

Abstract
Background
Predisposition to heavy or light human hookworm infection is consistently reported in treatment-reinfection studies. A significant role for host genetics in determining hookworm infection intensity has also been shown, but the relationship between host genetics and predisposition has not been investigated.
Methods
A treatment-reinfection study was carried out on 1302 individuals in Brazil. Bivariate variance components analysis was used to estimate heritability for pretreatment and reinfection intensity, and to estimate the contribution of genetic and household correlations between phenotypes to the overall phenotypic correlation (i.e. predisposition).
Results
Heritability for hookworm egg count was 17% pretreatment and 25% after reinfection. Predisposition to heavy or light hookworm infection was observed, with a phenotypic correlation of 0.34 between pretreatment and reinfection intensity. This correlation was reduced to 0.23 after including household and environmental covariates. Genetic and household correlations were 0.41 and 1 respectively, and explained 88% of the adjusted phenotypic correlation.
Conclusions
Predisposition to human hookworm infection in this area results from a combination of host genetics and consistent differences in exposure, with the latter explained by household and environmental factors. Unmeasured individual-specific differences in exposure did not contribute to predisposition.
Keywords: heritability, epidemiology, predisposition, resistance, variance components analysis, Necator americanus
INTRODUCTION
Predisposition to heavy or light infection with human helminths has been consistently observed [1]. Typically, predisposition has been demonstrated by a significant positive correlation between infection intensity (before treatment) and reinfection intensity (several months or years after treatment), adjusted for age and sex [2]. Predisposition implies that there are consistent differences between individuals in exposure or susceptibility to infection [1, 2]. Exposure to infection may be influenced by environmental, socio-economic or behavioral factors, while susceptibility may be affected by a range of known (immunological, physiological or nutritional) and unknown factors. Despite increasing knowledge of environmental and household risk factors [3-6] and protective immune responses [7, 8] in helminth infection, few studies have addressed their relative contributions to predisposition [9]. Understanding the factors responsible for predisposition will help determine whether control of helminths should focus on management of exposure or development of vaccines, and is of increasing relevance in understanding the efficacy of current school-based deworming programmes [10]. Recently, a significant role for host genetics in determining human helminth infection intensity has been reported, with heritability of up to 44% (reviewed by [11, 12]). Significant heritability for helminth infection suggests that host genetics may explain much of the observed predisposition to heavy or light infection. However, only one study has estimated heritability of initial and reinfection intensity; while both phenotypes were heritable, the extent to which predisposition was determined by host genetics was not examined [13].
The human hookworms, Necator americanus and Ancylostoma duodenale, are common intestinal nematodes, infecting around 740 million people worldwide and causing significant morbidity [14]. Predisposition to high or low hookworm burden [15-17] and genetic control of hookworm infection have been reported [18-20]. Here, we use data from a treatment-reinfection study in Brazil to examine the factors determining the intensity of hookworm infection (fecal egg counts) at initial survey and after reinfection. The study population has been extensively studied, with analyses demonstrating household clustering and spatial heterogeneity in infection, and the importance of exposure-related risk factors [3, 21]. Using a bivariate variance components approach, we first estimate the roles of environmental and household risk factors, shared household environment, and additive host genetics in determining variation in hookworm infection and reinfection intensity. We then estimate the contribution of these factors to predisposition to hookworm infection, by calculating the genetic, household and individual-specific correlations between initial and reinfection intensity.
METHODS
Study population
The study was conducted in Americaninhas, a rural community in northeast Minas Gerais state, Brazil [21-23]. The study design was a total population survey, with all individuals in a 10km2 area eligible for inclusion. Informed consent was obtained from all individuals or their parents and guardians. The study was approved by the ethical committees of Instituto René Rachou-FIOCRUZ, the Brazilian National Committee for Ethics in Research (CONEP), George Washington University Medical Center, and the London School of Hygiene and Tropical Medicine.
Household survey
Kinship information was collected by interviewing one adult member of every household. Name, age, sex and parents’ names were recorded for all residents, and names of first-degree (e.g. siblings) or second-degree relatives (e.g. grandparents, aunts or uncles) living in other households in the study area. Individuals were assembled into a pedigree if they were related to or married to anyone in a pedigree. Pedigrees were assembled and indexed using PEDSYS [24], and visualized using Cranefoot [25]. Doubtful pedigree relationships were confirmed by re-interviewing household members. Information on household risk factors was collected using a pre-tested, standardized household questionnaire [23]. Socioeconomic status (SES) was assessed by a wealth index as described [22]. All households in the study area were geo-referenced and remotely sensed environmental data (altitude and Normalized Difference Vegetation Index (NDVI)) were extracted as described [3].
