

Abstract
Gene transfer of the E. coli purine nucleoside phosphorylase (PNP) results in potent cytotoxicity after administration of the prodrug fludarabine phosphate (F-araAMP). Here we have tested whether application of this strategy in the context of replication-competent retrovirus (RCR) vectors, which can achieve highly efficient tumor-restricted transduction as well as persistent expression of transgenes, would result in effective tumor inhibition, or alternatively, would adversely affect viral replication. We found that RCR vectors could achieve high levels of PNP expression concomitant with the efficiency of their replicative spread, with significant cell killing activity in vitro and potent therapeutic effects in vivo. In U-87 xenograft models, replicative spread of the vector resulted in progressive transmission of the PNP transgene, as evidenced by increasing PNP enzyme activity with time following vector inoculation. Upon F-araAMP administration, high efficiency gene transfer of PNP by the RCR vector resulted in significant suppression of tumor growth and extended survival time. As the RCR mediates stable integration of the PNP gene and continuous expression, an additional round of F-araAMP administration resulted in further survival benefit. RCR-mediated PNP suicide gene therapy thus represents a highly efficient form of intracellular chemotherapy, and may achieve effective antitumor activity with less systemic toxicity.
Keywords: retrovirus, purine nucleoside phosphorylase, suicide gene, oncolytic therapy, brain tumor
Introduction
Adenine analogs can be employed as chemotoxins that are converted to nucleotides by endogenous cytosolic adenine phosphoribosyltransferase and incorporated into cellular RNA, resulting in disruption of both RNA and protein synthesis, and subsequent cell death 1. However, the potential for serious adverse effects including myelotoxicity generally precludes systemic administration, and tumor regressions cannot be attained simply by intratumoral injection of highly toxic adenine analogs 2. Expression of E. coli purine nucleoside phosphorylase (PNP) in cancer cells can achieve direct intracellular generation of such analogs by catalyzing the glycosidic cleavage of purine ribonucleoside prodrugs, such as 6-methylpurine 2′-deoxyriboside (MeP-dR) and fludarabine (converted from fludarabine phosphate, F-araAMP, in plasma), to 2′-deoxyribose-1-phosphate (or arabinose-1-phosphate) and free base compounds such as 6-methylpurine and 2-fluoroadenine, respectively. Both compounds are freely diffusible across cell membranes, allowing their spread from PNP-transduced to untransduced cells 1, 3, and are toxic to both proliferating and nonproliferating cells 1, thereby achieving a potent bystander effect.
A variety of replication-defective viral vectors, including retrovirus, lentivirus, and adenovirus-based systems, have been employed for E. coli PNP gene delivery to cancer cells ex vivo and in vivo, showing promising results in a variety of different tumor models 2, 4–7. However, clinical trials have demonstrated limited success of suicide gene therapy mediated by replication-defective vectors 8, and more recently, conditionally replication-competent strains of various viral agents have emerged as attractive tools for cancer therapy 9. These viral agents have been termed oncolytic, i.e., exhibiting some degree of tumor-selectivity and intrinsically cytolytic, and encompass attenuated strains of a large variety of different virus species including adenovirus, herpesvirus, reovirus, vesicular stomatitis virus, vaccinia, and Newcastle Disease virus. While transient tumor regression can be obtained with most of these agents, robust immune responses eventually result in viral clearance and disease relapse, hence efforts are now directed toward “arming” these viruses with additional cytotoxic genes 10.
We have previously shown the ability of murine leukemia virus (MLV)-based replication-competent retrovirus (RCR) vectors to achieve highly efficient gene transfer to tumor cells 11–16: with genomic integration of the virus, each infected tumor cell in effect permanently becomes a virus producer cell, resulting in progressive spread of the virus. Intratumoral injection of RCR vectors in an inoculum of as little as 1 × 104 total infectious units was found to be capable of spreading and transmitting an inserted transgene throughout entire solid tumor masses in vivo, achieving up to >99% transduction in breast cancer and glioma models 11, 12, 14. Notably, after direct intratumoral injection, systemic spread of vectors was undetectable by PCR assays in any of the normal tissues tested, suggesting that the inherent inability of retroviruses to infect non-dividing cells results in a significant degree of intrinsic tumor-selectivity.
To date, there have been few studies evaluating the therapeutic potential of E. coli PNP as a suicide gene to arm replicating virus vectors, and in particular, how its potent cytotoxic activity upon prodrug administration and its strong bystander effects on adjacent cells might potentiate or inhibit the dual processes of tumor destruction and viral proliferation. These issues are central to the very concept of arming replicating viruses with suicide genes. Indeed, in a previous study arming replicating vaccinia virus with PNP, it was found that intracellular conversion of MeP-dR prodrug into the active toxin completely abrogated replication of this DNA virus 17. Here, we developed an MLV-based RCR vector carrying the PNP gene, and examined its replicative ability, cytotoxicity, and therapeutic efficacy in vitro and in vivo in subcutaneous and intracranial glioma models.
Materials and methods
Virus vectors and cell lines
As described previously, plasmid pACE-GFP 18 encodes a replication-competent amphotropic MLV vector, in which the 5′ LTR U3 region has been replaced with the cytomegalovirus (CMV) promoter, and an internal ribosome entry site (IRES)-green fluorescent protein (GFP) marker gene cassette has been inserted between the amphotropic env gene and 3′ untranslated region (UTR). The PNP gene was amplified from E. coli genomic DNA by PCR using Pfu polymerase (Stratagene, La Jolla, CA), and used to replace the GFP sequence in pACE-GFP, generating plasmid pACE-PNP. The transformed human embryonic kidney cell line 293T 19 and U-87 human glioma cells 20 were grown in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum. Virus vectors were produced by transient transfection of 293T cells with plasmid pACE-PNP or pACE-GFP using Lipofectamine Plus reagent (Invitrogen, Carlsbad, CA). Conditioned medium harvested 48 hours post-transfection was filtered, frozen at −80 °C, and titered before use. For in vitro transduction experiments, 4 μg/ml polybrene (Sigma, St. Louis, MO) was added to the medium at the time of infection. Titer determination was performed on target cells in the presence of AZT (Sigma) to prevent secondary vector replication, as described previously 13. Briefly, viral supernatant was removed 24 hours post-infection, and cells were incubated in regular medium with 50 μM AZT for 24 hours and analyzed for GFP expression by using flow cytometry on a FACScan (Becton Dickinson, Franklin Lakes, NJ). Viral titer was represented as transducing units (TU)/ml.
