

Abstract
To determine the role of matrix metalloproteinase-8 (MMP-8) in acute lung injury (ALI), we delivered LPS or bleomycin by the intratracheal route to MMP-8−/− mice versus WT mice or subjected the mice to hyperoxia (95% O2) and measured lung inflammation and injury at intervals. MMP-8−/− mice with ALI had greater increases in lung PMN and macrophage counts, measures of alveolar capillary barrier injury, lung elastance, and mortality than WT mice with ALI. Bronchoalveolar lavage fluid (BALF) from LPS-treated MMP-8−/− mice had more macrophage inflammatory protein-1α (MIP-1α) than BALF from LPS-treated WT mice, but similar levels of other pro- and anti-inflammatory mediators. MIP-1α−/− mice with ALI had less acute lung inflammation and injury than WT mice with ALI, confirming that MIP-1α promotes acute lung inflammation and injury in mice. Genetically deleting MIP-1α in MMP-8−/− mice abrogated the increased lung inflammation and injury and mortality in MMP-8−/− mice with ALI. Soluble MMP-8 cleaved and inactivated MIP-1α in vitro, but membrane-bound MMP-8 on activated PMNs had greater MIP-1α-degrading activity than soluble MMP-8. High levels of membrane-bound MMP-8 were detected on lung PMNs from LPS-treated WT mice, but soluble, active MMP-8 was not detected in BALF samples. Thus, MMP-8 has novel roles in restraining lung inflammation and in limiting alveolar capillary barrier injury during ALI in mice by inactivating MIP-1α. In addition, membrane-bound MMP-8 on activated lung PMNs is likely to be the key bioactive form of the enzyme that limits lung inflammation and alveolar capillary barrier injury during ALI.
Keywords: rodent, neutrophil, inflammation, knockout mice, chemokine
Introduction
Matrix metalloproteinase-8 (MMP-82, collagenase-2, neutrophil collagenase) is a member of the interstitial collagenase subgroup of the MMP family of zinc-dependent, neutral proteinases [1]. PMNs are the main source of MMP-8 in humans and mice [2–4]. MMP-8 is not synthesized de novo by mature PMNs; rather it is synthesized by myelocyte PMN precursors in the bone marrow [5], and stored as a latent proenzyme (proMMP-8) in the specific granules of PMNs [6]. ProMMP-8 is rapidly released from PMNs activated to degranulate [2,7] and is then activated in the extracellular space via the cysteine switch mechanism [8–10]. Activation of PMNs in vitro also leads to translocation of MMP-8 from the specific granules of PMNs to the plasma membrane in a catalytically active form that is resistant to inhibition by tissue inhibitors of MMPs [TIMPs [2]]. However, it is not clear whether membrane-bound MMP-8 expressed on activated PMNs in vivo and whether this form of the enzyme contributes to physiologic or pathologic processes in the lung or other organs.
The main substrates for MMP-8 in vivo are widely believed to be the interstitial collagens. MMP-8 and other interstitial collagenases cleave interstitial collagens in vitro at a single locus generating ¾ and ¼ fragments, which is the initial and rate-limiting step in degradation of interstitial collagens. MMP-8 also cleaves a limited number of non-matrix proteins in vitro including serpins, bradykinin, angiotensin I, and substance P [4,11], but the biologic relevance of these activities is not clear. Studies of MMP-8−/− mice indicate that MMP-8 regulates neutrophilic inflammation in some tissues. In murine models of inflammation in the skin and dorsal air pouches, MMP-8 promotes early PMN recruitment by cleaving and activating the PMN chemokine, LPS-inducible CXC chemokine [LIX or CXCL5 [4,12]]. MMP-8 also increases PMN influx into the liver during TNF-α-mediated acute hepatitis by releasing LIX that is bound to hepatic extracellular matrix [13]. During LPS-mediated inflammation in the murine skin and cornea, MMP-8 also participates in a proteolytic cascade, along with MMP-9 and prolyl endopeptidase, that generates proline-glycine-proline (PGP) fragments of collagen that bind to CXCR1 and CXCR2 on PMNs to stimulate chemotaxis of PMNs [14–16]. However, MMP-8 reduces granulocyte numbers in the airways of mice with allergic airway inflammation and reduces late inflammation associated with skin wounds by increasing PMN apoptosis [17,18].