Parasitological survey and anthelminthic treatment
The initial parasitological survey was performed during April-July 2004. Subjects were asked to provide two fecal samples on two separate days, which were examined qualitatively by formalin-ether sedimentation. Helminth-positive samples were then examined by Kato–Katz fecal thick smear to quantify the intensity of infection, as eggs per gram of feces (epg). Two slides were counted from each day’s sample, i.e. 2-4 slides from each individual, as some individuals only provided one sample. Individuals egg-positive by sedimentation but negative by Kato-Katz were assigned a count of 3 epg, half the Kato-Katz detection limit. Hookworm was exclusively N. americanus [3]. Adults or children positive for gastrointestinal nematodes were offered a single 400 mg dose of albendazole. Egg-negative individuals were not treated. Treated individuals were examined post-treatment to confirm treatment efficacy, and offered repeat treatment(s) until egg-negative. Overall, 90% of hookworm-infected individuals received treatment. In December 2005 – March 2006 a follow-up parasitological survey was performed, with inclusion criteria of continued residence in the study area and willingness to participate. Treated individuals, and untreated individuals who were egg-negative at first survey, were included in the analysis of reinfection egg counts; 13 egg-positive but untreated subjects were excluded. Reinfection egg counts were performed a median of 14 months (interquartile range 12-16 months) after the last treatment date, or sample date if untreated. Individuals positive for gastrointestinal nematodes were treated with albendazole as above.
Pedigree structure
1302 individuals provided kinship and household information and at least one parasitological phenotype: 1294 with pretreatment egg counts and 605 with reinfection egg counts (597 sampled at both timepoints). 1266 individuals were assembled into 25 pedigrees, with one large multi-generational pedigree comprising 1157 phenotyped people, and 24 pedigrees of 2-11 phenotyped people; 36 individuals had no phenotyped relatives in the study area. The large pedigree spanned 4 generations of phenotyped individuals. Some inbreeding was present, with 50 phenotyped individuals resulting from 9 marriages between first cousins and 7 marriages between more distant relatives. There were 40,472 relative pairs for analysis, calculated using the program ‘Kinship’ in PEDSYS (Table 1).
Table 1.
Distribution of relative pairs by degree of relationship in the Americaninhas study population (Minas Gerais state, Brazil, n=1302)
| Relationship (degree) | Coefficient of relatedness |
Number of pairs |
|---|---|---|
| Identical twins | 1 | 4 |
| First | 0.5 – 0.5625 | 2853 |
| Second | 0.25 – 0.375 | 4062 |
| Third | 0.125 – 0.219 | 7246 |
| Fourth | 0.0625 – 0.117 | 9759 |
| Fifth | 0.031 – 0.055 | 9621 |
| Sixth | 0.0156 – 0.027 | 5237 |
| Seventh | 0.0078 – 0.017 | 1548 |
| Eighth | 0.0039 | 142 |
| Total | 40472 |
Statistical analysis
Hookworm infection intensity was analysed as ln(epg+1) to reduce skewness. Williams geometric means are presented for epg, i.e. geometric mean(epg+1)-1. Preliminary investigation of covariates was by linear regression, with standard errors adjusted for non-independence of individuals within households using robust Huber/White/Sandwich variance estimates, in Stata 9.0 (STATA Corporation, Houston, TX, USA). Covariates tested included age, sex, household characteristics (lack of toilet, crowding (>1 person/room), type of floor, SES) and environmental variables (sector, NDVI and altitude). Sector is a 6-level categorical variable summarizing the distribution of houses in relation to large-scale geographical features, with one large village (sector 1) and 5 rural sectors. Covariates were analyzed separately for infection and reinfection intensity. All covariates were included in an initial full model, and non-significant (p>0.1) covariates excluded sequentially to generate a minimal model. Excluded covariates were retested in the minimal model, and remained non-significant. SES and NDVI were analysed as continuous variables; for ease of interpretation, effect sizes are presented as the difference in average egg count between the highest and lowest quintiles.
Variance components analysis was used to estimate the amount of variation in infection intensity determined by additive genetic and household effects [26]. Univariate analysis compared four models, fitted by maximum-likelihood using SOLAR 4.2.0 [27]. In the sporadic model, the total variance (Vtot) of each phenotype was attributable to individual-specific error (Ve). In the polygenic model, Vtot was partitioned into variance due to additive genetic effects (Va) and error (Ve). In the household model, Vtot was divided into that due to common household environment Vc and error (Ve). In the saturated model, both additive genetic and household effects were fitted. Heritability was calculated as h2 = Va / Vtot and household effects as c2 = Vc / Vtot. Significance of variance components was calculated by likelihood-ratio tests. Covariates identified from the preliminary analysis as significant for either phenotype were included in all models. The amount of variation explained by covariates was estimated from the trait variance in models with and without covariates. The variance components analysis assumes multivariate normality, but has been shown to be relatively robust to departures from normality; residual kurtosis for each phenotype was within acceptable limits (<0.8).
To estimate the role of additive genetics and household effects in determining the correlation between initial and reinfection intensity (predisposition), bivariate variance components were fitted in SOLAR [28, 29]. A saturated model was fitted, estimating additive genetic, household and unexplained (individual-specific) variance components for each phenotype, and three additional parameters: the additive genetic correlation, ρG, shared-household correlation, ρC, and individual-specific correlation, ρE, between initial and reinfection intensity. The genetic correlation provides an estimate of shared genetic control of each trait: a high genetic correlation suggests that the same loci control initial and reinfection intensity. Similarly, the household correlation measures the degree to which the effects of shared household are common to each trait. Squaring each correlation component provides an estimate of the proportion of the associated variance component that is common to both phenotypes [30]. Thus a household correlation of 1 indicates that all of the effects of shared household on trait variance are common to both traits, while a correlation of 0.5 would show that 25% (=0.52) of the effects are in common.