Virus spread in human glioma cells
U-87 cells at 20–25% confluency in 6-cm dishes were grown in fresh medium containing ACE-GFP virus stock at various multiplicities of infection (MOI). At 3, 5, 7, and 10 days post-infection, the cells were analyzed for GFP expression by flow cytometry. This procedure was performed to ensure that the entire cell population exhibited GFP fluorescence. ACE-PNP infection to U-87 cells was performed in parallel, and examined by immunocytochemistry using monoclonal anti-viral envelope antibody 83A25 21, until full transduction was confirmed. In a separate experiment, ACE-GFP-transduced U-87 cells were mixed with uninfected U-87 cells at a ratio of 0, 0.1, 1, 10 or 100% of the cell population and seeded onto 6-cm dishes with complete medium containing 5 μM MeP-dR. Four and seven days after cell mixture, the cell population for each mixture ratio was analyzed for GFP expression.
Western blot analysis
To confirm the expression of PNP, cellular protein extracted from ACE-PNP-infected and uninfected U-87 cells was run on 4–20% gradient polyacrylamide gels (Invitrogen) and transferred onto PVDF membranes (Bio-Rad, Hercules, CA). The blots were blocked 1 hr in TBST (100 mM Tris-HCl [pH 7.5], 150 mM NaCl, 0.1% Tween 20) with 5% non-fat dry milk (Bio-Rad), and incubated 1 hr with a rabbit polyclonal antibody against bacterially expressed PNP at 1:10,000 dilutions. After being washed in TBST, the blots were incubated with goat anti-rabbit secondary antibody conjugated to horseradish peroxidase (Southern Biotech, Birmingham, AL) for 1 hour and then washed in TBST. Specific protein signals were detected with an enhanced chemiluminescence kit (Amersham Biosciences, Piscataway, NJ).
Determination of non-specific toxicity of prodrugs
Untransduced, ACE-GFP-transduced, and ACE-PNP-transduced U-87 cells were seeded in 12-well culture plates at 20% confluency 24 hours before treatment, and a dose finding study was performed by exposing the cells to MeP-dR (0, 5, 10, 25, 50, and 100 μM) (Invitrogen) or F-araAMP (0, 5, 10, 25, and 50 μM) (Berlex Laboratories, Richmond, CA) for 4 days at 37 °C in complete medium, washing twice with PBS, fixing in methanol/acetone (1:1 vol/vol) for 15 min, and staining the cells remaining on the plates with Giemsa (Sigma).
In vitro cytotoxicity experiments
ACE-PNP-transduced cells were mixed with uninfected cells at a ratio of 0, 0.1, 1, 10 or 100% of the cell population and seeded onto replicate 96-well plates (5000 cells/well). After overnight culture, the mixed cell populations were exposed to increasing concentrations of MeP-dR ranging from 10 to 50 μM, and cell viability was determined 4 days later by MTS assay using the CellTiter Aqueous One Solution Cell Proliferation Assay kit (Promega, Madison, WI). To examine the time course of cytotoxicity, the mixed cell populations (0, 1, 10 or 100% ACE-PNP-transduced) were seeded onto replicate 96-well plates (2000 cells/well), and after overnight culture, the cells were exposed to MeP-dR (5 μM) or F-araAMP (10 μM), and cell viability was determined daily by MTS as above.
Subcutaneous tumor models
For subcutaneous glioma models, 24 athymic nu/nu mice (Harlan Sprague Dawley, Indianapolis, IN) were inoculated with 1 × 106 U-87 cells into the right dorsal flank. When the tumor volumes reached about 250–400 mm3, 100 μl PBS vehicle (n=12) or 1 × 105 TU ACE-PNP (n=12) was injected into the center of each tumor. Fourteen days after virus vector injections, 6 mice in the PBS group and 6 mice in the ACE-PNP group received intraperitoneal injections of F-araAMP (80 mg/kg), while the remaining 12 mice received intraperitoneal PBS injections, once every other day, for a total of 5 treatments. In a second experiment, 1 × 106 U-87 cells were subcutaneously injected into 15 athymic mice to create xenograft tumors, and when the tumor volumes were about 30–50 mm3, 30 μl PBS vehicle or 3 × 104 TU ACE-PNP was injected into the center of each tumor. Eight days after virus vector injections, the mice received intraperitoneal injections of F-araAMP (80 mg/kg) or PBS, once every other day, for a total of 10 treatments. Fifty days after intratumoral virus vector injections, mice in the ACE-PNP:F-araAMP group again received injections of F-araAMP (80 mg/kg), once every other day, for a total of 5 treatments.
Intracranial tumor models
For the intracranial glioma model, 2 × 105 U-87 cells were injected into the right frontal lobe of athymic mice, using the following coordinates: 1 mm anterior and 1.5 mm lateral relative to the bregma, and 2.8 mm deep into the brain. Seven days later, the mice were stereotactically injected with 10 μl PBS vehicle or 1 × 104 TU ACE-PNP at the same coordinates. Eight days after virus vector transduction, the mice received intraperitoneal injections of F-araAMP (40 mg/kg) or PBS, once every other day, for a total of 8 treatments. In a second experiment, the ACE-PNP vector or PBS vehicle was injected 5 days after intracranial tumor inoculation. Sixteen days after virus vector transduction, the mice received intraperitoneal injections of F-araAMP (40 mg/kg) or PBS, once every other day, for a total of 8 treatments. Two weeks following the end of first round of F-araAMP treatments, mice in the ACE-PNP:F-araAMP group again received injections of F-araAMP (40 mg/kg), once every other day, for a total of 8 treatments.
Measurement of PNP enzymatic activity
Cell lysates were prepared as previously described 22 from cultured, subcutaneous, and intracranial U-87 tumor cells. The lysates were incubated with MeP-dR, and the formation of product was measured by reverse-phase high performance liquid chromatography (HPLC) analysis of the reaction mixture 23. PNP activity was expressed as conversion units, and one unit represents 1 nmole MeP-dR cleaved/mg cell lysate/hr.
Analysis of virus stability
Genomic DNA isolated from intracranial U-87 gliomas in vivo was examined for virus stability by PCR using PCR SuperMix (Invitrogen), an upstream primer hybridizing to the amphotropic env gene, and a downstream primer hybridizing to the 3′ UTR. After PCR amplification, the reaction products were resolved on agarose gels and visualized by ethidium bromide staining.
Statistical analysis
Student’s t-tests were performed for statistical analysis of in vitro cytotoxicity experiments and subcutaneous tumor volume. Survival analysis was performed using Kaplan-Meier curves and log-rank tests. All analyses were conducted using SAS software (SAS Institute, Cary, NC).
Results
Replicative spread of RCR vectors is not inhibited in the presence of prodrug
We have previously described the construction of an MLV-based RCR vector ACE-GFP 18 containing an IRES-GFP gene expression cassette (1.3-kb) inserted precisely between the amphotropic env gene and 3′ UTR (Figure 1a). To examine the ability of RCR vectors to replicate in cultured cells, we infected U-87 human glioma cells with ACE-GFP virus supernatant at different MOIs and monitored viral transduction by flow cytometry to detect GFP fluorescence. Consistent with our previous findings, we found that ACE-GFP can efficiently transduce U-87 cells and spread to entire cell populations (approaching 100% GFP positive) in culture within 7 days at MOIs as low as 0.01 12.
