyoshi M, Akagi M, Sawamura T, Nakamura T. Lectin-like oxidized low-density lipoprotein receptor 1 mediates matrix metalloproteinase 3 synthesis enhanced by oxidized low-density lipoprotein in rheumatoid arthritis cartilage. Arthritis Rheum. 2004;50:3495–3503. doi: 10.1002/art.20581. doi:10.1002/art.20581. [DOI] [PubMed] [Google Scholar]
- 37.Li D, Liu L, Chen H, Sawamura T, Ranganathan S, Mehta JL. LOX-1 mediates oxidized low-density lipoprotein-induced expression of matrix metalloproteinases in human coronary artery endothelial cells. Circulation. 2003;107:612–617. doi: 10.1161/01.cir.0000047276.52039.fb. doi:10.1161/01.CIR.0000047276.52039.FB. [DOI] [PubMed] [Google Scholar]
- 38.Li D, Williams V, Liu L, Chen H, Sawamura T, Antakli T, Mehta JL. LOX-1 inhibition in myocardial ischemia-reperfusion injury: modulation of MMP-1 and inflammation. Am J Physiol Heart Circ Physiol. 2002;283:H1795–H1801. doi: 10.1152/ajpheart.00382.2002. [DOI] [PubMed] [Google Scholar]
- 39.Li AC, Glass CK. The macrophage foam cell as a target for therapeutic intervention. Nat Med. 2002;8:1235–1242. doi: 10.1038/nm1102-1235. doi:10.1038/nm1102-1235. [DOI] [PubMed] [Google Scholar]
- 40.Kanters E, Pasparakis M, Gijbels MJ, Vergouwe MN, Partouns-Hendriks I, Fijneman RJ, Clausen BE, Forster I, Kockx MM, Rajewsky K, Kraal G, Hofker MH, de Winther MP. Inhibition of NF-kappaB activation in macrophages increases atherosclerosis in LDL receptor-deficient mice. J Clin Invest. 2003;112:1176–1185. doi: 10.1172/JCI18580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ferreira V, van Dijk KW, Groen AK, Vos RM, van der Kaa J, Gijbels MJ, Havekes LM, Pannekoek H. Macrophage-specific inhibition of NF-kappaB activation reduces foam-cell formation. Atherosclerosis. 2007;192:283–290. doi: 10.1016/j.atherosclerosis.2006.07.018. doi:10.1016/j.atherosclerosis.2006.07.018. [DOI] [PubMed] [Google Scholar]
- 42.Kanters E, Gijbels MJ, van der Made I, Vergouwe MN, Heeringa P, Kraal G, Hofker MH, de Winther MP. Hematopoietic NF-kappaB1 deficiency results in small atherosclerotic lesions with an inflammatory phenotype. Blood. 2004;103:934–940. doi: 10.1182/blood-2003-05-1450. doi:10.1182/blood-2003-05-1450. [DOI] [PubMed] [Google Scholar]
- 43.Cheong R, Hoffmann A, Levchenko A. Understanding NF-kappaB signaling via mathematical modeling. Mol Syst Biol. 2008;4:192. doi: 10.1038/msb.2008.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gilmore TD. Introduction to NF-kappaB: players, pathways, perspectives. Oncogene. 2006;25:6680–6684. doi: 10.1038/sj.onc.1209954. doi:10.1038/sj.onc.1209954. [DOI] [PubMed] [Google Scholar]
- 45.Ahn KS, Sethi G, Aggarwal BB. Nuclear factor-kappa B: from clone to clinic. Curr Mol Med. 2007;7:619–637. doi: 10.2174/156652407782564363. doi:10.2174/156652407782564363. [DOI] [PubMed] [Google Scholar]
- 46.Li X, Zhang S, Blander G, Tse JG, Krieger M, Guarente L. SIRT1 deacetylates and positively regulates the nuclear receptor LXR. Mol Cell. 2007;28:91–106. doi: 10.1016/j.molcel.2007.07.032. doi:10.1016/j.molcel.2007.07.032. [DOI] [PubMed] [Google Scholar]
- 47.Milne JC, Lambert PD, Schenk S, Carney DP, Smith JJ, Gagne DJ, Jin L, Boss O, Perni RB, Vu CB, Bemis JE, Xie R, Disch JS, Ng PY, Nunes JJ, Lynch AV, Yang H, Galonek H, Israelian K, Choy W, Iffland A, Lavu S, Medvedik O, Sinclair DA, Olefsky JM, Jirousek MR, Elliott PJ, Westphal CH. Small molecule activators of SIRT1 as therapeutics for the treatment of type 2 diabetes. Nature. 2007;450:712–716. doi: 10.1038/nature06261. doi:10.1038/nature06261. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.