The overall phenotypic correlation (ρP) between traits is thus partitioned into additive genetic, household and individual-specific correlation components. An unbiased estimate of ρP can be calculated from these correlations as:
where sub-scripts 1 and 2 represent variance component estimates for each trait [28]. The significance of each correlation component was evaluated by likelihood-ratio tests comparing the general model, in which the covariance parameter is estimated, with a restricted model in which the covariance parameter is constrained to zero (i.e. no correlation between traits for that effect).
RESULTS
Parasitological and demographic data
These data are summarized in Table 2. The prevalence and intensity of hookworm infection after reinfection were much lower than pretreatment. Hookworm burden increased in childhood to a plateau by the age of 5-10 years, with higher intensities in adult males (Fig. 1). This relationship was well described by a sex-dependent linear increase with age in 0-5 year olds, with no change with age in older hosts. There was significant predisposition to heavy or light hookworm infection, with a correlation coefficient between initial and reinfection intensity of 0.376 (n=597, P<0.0001), adjusting for age and sex.
Table 2.
Parasitological, demographic and socio-economic characteristics of the Americaninhas study population (Minas Gerais state, Brazil), at initial survey and at follow-up (a median of 14 months after anthelmintic treatment)
| Initial survey | Reinfection survey | |
|---|---|---|
| N | 1294 | 605 |
| Hookworm infection | ||
| Prevalence, % (n/total) | 70.9% (918/1294) | 29.4% (178/605) |
| egg count, mean, epg a | 1441 | 72 |
| median (interquartile range) a | 198 (0-1080) | 0 (0-12) |
| geometric mean, epg a | 78 | 2.5 |
| Sex, % male (n/total) | 49.5% (641/1294) | 45.8% (277/605) |
| Age, mean, years b | 25.4 (median 17) | 31.1 (median 26) |
| Age, range, years b | 0-95 | 3-87 |
| Households | ||
| Number | 303 | 226 |
| Size, range, people c | 1 to 12 | 1 to 8 |
| Size, mean, people c | 4.3 | 2.7 |
| Toilet, % (n/total) | 49.5% (150/303) | 54.4% (123/226) |
| Crowded d, % (n/total) | 34.7% (105/303) | 36.7% (83/226) |
| NDVI e, mean (SD) | 0.314 (0.166) | 0.289 (0.172) |
includes individuals with egg count of zero
on sampling date
number of phenotyped individuals
>1 person/room
Normalized Difference Vegetation Index
Figure 1.
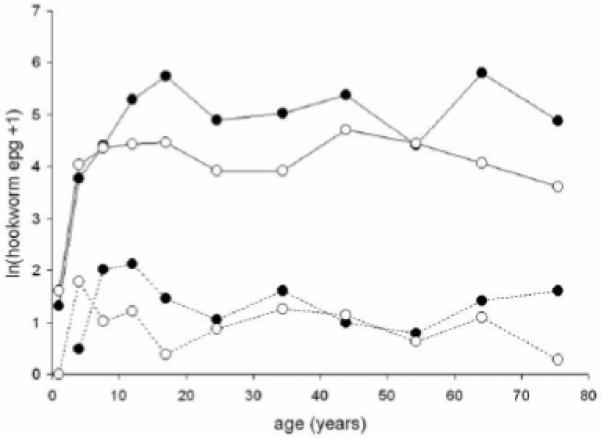
Age-intensity profiles for hookworm (Necator americanus) infection, expressed as ln(epg+1), at initial survey (solid lines), and at follow-up (14 months after treatment)(dotted lines). Solid points males, open points females.
Univariate variance components analysis
In univariate analysis pretreatment and reinfection egg counts were analysed in separate models. Pretreatment burden was best described by a saturated model including both additive genetic and household effects. The heritability h2 (the proportion of variance in egg counts explained by relatedness, after accounting for covariates) was 0.234. An additional 0.121 of variance was explained by shared household environment (c2). Both variance components were significant when compared to models including only h2 or c2 (P≤0.0003). Heritability for reinfection intensity was slightly higher (h2=0.327), while the proportion of variance explained by shared household was lower (c2=0.046); heritability was significant when compared to a model including only household effects (P=0.0008), but household effects were not significant when compared to the polygenic-only model (P=0.26). Analyses were adjusted for significant covariates identified in the preliminary data analysis.