Figure 1.

(a) Structure of RCR vectors carrying transgenes. RCR vector contains an IRES-GFP or IRES-PNP gene expression cassette (1.3-kb) inserted precisely between env and the 3′-untranslated region. CMV: cytomegalovirus promoter.ψ :packaging signal. (b) Replicative spread of ACE-GFP in tumor cells in the presence of prodrug. ACE-GFP-transduced and untransduced U-87 cells were mixed at different ratios and cultured in the presence of 5 μM MeP-dR. Four and seven days later, the cells were analyzed for GFP expression. X-axis: days after cell mixture. Y-axis: % of cells expressing GFP. (c) Western blot analysis of PNP protein. Cellular proteins from U-87 cells were detected by Western blotting with anti-PNP antibody. The 30-kDa band represents the PNP protein. 1: untransduced U-87 cells. 2: ACE-PNP-transduced U-87 cells. (d) In vitro cytotoxicity achieved by ACE-PNP plus prodrugs. ACE-PNP-transduced and untransduced U-87 cells were mixed at various ratios (0, 0.1, 1, 10 or 100% ACE-PNP-transduced cells) and exposed to increasing concentrations of MeP-dR ranging from 10 to 50 μM, and cell viability was determined 4 days later by MTS assay.
Although the marker gene vector ACE-GFP itself appears to have no noticeable effect on cell growth, it is conceivable that concurrent viral infection might alter cellular sensitivity to non-specific toxicity after exposure to prodrug. Therefore, we first compared the overall toxicity at different concentrations of MeP-dR (5, 10, 25, 50, and 100 μM) or F-araAMP (5, 10, 25, and 50 μM) on untransduced U-87 cells, as compared to U-87 cells that had been fully transduced with ACE-GFP. After 4 days of prodrug exposure, Giemsa staining was employed for visualization of remaining viable cells. Both untransduced U-87 cells as well as those transduced with ACE-GFP showed negligible cytotoxicity after exposure to either MeP-dR or F-araAMP at concentrations of 10 μM or less and 25 μM or less, respectively. At higher concentrations of either prodrug, progressive loss of cell viability was observed but this was the same in both transduced and untransduced cells, indicating that sensitivity to non-specific prodrug toxicity had not been altered by retroviral infection (data not shown).
Retroviruses replicate through a process involving reverse transcription of RNA to proviral DNA, and many anti-viral agents are nucleoside analogs that disrupt this process. Accordingly, we then sought to determine whether the presence of an adenine analog prodrug, in concentrations that do not appreciably affect overall cell viability, would exert any effect on the ability of the RCR vector to replicate. Therefore, ACE-GFP-transduced and uninfected U-87 cells were mixed at various ratios and cultured in the presence of 5 μM MeP-dR, a concentration of prodrug that did not cause non-specific cytotoxicity but which is sufficient to achieve highly effective killing of U-87 cells expressing PNP (see below). The subsequent spread of GFP fluorescence was monitored by flow cytometry over 7 days of continuous exposure to MeP-dR, which was replenished every other day in fresh medium. As observed previously in U-87 cells without exposure to MeP-dR, transmission of GFP fluorescence was highly robust and reached 100% within 7 days even when the initial percentage of transduced cells was as low as 1%, indicating that ACE-GFP replication was not significantly attenuated by the presence of the prodrug (Figure 1b).
RCR vectors achieve efficient expression of PNP in tumor cells, and achieve potent cell killing activity
To convert the RCR vector into a therapeutic reagent, the PNP coding sequence was cloned from E. coli and used to replace the GFP gene in ACE-GFP, producing ACE-PNP (Figure 1a). Infection of U-87 cells with ACE-PNP was performed in parallel with ACE-GFP, and protein lysates from untransduced control cells and ACE-PNP-transduced cells were examined by Western blot analysis using an antibody specific for E. coli PNP. While only non-specific signals were observed in protein lysates from untransduced cells, immunoblot of ACE-PNP-transduced U-87 cells showed a specific band at the expected molecular size, indicating efficient expression of the PNP gene product (Figure 1c).
In order to determine the functional dose range for the prodrugs employed, U-87 cells that had been fully transduced with ACE-PNP, as confirmed by immunocytochemistry with an MLV envelope-specific antibody, were incubated with MeP-dR or F-araAMP at various concentrations as above, and examined the remaining viable cells 4 days later by Giemsa staining or MTS assay. Transduction by ACE-PNP alone, without addition of prodrug, had no significant effect on cell viability. However, exposure of cells to MeP-dR or F-araAMP resulted in potent killing of ACE-PNP-transduced cells at all concentrations tested (Figure 1d and data not shown).
To further quantitate the sensitivity of transduced cells upon prodrug administration, ACE-PNP-transduced and uninfected U-87 cells were mixed at various ratios ranging from 0.1% to 100% transduced cells. These mixed cell populations were then exposed to increasing concentrations of MeP-dR ranging from 10 to 50 μM, and cell viability was quantitated 4 days later. As noted previously, at higher prodrug concentrations, some degree of cytotoxicity was seen even in the absence of ACE-PNP transduction. However, addition of PNP-expressing cells, even at low percentages, resulted in enhanced cytotoxicity upon addition of prodrug at all concentrations tested. Progressive enhancement of cytotoxicity was observed to correlate well with increasing percentage of ACE-PNP transduced cells. While there was a trend correlating higher prodrug concentration with slightly better cell killing, prodrug concentration had much less effect on overall cell killing than the percentage of transduced cells (Figure 1d).
Time course of cell killing achieved by ACE-PNP followed by prodrug administration is dependent on initial transduction level
We next examined the time course of enhanced cell killing mediated by ACE-PNP transduction at various ratios of transduced and untransduced U-87 cells. These mixed cell populations were exposed to 5 μM MeP-dR, and cell viability was quantitated daily. Exposure of 100% transduced cells to MeP-dR led to a dramatic drop in cell viability within 2 to 3 days, followed by continuing decrease in viability until complete cell clearance was achieved in 5 to 7 days. With initial inoculation of 1% or 10% ACE-PNP-expressing cells, slower but still progressive cell killing was observed over time with exposure to MeP-dR (Figure 2a). These results indicate that an ACE-PNP transduction level of even 1% is sufficient to achieve significant cell killing over time, but the rapidity of cell killing is highly dependent on the initial level of transduction. In contrast, the viability of uninfected negative control cells was not measurably affected by exposure to the MeP-dR prodrug alone (Figure 2a, 0% PNP: +MeP-dR), and the viability of fully transduced control cells were also largely unaffected in the absence of prodrug (Figure 2a, 100% PNP: – MeP-dR). Similar to MeP-dR, exposure to F-araAMP at a concentration of 10 μM was equally as efficient in killing U-87 cell populations containing ACE-PNP-transduced cells at various ratios, yet showed no cytotoxicity on uninfected cells (Figure 2b). Again, the rapidity of cell killing correlated with the initial percentage of ACE-PNP-transduced cells. As it was as efficient as MeP-dR in killing tumor cells expressing PNP, and is a clinically approved drug currently used in the treatment of hematological malignancies, F-araAMP was chosen as the prodrug for use in subsequent in vivo studies.