Bivariate variance components analysis
Bivariate analysis allows joint modeling of pretreatment and reinfection hookworm intensity, and the estimation of genetic and household correlations between phenotypes. Fixed effects (covariates) accounted for 25% and 13% of the variation in infection and reinfection intensity respectively, of which 9% and 2% was accounted for by age/sex. The heritability (h2) of hookworm intensity was 0.166 (SE 0.063) at initial survey and 0.245 (0.089) after reinfection, adjusting for covariates (Table 3). Shared household environment (c2) accounted for a further 0.163 (0.043) and 0.095 (0.049) of residual variance, with a high proportion of variance (0.66-0.67) remaining unexplained. Both heritability and shared household effects were higher in models adjusted for only age and sex (Table 3). Parameter estimates for covariates are shown in Table 4, as effects on ln(epg+1). Back-transforming these estimates (i.e. eb) shows that intensity (epg+1) was 1.6-1.8× higher in households without a toilet, 1.5-1.7× higher in crowded households, and 1.8-3.0× higher in the lowest compared to the highest quintile for SES. NDVI was associated with higher intensity at initial survey only, with a 3.1× higher intensity in the highest versus lowest quintile. Intensity was lower in the village area (sector 1), and generally higher in rural areas (sectors 3-7).
Table 3.
Variance proportions (SE) from bivariate analysis of hookworm infection intensity (ln(epg+1)) at initial survey and after reinfection
| (a) adjusted for age and sex | ||
|---|---|---|
|
| ||
| Pretreatment | Reinfection | |
|
| ||
| Heritability (h2) | 0.260 (0.066) | 0.300 (0.083) |
| Shared household (c2) | 0.234 (0.052) | 0.146 (0.057) |
| Unexplained variation | 0.506 (0.041) | 0.554 (0.060) |
| (b) adjusted for all covariates a | ||
|---|---|---|
|
| ||
| Pretreatment | Reinfection | |
|
| ||
| Heritability (h2) | 0.166 (0.063) | 0.245 (0.089) |
| Shared household (c2) | 0.163 (0.043) | 0.095 (0.049) |
| Unexplained variation | 0.671 (0.046) | 0.660 (0.071) |
adjusted for age, sex, sector, toilet facilities, crowding, SES, NDVI
Table 4.
Parameter estimates (SE) for covariates affecting hookworm infection intensity (ln(epg+1)) at initial survey and after reinfection
| Pretreatment | Reinfection | |
|---|---|---|
| Age a | 1.11 (0.094) | 0.77 (0.65) |
| Sex (female vs male) | −0.50 (0.14) | −0.43 (0.31) |
| Sex × age a | −0.27 (0.13) | −0.31 (0.81) |
| Toilet (yes vs no) | −0.59 (0.32) | −0.44 (0.28) |
| Crowding (yes vs no) | 0.55 (0.22) | 0.41 (0.21) |
| Socio-economic Status b | −0.26 (0.089) | −0.13 (0.076) |
| NDVI b | 2.44 (0.78) | 0.46 (0.67) |
| Sector (vs sector 1) | ||
| Sector 3 | 1.49 (0.37) | 0.97 (0.33) |
| Sector 4 | 1.24 (0.45) | 0.31 (0.43) |
| Sector 5 | −1.01 (0.56) | −0.97 (0.51) |
| Sector 6 | 1.48 (0.62) | 0.88 (0.60) |
| Sector 7 | 0.26 (0.41) | 0.07 (0.37) |
age modeled as a linear increase up to 5 years old
modeled as a continuous variable
Predisposition to hookworm infection
Bivariate variance components models were used to estimate the contribution of covariates, additive genetics and household effects to predisposition for hookworm infection, i.e. the correlation between pretreatment and reinfection intensity. The overall phenotypic correlation was calculated as 0.336, controlling for age and sex (Table 5). Adjustment for other covariates reduced this correlation by 30% to 0.234, as expected from the similar effects of covariates on each phenotype. There was some evidence for common genetic control of both phenotypes, with a genetic correlation ρG=0.407 (SE 0.237); however ρG did not differ significantly from zero (P=0.15), and was significantly <1 (P=0.01). The relatively low genetic correlation indicated that only 17% (=0.4072) of the additive genetic factors were common to each phenotype. There was strong evidence that shared household effects contributed to predisposition: the household correlation converged to the upper limit of 1, i.e. all the shared household effect on intensity was common to both phenotypes. Genetic and household effects together explained the majority of predisposition, accounting for 88% of the adjusted phenotypic correlation. In contrast, there was no evidence that unmeasured variation in exposure between individuals contributed to predisposition. The individual-specific correlation was very low, ρE=0.04 (SE 0.07), showing that unmeasured individual-specific effects were not consistent across phenotypes. To explore any effect of confounding between genetic and household effects on these results, analysis was repeated using a polygenic-only model. The genetic correlation in this model was 0.638 (SE 0.117), significantly different from both 0 (P<0.0001) and 1 (P=0.0008). The individual-specific correlation remained very low, ρE=−0.001 (SE 0.068).
Table 5.