Figure 2.
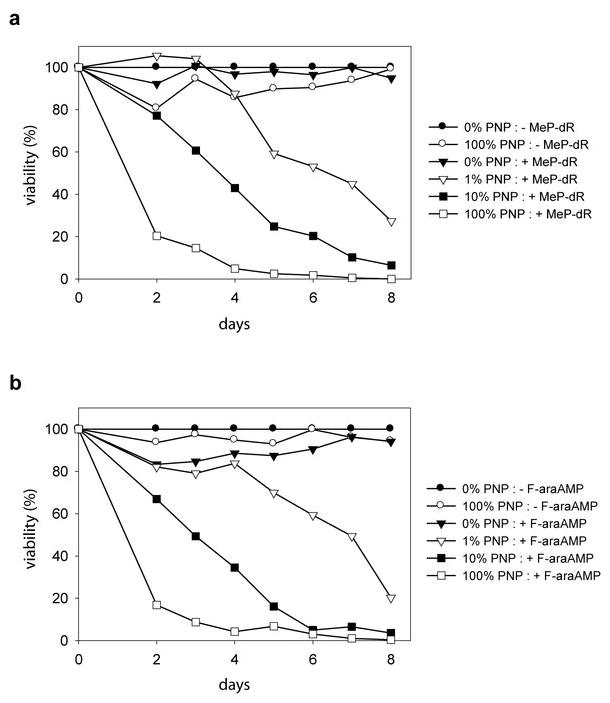
Time course of cell killing achieved by ACE-PNP followed by prodrug administration. ACE-PNP-transduced U-87 cells and untransduced U-87 cells were mixed at various ratios (0, 1, 10 or 100% ACE-PNP-transduced cells) and exposed to 5 μM MeP-dR (a) or 10 μM F-araAMP (b), and cell viability was determined daily by MTS assay. Shown values of viability % were obtained from quadruplicate repeat experiments. X-axis: days after prodrug treatment. Y-axis: % of viability of the cells normalized to that of negative control 0% PNP:- MeP-dR or 0% PNP:- F-araAMP.
ACE-PNP suppresses growth of subcutaneous tumors after treatment with F-araAMP
To determine whether delivery of the PNP gene mediated by inoculation of replicating retrovirus vectors could suppress tumor growth, we injected a single dose of 1 × 105 total transducing units of ACE-PNP virus supernatant into a naïve pre-established subcutaneous U-87 tumor model. When mean tumor volume reached about 2000 mm3, the mice received intraperitoneal injections of F-araAMP every other day over a period of 10 days, and tumor size was measured by caliper twice a week. Our results show that, immediately after F-araAMP administration, the growth of ACE-PNP-injected tumors was significantly inhibited, indicating the therapeutic effect achieved by RCR-mediated replicative spread of the PNP transgene within the tumors (Figure 3a). In the second experiment, the mice started receiving intraperitoneal injections of F-araAMP when tumor sizes reached about 250 mm3 in average. Similarly, a single injection of ACE-PNP virus followed by a single cycle of F-araAMP administration resulted in significant suppression of tumor growth, and reduced tumor volume to one-fourth that of control tumors by Day 38 to 41 (P<0.01) (Figure 3b). These tumors subsequently grew to an average of about 2000 mm3 in size by Day 51 after the first cycle of prodrug administration, therefore a second cycle of F-araAMP treatment was initiated. The second cycle of prodrug administration again efficiently inhibited tumor growth, indicating that on-going PNP expression in tumors from stably integrated and continuously spreading RCR vectors catalyzed functional conversion of F-araAMP to toxic metabolites within tumors, and contributed to the antitumor effect.
Figure 3.
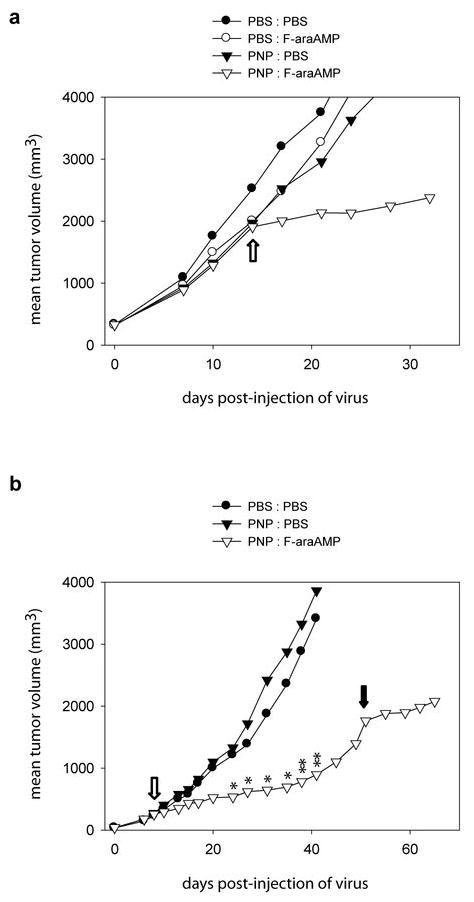
Anti-tumor effect achieved by RCR suicide gene therapy. (a) Growth of subcutaneous U-87 tumor in response to ACE-PNP and F-araAMP treatments. Pre-established U-87 tumors with mean tumor volume 320 mm3 were injected with ACE-PNP or PBS, and after 14 days the mice received intraperitoneal injections of F-araAMP (80 mg/kg) or PBS, once every other day, for a total of 5 treatments. Open arrow indicates the first day of F-araAMP treatment. (b) Growth of subcutaneous tumor in response to additional round of F-araAMP treatments. Pre-established U-87 tumors with mean tumor volume about 40 mm3 were injected with ACE-PNP or PBS, and after 8 days the mice received intraperitoneal injections of F-araAMP (80 mg/kg) or PBS, once every other day, for a total of 10 treatments. At fifty days post-injection of vectors, mice in the PNP:F-araAMP group received injections of F-araAMP (80 mg/kg), once every other day, for a total of 5 treatments. At 24, 27, 31, 35, 38, and 41 days post-injection of vectors, statistically significant differences in tumor size between PNP:F-araAMP group and control groups were observed. * P<0.05. ** P<0.01. Open arrow: beginning of the first round of F-araAMP treatments. Solid arrow: beginning of the second round of F-araAMP treatments.
RCR-mediated PNP suicide gene delivery significantly improves survival
To test if high transduction efficiency and significant tumor inhibition achieved by RCR vectors has the potential to improve survival, we then tested ACE-PNP in an intracranial U-87 glioma model. After establishment of intracranial U-87 gliomas by stereotactic implantation, 1 × 104 total transducing units of ACE-PNP virus supernatant were inoculated by stereotactic intratumoral injection. Treatment with the ACE-PNP virus followed by systemic F-araAMP administration resulted in a significant survival advantage with a median survival time of 59 days, as compared to both control groups which were treated with either virus alone or prodrug alone, with a median survival time of 30 days in either case (P<0.0001) (Figure 4a).
Figure 4.
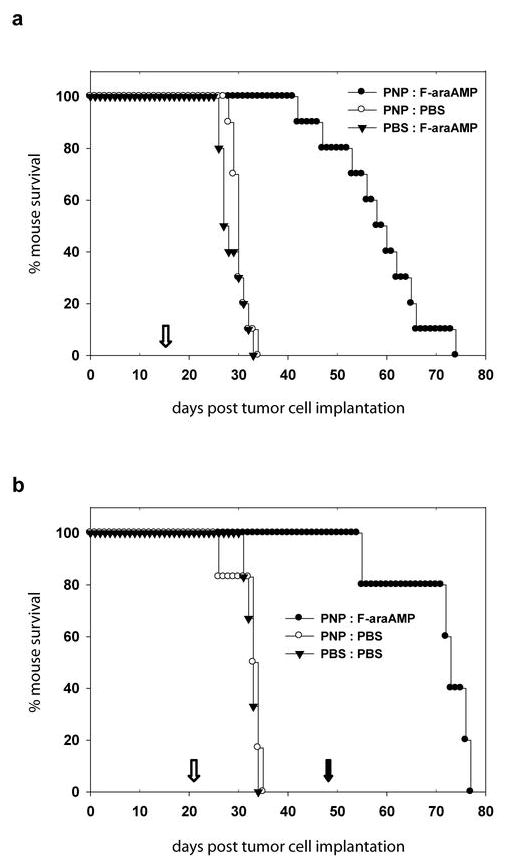
Survival analysis of athymic mice bearing intracranial U-87 glioma. (a) ACE-PNP or PBS was stereotactically injected into pre-established intracranial U-87 xenografts seven days after tumor inoculation. Eight days after vector administration, the mice received intraperitoneal injections of F-araAMP (40 mg/kg) or PBS, once every other day, for a total of 8 treatments. The median survival times for mice in the groups of PNP:F-araAMP, PNP:PBS, and PBS:F-araAMP are 59, 30, and 28 days, respectively. Open arrow: beginning of F-araAMP treatments. (b) ACE-PNP or PBS was injected into intracranial U-87 xenografts five days after tumor inoculation. Sixteen days after vector administration, the mice received intraperitoneal injections of F-araAMP (40 mg/kg) or PBS, once every other day, for a total of 8 treatments. Two weeks after the end of first round of F-araAMP treatments, mice in the PNP:F-araAMP group received injections of F-araAMP (40 mg/kg), once every other day, for a total of 8 treatments. The median survival times for mice in the groups of PNP:F-araAMP, PNP:PBS, and PBS:PBS are 73, 33, and 33 days, respectively. Open arrow: beginning of the first round of F-araAMP treatments. Solid arrow: beginning of the second round of F-araAMP treatments.
As noted above, a unique characteristic of RCR vectors compared to other replicating vector systems is the ability to mediate stable integration and long-term expression of transgenes that are transmitted from cell to cell as the virus replicates. We therefore tested whether additional therapeutic benefit might be achieved by an additional cycle of F-araAMP administration, as observed in the subcutaneous tumor model above, thereby activating PNP suicide gene activity in tumor cells that had escaped prior treatment or had been newly infected from residual transduced cells. Hence, in the second experiment, ACE-PNP-infected mice received a second cycle of F-araAMP administration two weeks after the end of first round of F-araAMP treatments. As postulated, these mice showed significant therapeutic benefit with 100% survival for more than 50 days (median survival time 73 days), compared to the mice in control groups receiving no treatment, or treatment with virus alone without prodrug, which showed 0% survival in less than 35 days (median survival time 33 days) (P<0.005) (Figure 4b). In fact, the median survival time of ACE-PNP-infected mice receiving the second cycle of F-araAMP (73 days) was even more prolonged than that observed in the first experiment with a single round of F-araAMP (59 days), demonstrating the potential for additional survival benefit from multiple cycles of prodrug administration.
PNP activity in vitro and in vivo
PNP activity in cultured cells, intracranial tumors, and subcutaneous tumors following ACE-PNP inoculation is shown in Table 1. The PNP activity in subcutaneous tumors increased with time after virus inoculation, indicating progressive transmission of the PNP transgene with replicative spread of the virus, and by 6 weeks after vector transduction the activity reached a level close to that observed in cultured U-87 cells that had been 100% transduced with ACE-PNP. Intracranial tumors also expressed a similarly high level of PNP activity by the third week after a single dose of the ACE-PNP virus. Uninfected control tumors showed no detectable E. coli PNP activity.
Table 1.
PNP activity following ACE-PNP inoculation
| Model | Treatment | Time post virus inoculation (wks) | PNP activity unitsa |
|---|---|---|---|
| Cultured U-87 cells | ACE-PNP | 1 | 4500 |
| untreated | - | 0 | |
| Intracranial U-87 tumors | ACE-PNP | 3 | 2900 ± 300 |
| PBS | 3 | 5 | |
| Subcutaneous U-87 tumors | ACE-PNP | 1.5 | 63 |
| ACE-PNP | 4 | 1800 | |
| ACE-PNP | 6 | 3300 | |
| PBS | 6 | 1 | |
| ACE-PNP plus F-araAMP | 6 | 2300 |
One PNP activity unit = 1 nmole MeP-dR cleaved/mg cell lysate/hr.
Notably, significant levels of PNP activity were measured after ACE-PNP injection even in subcutaneous tumors that had been treated with F-araAMP until 3 weeks prior to harvest (Table 1). The activity level in this instance was approximately two-thirds that of tumors injected with ACE-PNP but without exposure to prodrug at the same time point, suggesting that F-araAMP treatment had eliminated a significant fraction of the infected tumor cells, while the stably integrated PNP gene and/or on-going RCR replication still rendered residual cells susceptible to prodrug administration even after re-growth of the tumor.
IRES-PNP sequence is stably retained during prolonged RCR replication in vivo
To examine whether the inserted IRES-PNP sequence was stably retained in the replication-competent virus genome after long-term propagation in vivo, we performed PCR analysis to detect this sequence in genomic DNA isolated from intracranial tumors in each group of mice. As shown in Figure 5, the full-length IRES-PNP sequence could be detected in ACE-PNP-injected gliomas by PCR amplification. As expected, the IRES-PNP sequence was also detected in gliomas from mice in ACE-PNP plus F-araAMP treated group, again implying that persistent expression of the therapeutic gene was achieved by the replicating retrovirus vector.