Correlation components from bivariate variance components analysis of hookworm infection intensity. The phenotypic correlation coefficient between initial and reinfection hookworm burden, and the correlation component (r), proportion of phenotypic correlation explained by each component (%), and significance of each correlation component (P) compared to a model with that correlation constrained to 0
| Correlation | r | ||
|---|---|---|---|
| Overall phenotypic correlationa (ρP) | 0.336 | ||
| Adjusted phenotypic correlation b (ρP) | 0.234 | ||
|
| |||
| Correlation components | r (SE) | % | P |
|
| |||
| Genetic correlation b (ρG) | 0.407 (0.237) | 35% | 0.15 |
| Household correlation b (ρC) | 1 | 53% | -c |
| Unexplained correlation b (ρE) | 0.042 (0.065) | 12% | 0.52 |
adjusted for age and sex
adjusted for age, sex, sector, toilet facilities, crowding, SES, NDVI
not tested
DISCUSSION
The current study is the most detailed to date on the genetic epidemiology of human helminth infection. Joint modeling of initial and reinfection intensity estimated the heritability of hookworm infection as 17% at initial survey, and 25% after 14 months interval for reinfection. A smaller percentage of variance was explained by shared household environment, 16% and 10% respectively. Significant predisposition to heavy or light infection was observed, and an analysis of the correlation components allowed the roles of genetics and shared household in generating this phenotypic correlation to be quantified. Host genetics, shared household effects, and measured risk factors together accounted for nearly all the observed predisposition, with no role for individual-specific differences in exposure.
Previous studies of the factors determining hookworm burden have examined household or environmental risk factors [3-6] or genetic factors (heritability) [18-20]. This is the first study to assess the influence of all three factors simultaneously, which should allow more accurate estimates of their importance. Heritabilities are population-specific, as they depend on both genetic and environmental variances; our heritability estimate depends on a single large pedigree, which may result in lower genetic and/or environmental variance. However, our estimate of 17% is comparable to hookworm studies elsewhere which estimate heritability as 10-25% [18, 20], or 15-37% when not modeling household effects [18, 19]. Estimates for other human helminths vary from 9-44% [11, 12, 31]; one study of Schistosoma japonicum infection did not find significant heritability [32]. Most previous studies have looked at pretreatment intensity only. Here, the heritability of reinfection intensity (25%) was somewhat higher than that of initial intensity, as also seen for Ascaris in Nepal [13]. This higher heritability may reflect the reduced variation in duration of exposure to infection at follow-up [13]. However, a limitation of our study is that reinfection was assessed at a single time-point, when the extent of reinfection was low. Reinfection intensity was lower than expected [20], only 5% of initial intensity, which may be explained by the high coverage and efficacy of anthelminthic treatment. Heritability is likely to vary with time since reinfection, as the extent of reinfection increases, and studies with a longer period of reinfection would be useful.
Heritability estimates can be confounded by household and environmental risk factors, since related individuals are likely to share the same residence and hence have similar risk factors. Here, the analysis of detailed household and environmental information for a large, multi-household pedigree will have reduced confounding. Analysis of risk factors reduced, but did not remove, the effects of shared household, indicating the existence of other important unmeasured household risk factors (Table 3). Accounting for risk factors also reduced estimates of heritability. This suggests that relatives share risk factors even when controlling for shared household, and that heritability may be overestimated if this is not taken into account. Family ties may influence exposure to infection in a variety of ways, such as shared activities outside the household [33]. Similarly, low socio-economic status is a risk factor for helminth infection [34], and has a high heritability [35]. Such considerations highlight the difficulties in interpreting heritability: heritability estimates the proportion of variance in a phenotype that is correlated with relatedness rather than a causative relationship between genotype and phenotype. Molecular genetic studies to identify loci underlying resistance to hookworm infection will be important to further understand the role of host genetics.
We found significant predisposition to heavy or light hookworm infection, as generally reported by studies of human helminths [1]. Our bivariate analysis allowed the estimation of the relative contribution of shared genes, shared environment and risk factors to predisposition. Household and environmental risk factors explained about a third of predisposition. Risk factors for high hookworm intensity were relative poverty, household crowding, lack of a toilet and higher NDVI. These are similar to those previously reported from the study site for hookworm prevalence [3], and from studies elsewhere [4-6]. These risk factors would be expected to be relatively constant during reinfection, illustrated by their generally consistent effects on infection and reinfection intensity. After accounting for risk factors, the majority of the remaining predisposition was explained by genetic (35%) and household (53%) effects. The household correlation was 1, suggesting that household determinants of exposure were common to both phenotypes, i.e. consistent through time, while the genetic correlation was lower. Despite a high proportion of unexplained (individual-specific) variance in intensity at both time points, the individual-specific correlation was very low (ρE=0.04). Unmeasured variation in exposure between individuals is likely to explain much of this unexplained variance in egg counts, but these results indicate that this variation in exposure was not consistent between individuals through time, and thus did not contribute to predisposition. Unmeasured variation may thus reflect chance exposure to infective stages, amplified by the aggregated distribution of infective stages [36, 37]. Another important factor generating unexplained variation in egg counts is likely to be measurement error. Also, fecal egg counts are an indirect measure of underlying worm burdens and, although there is a positive relationship between fecal egg counts and hookworm burden [38], density-dependent effects may reduce egg count at high burdens [39].