Figure 5.
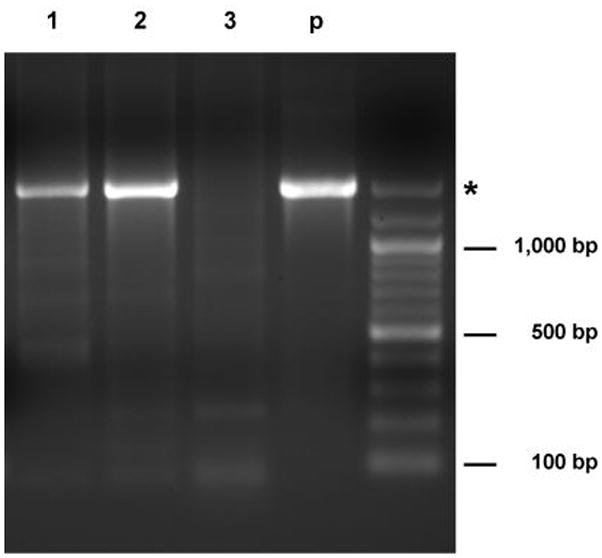
Genetic stability of ACE-PNP after long-term propagation in tumors. Genomic DNA isolated from intracranial gliomas was amplified by PCR using primers hybridizing to virus sequences flanking the IRES-PNP insert. Asterisk indicates the expected size of PCR products for undeleted ACE-PNP virus. 1–3: glioma genomic DNA from mice in the groups of PNP:F-araAMP (day 74), PNP:PBS (day 30), and PBS:F-araAMP (day 29), respectively. p: plasmid pACE-PNP.
As the primers employed in this analysis hybridized to viral sequences flanking the IRES-PNP insert, viral revertants that had deleted the insert sequence would have been detected as shorter-sized bands upon PCR amplification. However, no such bands were observed, even in the ACE-PNP plus F-araAMP treated group, suggesting that suicide gene-mediated cell killing did not exert sufficient selective pressure that would predispose to the emergence of deletion mutants within the time frame of the experiment with the treatment protocol employed.
Discussion
Conventional replication-defective retrovirus vectors have largely failed to achieve significant transduction efficiencies or therapeutic benefit, despite initially promising results in animal models 24. In the largest clinical trial of cancer gene therapy to date, transduction levels after intratumoral injection of replication-defective retroviral vector producer cells were invariably as low as 0.002% 8. In contrast, we have previously demonstrated that RCR vectors can stably propagate in human tumors, achieving tremendously high transduction efficiencies in vivo. We have now shown that replicative spread of RCR vectors armed with a suicide gene is compatible with on-going cell killing induced by exposure to the prodrug. In vitro studies showed extremely efficient cell killing by ACE-PNP in combination with MeP-dR and F-araAMP even when only 1% of the cell population was transduced initially.
PNP activity assays demonstrated that RCR vector spread in vivo was highly efficient and progressive, and the levels of PNP activity achieved after several weeks in intracranial and subcutaneous tumors were comparable to that in cultured tumor cells which were fully transduced with ACE-PNP. Indeed, the level of PNP activity achieved in tumors by RCR-mediated gene transfer was 10-fold higher than that previously reported in tumors pre-transduced by conventional replication-defective retrovirus vectors carrying the PNP gene 2, 25, and similar to activity levels reported in tumors transduced with substantially higher doses of PNP-expressing adenovirus vector 2.
Suicide gene transfer by ACE-PNP followed by F-araAMP prodrug administration resulted in a significant tumor inhibitory effect on subcutaneous U-87 xenografts, although tumors were not completely suppressed. Based on our experience from previous studies 12, 14, 26, it is likely that these animals were treated with F-araAMP while their tumors were still only partially transduced with ACE-PNP; therefore, any untransduced tumor that escaped bystander effects presumably resumed growth. This suggests that optimally effective doses of the RCR vector may not have been delivered to achieve a sufficient level of transduction throughout the tumor mass before initiating F-araAMP administration. As observed in the PNP activity assays, after RCR inoculation, subcutaneous U-87 tumors took a much longer time to reach PNP activity levels comparable to those achieved in intracranial tumors at an earlier time point. Therefore, a higher vector dosage might indeed be necessary to achieve more efficient tumor inhibition in the subcutaneous U-87 tumor model. Furthermore, in this study we employed relatively low concentrations of F-araAMP, similar to low-dose regimens that have been reported to achieve significant antitumor effects with adenovirus-mediated gene transfer of PNP 4, 27. However, these F-araAMP concentrations are substantially below the doses that have been used for PNP-mediated suicide gene therapy in the majority of previous reports 2, 6, 7, 28, suggesting that a higher F-araAMP dosage might help to achieve even more potent oncolytic effects.
Survival analysis demonstrated that RCR-mediated suicide gene therapy was indeed able to prolong mouse survival after only a single dose of vector supernatant at low multiplicity of infection followed by administration of F-araAMP. Such survival benefit achieved by RCR is not only observed in our current nude mouse tumor model, but also in an immunocompetent intracranial glioma model 29. Although we did not detect overt evidence of an immune response against RCR in the brain of immunocompetent animal, the fact that we did not detect RCR systemically in the immunocompetent model suggests that the immune system is capable of interacting with RCR, which might favorably contribute additional safety for the use of RCR for glioma gene therapy by reducing unwanted systemic spread of viruses. Continuous prodrug administration may further prolong survival due to the long-term persistence and stable expression of the RCR, as observed from PNP activity assays; this degree of viral persistence is unique among oncolytic viruses and is presumably due to the ability of MLV to stably integrate into the genome of its host cell. Stable integration of the vector represents a characteristic that may be especially advantageous in the treatment of aggressively invasive or highly metastatic tumors, as any infected tumor cell will be converted into a persistent source of on-going virus production that will spread the vector to ectopic sites as those cells migrate or metastasize. Furthermore, a major advantage of this strategy is that F-araAMP is relatively well-tolerated by mammalian cells prior to intracellular conversion by the PNP transgene product, so that systemic prodrug administration can be performed without incurring systemic toxicity, and the cell killing effects will largely be limited to transduced tumor tissues.