The genetic correlation between initial and reinfection intensity (ρG=0.41) was unexpectedly low. It is possible that confounding between household and genetic effects reduced this estimate, given the very high household correlation, but the genetic correlation from a polygenic model, ρG=0.64, was still significantly <1. A genetic correlation close to 1 would suggest that the same loci control both infection and reinfection intensity, and high genetic correlations have been reported between gastrointestinal nematode intensities at different time-points in domestic and wild sheep [40-42]. However, the genetic correlation for human malaria between years was also around 0.4 [43]. Our results imply that only 17-41% of genetic effects were common to both phenotypes. This suggests that infection and reinfection intensity may be partly controlled by distinct loci; it is possible that different loci control anti-adult and anti-larval immune responses, and anti-adult responses are likely to have a greater role in determining pretreatment intensity. Immunoepidemiological studies have shown that different immune responses are correlated with initial and reinfection intensity [44, 45]. Immunomodulation by adult hookworms may suppress protective responses [44, 46-48], so host genes predisposing to suppression may have a greater effect pretreatment, when adult worm burdens are higher. There may also be statistical explanations: the low degree of reinfection may have limited the power of the study, while Carey [49] demonstrated that low genetic correlations between phenotypes can result even if the same loci control each phenotype.
Further studies of the genetic epidemiology of human helminth infection will be useful, particularly treatment-reinfection studies with a longer period of reinfection. Our results raise the possibility that some of the loci controlling infection and reinfection intensity may be different, but confirmation awaits identification of the genes involved by linkage and association studies. To date, only one candidate gene for human hookworm infection has been investigated [50]. More generally, the demonstration that predisposition is dependent on factors acting at the family and household level suggests that public health interventions against hookworm infection would be usefully focused on household risk factors.
Acknowledgements
We thank the inhabitants of Americaninhas, particularly the local field technicians, and Renata Diniz and other technicians from Instituto René Rachou. Neal Alexander commented on the manuscript.
Financial support: Fieldwork was supported by the Human Hookworm Vaccine Initiative of the Sabin Vaccine Institute, which receives support from the Bill and Melinda Gates Foundation as well as the NIH/NIAID. SB is supported by a Wellcome Trust Career Development Fellowship (081673), and RLP by a UK Medical Research Council studentship. LPB was supported by a European Union Marie Curie EST Programme award.
Footnotes
Potential conflicts of interest: the authors declare no conflicts of interest
References
- 1.Anderson RM, May RM. Infectious Diseases of Humans. Dynamics and Control. Oxford University Press; Oxford: 1991. [Google Scholar]
- 2.Keymer AE, Pagel M. Predisposition to helminth infection. In: Schad GA, Warren KS, editors. Hookworm Disease: Current Status and New Directions. Taylor and Francis; London: 1990. pp. 177–209. [Google Scholar]
- 3.Pullan RL, Bethony JM, Geiger SM, et al. Human helminth co-infection: analysis of spatial patterns and risk factors in a Brazilian community. PLoS Neglect Trop Dis. 2008;2:e352. doi: 10.1371/journal.pntd.0000352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Olsen A, Samuelsen H, Onyango-Ouma W. A study of risk factors for intestinal helminth infections using epidemiological and anthropological approaches. J Biosoc Sci. 2001;33:569–84. doi: 10.1017/s0021932001005697. [DOI] [PubMed] [Google Scholar]
- 5.Traub RJ, Robertson ID, Irwin P, Mencke N, Thompson RCA. The prevalence, intensities and risk factors associated with geohelminth infection in tea-growing communities of Assam, India. Trop Med Int Health. 2004;9:688–701. doi: 10.1111/j.1365-3156.2004.01252.x. [DOI] [PubMed] [Google Scholar]
- 6.Raso G, Vounatsou P, Gosoniu L, Tanner M, N’Goran EK, Utzinger J. Risk factors and spatial patterns of hookworm infection among schoolchildren in a rural area of western Cote d’ Ivoire. Int J Parasitol. 2006;36:201–10. doi: 10.1016/j.ijpara.2005.09.003. [DOI] [PubMed] [Google Scholar]
- 7.Quinnell RJ, Bethony J, Pritchard DI. The immunoepidemiology of human hookworm infection. Parasite Immunol. 2004;26:443–54. doi: 10.1111/j.0141-9838.2004.00727.x. [DOI] [PubMed] [Google Scholar]
- 8.Hoffmann KF, Wynn TA, Dunne DW. Cytokine-mediated host responses during schistosome infections; walking the fine line between immunological control and immunopathology. Adv Parasitol. 2002;52:265–307. doi: 10.1016/s0065-308x(02)52014-5. [DOI] [PubMed] [Google Scholar]
- 9.Holland CV. Predisposition to ascariasis: patterns, mechanisms and implications. Parasitology. 2009;136:1537–47. doi: 10.1017/S0031182009005952. [DOI] [PubMed] [Google Scholar]
- 10.Bundy DAP, Medley GF. Immuno-epidemiology of human geohelminthiasis - ecological and immunological determinants of worm burden. Parasitology. 1992;104:S105–S119. doi: 10.1017/s0031182000075284. [DOI] [PubMed] [Google Scholar]
- 11.Quinnell RJ. Genetics of susceptibility to human helminth infection. Int J Parasitol. 2003;33:1219–31. doi: 10.1016/s0020-7519(03)00175-9. [DOI] [PubMed] [Google Scholar]
- 12.Bethony JM, Quinnell RJ. Genetic epidemiology of human schistosomiasis in Brazil. Acta Trop. 2008;108:166–74. doi: 10.1016/j.actatropica.2007.11.008. [DOI] [PubMed] [Google Scholar]
- 13.Williams-Blangero S, Subedi J, Upadhayay RP, et al. Genetic analysis of susceptibility to infection with Ascaris lumbricoides. Am J Trop Med Hyg. 1999;60:921–6. doi: 10.4269/ajtmh.1999.60.921. [DOI] [PubMed] [Google Scholar]
- 14.de Silva NR, Brooker S, Hotez PJ, Montresor A, Engels D, Savioli L. Soil-transmitted helminth infections: updating the global picture. Trends Parasitol. 2003;19:547–51. doi: 10.1016/j.pt.2003.10.002. [DOI] [PubMed] [Google Scholar]
- 15.Schad GA, Anderson RM. Predisposition to hookworm infection in humans. Science. 1985;228:1537–40. doi: 10.1126/science.4012307. [DOI] [PubMed] [Google Scholar]
- 16.Bradley M, Chandiwana SK. Age dependency in predisposition to hookworm infection in the Burma valley area of Zimbabwe. Trans Roy Soc Trop Med Hyg. 1990;84:826–8. doi: 10.1016/0035-9203(90)90096-w. [DOI] [PubMed] [Google Scholar]
- 17.Quinnell RJ, Griffin J, Nowell MA, Raiko A, Pritchard DI. Predisposition to hookworm infection in Papua New Guinea. Trans Roy Soc Trop Med Hyg. 2001;95:139–42. doi: 10.1016/s0035-9203(01)90138-5. [DOI] [PubMed] [Google Scholar]
- 18.Breitling LP, Wilson AJ, Raiko A, et al. Heritability of human hookworm infection in Papua New Guinea. Parasitology. 2008;135:1407–15. doi: 10.1017/S0031182008004976. [DOI] [PubMed] [Google Scholar]
- 19.Williams-Blangero S, Blangero J, Bradley M. Quantitative genetic analysis of susceptibility to hookworm infection in a population from rural Zimbabwe. Hum Biol. 1997;69:201–8. [PubMed] [Google Scholar]
- 20.Brooker S, Bethony J, Hotez PJ. Human hookworm infection in the 21st century. Adv Parasitol. 2004;58:197–288. doi: 10.1016/S0065-308X(04)58004-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brooker S, Alexander N, Geiger S, et al. Contrasting patterns in the small-scale heterogeneity of human helminth infections in urban and rural environments in Brazil. Int J Parasitol. 2006;36:1143–51. doi: 10.1016/j.ijpara.2006.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brooker S, Jardim-Botelho A, Quinnell RJ, et al. Age-related changes in hookworm infection, anaemia and iron deficiency in an area of high Necator americanus hookworm transmission in south-eastern Brazil. Trans Roy Soc Trop Med Hyg. 2007;101:146–54. doi: 10.1016/j.trstmh.2006.05.012. [DOI] [PubMed] [Google Scholar]
- 23.Fleming FM, Brooker S, Geiger SM, et al. Synergistic associations between hookworm and other helminth species in a rural community in Brazil. Trop Med Int Health. 2006;11:56–64. doi: 10.1111/j.1365-3156.2005.01541.x. [DOI] [PubMed] [Google Scholar]
- 24.Dyke B. PEDSYS: A Pedigree Data Management System. Southwest Foundation for Biomedical Research; San Antonio, TX: 1999. [Google Scholar]
- 25.Makinen VP, Parkkonen M, Wessman M, Groop PH, Kanninen T, Kaski K. High-throughput pedigree drawing. Eur J Hum Genet. 2005;13:987–9. doi: 10.1038/sj.ejhg.5201430. [DOI] [PubMed] [Google Scholar]
- 26.Lynch M, Walsh B. Genetics and Analysis of Quantitative Traits. Sinauer; Sunderland, Ma: 1998. [Google Scholar]
- 27.Almasy L, Blangero J. Multipoint quantitative-trait linkage analysis in general pedigrees. Am J Hum Genet. 1998;62:1198–211. doi: 10.1086/301844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hopper JL, Mathews JD. Extensions to multivariate normal models for pedigree analysis. Ann Hum Genet. 1982;46:373–83. doi: 10.1111/j.1469-1809.1982.tb01588.x. [DOI] [PubMed] [Google Scholar]
- 29.Lange K, Boehnke M. Extensions to pedigree analysis. IV. Covariance components models for multivariate traits. Am J Med Genet. 1983;14:513–24. doi: 10.1002/ajmg.1320140315. [DOI] [PubMed] [Google Scholar]
- 30.Boehnke M, Moll PP, Lange K, Weidman WH, Kottke BA. Univariate and bivariate analyses of cholesterol and triglyceride levels in pedigrees. Am J Med Genet. 1986;23:775–92. doi: 10.1002/ajmg.1320230306. [DOI] [PubMed] [Google Scholar]