While previous safety studies examining the effect of systemically administering wild type MLV to primates showed no evidence of retrovirus-induced pathology 30, 31, and we have previously demonstrated that the RCR proviral genome cannot be detected in any systemic organs examined after direct intratumoral injection and replicative vector spread in both subcutaneous and intracranial tumors 11, 12, it is conceivable that low levels of systemic RCR dissemination might occur below the detection limit of our assay, and the potential for retrovirus vectors to cause insertional activation events certainly remains a major concern. Nonetheless, the setting in which retroviral insertion has been verified to cause clonal proliferation has been in the course of gene replacement therapy, involving transplantation of hematopoietic progenitor cells that have been directly transduced ex vivo under heavy cytokine stimulation with genes that confer a selective growth advantage 32–35. In contrast, when contemplating the use of RCR vectors as a cancer therapeutic agent, these concerns are mitigated, as incorporation of a suicide gene not only arms the virus against tumor cells, but also represents an inherent self-destruct mechanism that would help to eliminate any inadvertently transduced normal cells that might potentially become transformed.
In summary, the use of RCR vectors may represent a promising experimental treatment strategy because of its ability to efficiently and selectively transduce tumor cells in vivo, achieving stable integration and persistent transgene expression concomitant with progressive virus replication within the tumor. Arming RCR vectors with the PNP suicide gene does not appear to inhibit the ability of the virus to effectively replicate within solid tumors and mediate significant tumor growth inhibition and prolong survival. Thus, RCR-mediated PNP suicide gene therapy represents a highly efficient form of tumor-selective intracellular chemotherapy that can be activated in situ by prodrug administration at any time once stable integration has been achieved, and can lead to efficient antitumor effects without the toxicity due to systemic administration of antitumor agents.
Acknowledgments
Disclosure of source of funds: This work was supported by grant NSC95-2320-B-194-004-MY3 (CKT) from the National Science Council, Taiwan and NIH grants P01 CA59318 (NK), R01 CA105171 (NK) and U19 CA67763 (WBP and EJS).
Footnotes
Disclosure of financial arrangements with companies: None.
Conflict of interest
The authors declare no conflict of interest.
References
- 1.Parker WB, Allan PW, Shaddix SC, Rose LM, Speegle HF, Gillespie GY, et al. Metabolism and metabolic actions of 6-methylpurine and 2-fluoroadenine in human cells. Biochem Pharmacol. 1998;55(10):1673–1681. doi: 10.1016/s0006-2952(98)00034-3. [DOI] [PubMed] [Google Scholar]
- 2.Hong JS, Waud WR, Levasseur DN, Townes TM, Wen H, McPherson SA, et al. Excellent in vivo bystander activity of fludarabine phosphate against human glioma xenografts that express the escherichia coli purine nucleoside phosphorylase gene. Cancer Res. 2004;64(18):6610–6615. doi: 10.1158/0008-5472.CAN-04-0012. [DOI] [PubMed] [Google Scholar]
- 3.Sorscher EJ, Peng S, Bebok Z, Allan PW, Bennett LL, Jr, Parker WB. Tumor cell bystander killing in colonic carcinoma utilizing the Escherichia coli DeoD gene to generate toxic purines. Gene Ther. 1994;1(4):233–238. [PubMed] [Google Scholar]
- 4.Mohr L, Shankara S, Yoon SK, Krohne TU, Geissler M, Roberts B, et al. Gene therapy of hepatocellular carcinoma in vitro and in vivo in nude mice by adenoviral transfer of the Escherichia coli purine nucleoside phosphorylase gene. Hepatology. 2000;31(3):606–614. doi: 10.1002/hep.510310310. [DOI] [PubMed] [Google Scholar]
- 5.Voeks D, Martiniello-Wilks R, Madden V, Smith K, Bennetts E, Both GW, et al. Gene therapy for prostate cancer delivered by ovine adenovirus and mediated by purine nucleoside phosphorylase and fludarabine in mouse models. Gene Ther. 2002;9(12):759–768. doi: 10.1038/sj.gt.3301698. [DOI] [PubMed] [Google Scholar]
- 6.Parker WB, King SA, Allan PW, Bennett LL, Jr, Secrist JA, 3rd, Montgomery JA, et al. In vivo gene therapy of cancer with E. coli purine nucleoside phosphorylase. Hum Gene Ther. 1997;8(14):1637–1644. doi: 10.1089/hum.1997.8.14-1637. [DOI] [PubMed] [Google Scholar]
- 7.Martiniello-Wilks R, Dane A, Voeks DJ, Jeyakumar G, Mortensen E, Shaw JM, et al. Gene-directed enzyme prodrug therapy for prostate cancer in a mouse model that imitates the development of human disease. J Gene Med. 2004;6(1):43–54. doi: 10.1002/jgm.474. [DOI] [PubMed] [Google Scholar]
- 8.Rainov NG. A phase III clinical evaluation of herpes simplex virus type 1 thymidine kinase and ganciclovir gene therapy as an adjuvant to surgical resection and radiation in adults with previously untreated glioblastoma multiforme. Hum Gene Ther. 2000;11(17):2389–2401. doi: 10.1089/104303400750038499. [DOI] [PubMed] [Google Scholar]
- 9.Kirn D, Martuza RL, Zwiebel J. Replication-selective virotherapy for cancer: Biological principles, risk management and future directions. Nat Med. 2001;7(7):781–787. doi: 10.1038/89901. [DOI] [PubMed] [Google Scholar]
- 10.Hermiston TW, Kuhn I. Armed therapeutic viruses: Strategies and challenges to arming oncolytic viruses with therapeutic genes. Cancer Gene Ther. 2002;9(12):1022–1035. doi: 10.1038/sj.cgt.7700542. [DOI] [PubMed] [Google Scholar]
- 11.Logg CR, Tai CK, Logg A, Anderson WF, Kasahara N. A uniquely stable replication-competent retrovirus vector achieves efficient gene delivery in vitro and in solid tumors. Hum Gene Ther. 2001;12(8):921–932. doi: 10.1089/104303401750195881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang WJ, Tai CK, Kasahara N, Chen TC. Highly efficient and tumor-restricted gene transfer to malignant gliomas by replication-competent retroviral vectors. Hum Gene Ther. 2003;14(2):117–127. doi: 10.1089/104303403321070810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tai CK, Logg CR, Park JM, Anderson WF, Press MF, Kasahara N. Antibody-mediated targeting of replication-competent retroviral vectors. Hum Gene Ther. 2003;14(8):789–802. doi: 10.1089/104303403765255174. [DOI] [PubMed] [Google Scholar]