- 31.Pullan RL, Bethony JM, Geiger SM, Correa-Oliveira R, Brooker S, Quinnell RJ. Human helminth co-infection: no evidence of common genetic control of hookworm and Schistosoma mansoni infection intensity in a Brazilian community. Int J Parasitol. 2010;40:299–306. doi: 10.1016/j.ijpara.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Seto EYW, Zhong B, Kouch J, Hubbard A, Spear RC. Genetic and household risk factors for Schistosoma japonicum infection in the presence of larger scale environmental differences in the mountainous transmission areas of China. Am J Trop Med Hyg. 2005;73:1145–50. [PubMed] [Google Scholar]
- 33.Bethony J, Williams JT, Brooker S, et al. Exposure to Schistosoma mansoni infection in a rural area in Brazil. Part III: household aggregation of water-contact behaviour. Trop Med Int Health. 2004;9:381–9. doi: 10.1111/j.1365-3156.2004.01203.x. [DOI] [PubMed] [Google Scholar]
- 34.Hotez P. Hookworm and poverty. Ann N Y Acad Sci. 2008;1136:38–44. doi: 10.1196/annals.1425.000. [DOI] [PubMed] [Google Scholar]
- 35.Rowe DC, Rodgers JL. Poverty and behavior: Are environmental measures nature and nurture? Dev Rev. 1997;17:358–75. [Google Scholar]
- 36.Hominick WM, Dean CG, Schad GA. Population biology of hookworms in West Bengal - analysis of numbers of infective larvae recovered from damp pads applied to the soil surface at defecation sites. Trans Roy Soc Trop Med Hyg. 1987;81:978–86. doi: 10.1016/0035-9203(87)90371-3. [DOI] [PubMed] [Google Scholar]
- 37.Quinnell RJ, Grafen A, Woolhouse MEJ. Changes in parasite aggregation with age: A discrete infection model. Parasitology. 1995;111:635–44. [Google Scholar]
- 38.Stoll NR. Investigations on the control of hookworm disease. XXXIII. The significance of egg-count data in necator infestations. Am J Hyg. 1924;4:466–500. [Google Scholar]
- 39.Anderson RM, Schad GA. Hookworm burdens and fecal egg counts - an analysis of the biological basis of variation. Trans Roy Soc Trop Med Hyg. 1985;79:812–25. doi: 10.1016/0035-9203(85)90128-2. [DOI] [PubMed] [Google Scholar]
- 40.Coltman DW, Pilkington J, Kruuk LEB, Wilson K, Pemberton JM. Positive genetic correlation between parasite resistance and body size in a free-living ungulate population. Evolution. 2001;55:2116–25. doi: 10.1111/j.0014-3820.2001.tb01326.x. [DOI] [PubMed] [Google Scholar]
- 41.Woolaston RR, Windon RG. Selection of sheep for response to Trichostrongylus colubriformis larvae: genetic parameters. Anim Sci. 2001;73:41–8. [Google Scholar]
- 42.Morris CA, Bisset SA, Vlassoff A, West CJ, Wheeler M. Faecal nematode egg counts in lactating ewes from Romney flocks selectively bred for divergence in lamb faecal egg count. Anim Sci. 1998;67:283–8. [Google Scholar]
- 43.Mackinnon MJ, Mwangi TW, Snow RW, Marsh K, Williams TN. Heritability of malaria in Africa. PLoS Med. 2005;2:1253–9. doi: 10.1371/journal.pmed.0020340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Quinnell RJ, Pritchard DI, Raiko A, Brown AP, Shaw MA. Immune responses in human necatoriasis: association between interleukin-5 responses and resistance to reinfection. J Infect Dis. 2004;190:430–8. doi: 10.1086/422256. [DOI] [PubMed] [Google Scholar]
- 45.Quinnell RJ, Woolhouse MEJ, Walsh EA, Pritchard DI. Immunoepidemiology of human necatoriasis - correlations between antibody responses and parasite burdens. Parasite Immunol. 1995;17:313–8. doi: 10.1111/j.1365-3024.1995.tb00897.x. [DOI] [PubMed] [Google Scholar]
- 46.Geiger SM, Caldas IR, McGlone BE, et al. Stage-specific immune responses in human Necator americanus infection. Parasite Immunol. 2007;29:347–58. doi: 10.1111/j.1365-3024.2007.00950.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Geiger SM, Massara CL, Bethony J, Soboslay PT, Correa-Oliveira R. Cellular responses and cytokine production in post-treatment hookworm patients from an endemic area in Brazil. Clin Exp Immunol. 2004;136:334–40. doi: 10.1111/j.1365-2249.2004.02449.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hsieh GCF, Loukas A, Wahl AM, et al. A secreted protein from the human hookworm Necator americanus binds selectively to NK cells and induces IFN-γ production. J Immunol. 2004;173:2699–704. doi: 10.4049/jimmunol.173.4.2699. [DOI] [PubMed] [Google Scholar]
- 49.Carey G. Inference about genetic correlations. Behav Genet. 1988;18:329–338. doi: 10.1007/BF01260933. [DOI] [PubMed] [Google Scholar]
- 50.Hall AJ, Quinnell RJ, Raiko A, et al. Chitotriosidase deficiency is not associated with human hookworm infection in a Papua New Guinean population. Infect Genet Evol. 2007;7:743–7. doi: 10.1016/j.meegid.2007.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]