- 14.Tai CK, Wang WJ, Chen TC, Kasahara N. Single-shot, multicycle suicide gene therapy by replication-competent retrovirus vectors achieves long-term survival benefit in experimental glioma. Mol Ther. 2005;12(5):842–851. doi: 10.1016/j.ymthe.2005.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hiraoka K, Kimura T, Logg CR, Kasahara N. Tumor-selective gene expression in a hepatic metastasis model after locoregional delivery of a replication-competent retrovirus vector. Clin Cancer Res. 2006;12(23):7108–7116. doi: 10.1158/1078-0432.CCR-06-1452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hiraoka K, Kimura T, Logg CR, Tai CK, Haga K, Lawson GW, et al. Therapeutic efficacy of replication-competent retrovirus vector-mediated suicide gene therapy in a multifocal colorectal cancer metastasis model. Cancer Res. 2007;67(11):5345–5353. doi: 10.1158/0008-5472.CAN-06-4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Puhlmann M, Gnant M, Brown CK, Alexander HR, Bartlett DL. Thymidine kinase-deleted vaccinia virus expressing purine nucleoside phosphorylase as a vector for tumor-directed gene therapy. Hum Gene Ther. 1999;10(4):649–657. doi: 10.1089/10430349950018724. [DOI] [PubMed] [Google Scholar]
- 18.Logg CR, Logg A, Matusik RJ, Bochner BH, Kasahara N. Tissue-specific transcriptional targeting of a replication-competent retroviral vector. J Virol. 2002;76(24):12783–12791. doi: 10.1128/JVI.76.24.12783-12791.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.DuBridge RB, Tang P, Hsia HC, Leong PM, Miller JH, Calos MP. Analysis of mutation in human cells by using an Epstein-Barr virus shuttle system. Mol Cell Biol. 1987;7(1):379–387. doi: 10.1128/mcb.7.1.379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ponten J, Macintyre EH. Long term culture of normal and neoplastic human glia. Acta Pathol Microbiol Scand. 1968;74(4):465–486. doi: 10.1111/j.1699-0463.1968.tb03502.x. [DOI] [PubMed] [Google Scholar]
- 21.Evans LH, Morrison RP, Malik FG, Portis J, Britt WJ. A neutralizable epitope common to the envelope glycoproteins of ecotropic, polytropic, xenotropic, and amphotropic murine leukemia viruses. J Virol. 1990;64(12):6176–6183. doi: 10.1128/jvi.64.12.6176-6183.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Parker WB, Allan PW, Hassan AE, Secrist JA, 3rd, Sorscher EJ, Waud WR. Antitumor activity of 2-fluoro-2′-deoxyadenosine against tumors that express Escherichia coli purine nucleoside phosphorylase. Cancer Gene Ther. 2003;10(1):23–29. doi: 10.1038/sj.cgt.7700520. [DOI] [PubMed] [Google Scholar]
- 23.Gadi VK, Alexander SD, Kudlow JE, Allan P, Parker WB, Sorscher EJ. In vivo sensitization of ovarian tumors to chemotherapy by expression of E. coli purine nucleoside phosphorylase in a small fraction of cells. Gene Ther. 2000;7(20):1738–1743. doi: 10.1038/sj.gt.3301286. [DOI] [PubMed] [Google Scholar]
- 24.Kruse CA, Roper MD, Kleinschmidt-DeMasters BK, Banuelos SJ, Smiley WR, Robbins JM, et al. Purified herpes simplex thymidine kinase Retrovector particles. I. In vitro characterization, in situ transduction efficiency, and histopathological analyses of gene therapy-treated brain tumors. Cancer Gene Ther. 1997;4(2):118–128. [PubMed] [Google Scholar]
- 25.Gadi VK, Alexander SD, Waud WR, Allan PW, Parker WB, Sorscher EJ. A long-acting suicide gene toxin, 6-methylpurine, inhibits slow growing tumors after a single administration. J Pharmacol Exp Ther. 2003;304(3):1280–1284. doi: 10.1124/jpet.102.044743. [DOI] [PubMed] [Google Scholar]
- 26.Kikuchi E, Menendez S, Ozu C, Ohori M, Cordon-Cardo C, Logg CR, et al. Delivery of replication-competent retrovirus expressing Escherichia coli purine nucleoside phosphorylase increases the metabolism of the prodrug, fludarabine phosphate and suppresses the growth of bladder tumor xenografts. Cancer Gene Ther. 2007;14(3):279–286. doi: 10.1038/sj.cgt.7701013. [DOI] [PubMed] [Google Scholar]
- 27.Wang XY, Martiniello-Wilks R, Shaw JM, Ho T, Coulston N, Cooke-Yarborough C, et al. Preclinical evaluation of a prostate-targeted gene-directed enzyme prodrug therapy delivered by ovine atadenovirus. Gene Ther. 2004;11(21):1559–1567. doi: 10.1038/sj.gt.3302308. [DOI] [PubMed] [Google Scholar]
- 28.Martiniello-Wilks R, Wang XY, Voeks DJ, Dane A, Shaw JM, Mortensen E, et al. Purine nucleoside phosphorylase and fludarabine phosphate gene-directed enzyme prodrug therapy suppresses primary tumour growth and pseudo-metastases in a mouse model of prostate cancer. J Gene Med. 2004;6(12):1343–1357. doi: 10.1002/jgm.629. [DOI] [PubMed] [Google Scholar]
- 29.Wang W, Tai CK, Kershaw AD, Solly SK, Klatzmann D, Kasahara N, et al. Use of replication-competent retroviral vectors in an immunocompetent intracranial glioma model. Neurosurg Focus. 2006;20(4):E25. doi: 10.3171/foc.2006.20.4.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cornetta K, Morgan RA, Gillio A, Sturm S, Baltrucki L, O’Reilly R, et al. No retroviremia or pathology in long-term follow-up of monkeys exposed to a murine amphotropic retrovirus. Hum Gene Ther. 1991;2(3):215–219. doi: 10.1089/hum.1991.2.3-215. [DOI] [PubMed] [Google Scholar]
- 31.Cornetta K, Moen RC, Culver K, Morgan RA, McLachlin JR, Sturm S, et al. Amphotropic murine leukemia retrovirus is not an acute pathogen for primates. Hum Gene Ther. 1990;1(1):15–30. doi: 10.1089/hum.1990.1.1-15. [DOI] [PubMed] [Google Scholar]
- 32.Donahue RE, Kessler SW, Bodine D, McDonagh K, Dunbar C, Goodman S, et al. Helper virus induced T cell lymphoma in nonhuman primates after retroviral mediated gene transfer. J Exp Med. 1992;176(4):1125–1135. doi: 10.1084/jem.176.4.1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Marshall E. Clinical research. Gene therapy a suspect in leukemia-like disease. Science. 2002;298(5591):34–35. doi: 10.1126/science.298.5591.34. [DOI] [PubMed] [Google Scholar]
- 34.Hacein-Bey-Abina S, Von Kalle C, Schmidt M, McCormack MP, Wulffraat N, Leboulch P, et al. LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science. 2003;302(5644):415–419. doi: 10.1126/science.1088547. [DOI] [PubMed] [Google Scholar]
- 35.Li Z, Dullmann J, Schiedlmeier B, Schmidt M, von Kalle C, Meyer J, et al. Murine leukemia induced by retroviral gene marking. Science. 2002;296(5567):497. doi: 10.1126/science.1068893. [DOI] [PubMed] [Google Scholar]