Little is currently known about the roles of MMP-8 and other interstitial collagenases in acute lung injury (ALI) syndromes which are characterized by rapid influx of PMNs into the lung and alveolar capillary barrier dysfunction. The levels of immunoreactive MMP-8 are increased in tracheal aspirates from preterm infants with the acute respiratory distress syndrome [ARDS [19]]. Interstitial collagenase activity is also increased in BALF from adult patients with ARDS [20,21]. However, it is not clear which enzymes contribute to this interstitial collagenase activity in ARDS patients since several proteinases cleave interstitial collagens [1,22–25]. It is also not clear whether the elevated interstitial collagenase activity in the lung contributes to pathologic processes occurring in the lungs of ARDS patients. To determine whether MMP-8 plays pathogenic roles during ALI, we compared lung inflammation and injury in MMP-8−/− mice versus WT mice in several models of ALI that are associated with robust acute lung inflammation and/or alveolar capillary barrier injury (two key pathologic features of human ARDS) which enabled us to determine the contributions of MMP-8 to both processes.
We now report several novel activities for MMP-8 during ALI. First, MMP-8 potently reduces the accumulation of PMNs and macrophages in the lungs of mice with ALI. Second, MMP-8 unexpectedly limits injury to the alveolar capillary barrier during LPS- and hyperoxia-mediated ALI and also limits mortality during hyperoxia-induced ALI. Third, MMP-8 cleaves and inactivates a CC chemokine (MIP-1α) and thereby dampens lung inflammation and limits alveolar capillary barrier injury in mice. Our study is the first to demonstrate that a PMN-derived proteinase regulates lung inflammation and injury to the alveolar capillary barrier during ALI in mice. We also demonstrated for the first time that MMP-8 is abundantly expressed on the surface of activated PMNs in vivo. Since the current study showed that membrane-bound MMP-8 is a more potent MIP-1α-degrading enzyme than soluble MMP-8 and our previous studies showed that membrane-bound MMP-8 is resistant to inhibition by TIMPs [2], our results indicate that membrane-bound MMP-8 on lung PMNs is the key bioactive form of the enzyme mediating its activities during ALI.
Materials and Methods
Materials
Kits for annexin V & propidium iodide staining and quenched DQ FITC-conjugated type I collagen were obtained from Invitrogen (Carlsbad, CA). The caspase-3 fluorogenic assay, purified caspase-3 assay, and ELISA kits for quantifying murine cytokines and chemokines were purchased from R & D Systems (Minneapolis, MN). The QuantiChrom™ Hemoglobin Assay kit was purchased from BioAssay Systems (Hayward, CA). Recombinant MIP-1α (rMIP-1α) was purchased from Peprotech (Rocky Hill, NJ). The hamster anti-CD95 antibody (BD 554254, clone Jo2) and non-immune hamster IgG control antibodies were purchased from BD Biosciences (San Jose, CA). All other reagents were purchased from Sigma Chemical Co. (St. Louis, MO).
Animals
All procedures performed on mice were approved by the Harvard Medical School Animal Care and Use Committee. Mice were housed in a barrier facility under specific pathogen-free conditions. MMP-8−/− mice were generated and initially studied in the mixed SVeV129 X C57BL/6J strain [4]. The mice were backcrossed 10 generations into the pure C57BL/6J strain and then studied in this pure genetic background. The MMP-8−/− mice (in both genetic strains) have normal lifespan, fertility, and lung development, and no abnormality in the unchallenged state [4]. We initially studied F10 C57BL/6J wild type littermate mice as controls for C57BL6/J MMP-8−/− mice. In subsequent experiments, we studied C57BL/6J WT mice purchased from The Jackson Laboratory (Bar Harbor, ME) as controls since our studies showed that C57BL/6J wild type littermate control mice and C57BL/6J WT mice obtained from The Jackson Laboratory had similar lung inflammatory and injury responses to IT LPS. MIP-1α−/− mice in the pure C57BL/6J strain were purchased from The Jackson Laboratory. MMP-8−/− X MIP-1α−/− mice were generated by crossing MMP-8−/− and MIP-1α−/− mice. The genotypes of the mice were confirmed on genomic DNA extracted from murine tail biopsies using PCR-based genotyping protocols.
LPS-mediated ALI in mice
Age- and gender-matched adult mice (10–16 weeks old) were anesthetized with ketamine (50 mg/Kg) and xylazine (5 mg/Kg) and then given 10 μg LPS from E. coli 0111:B4 in 30 μl of endotoxin-free PBS, or 30 μl PBS alone by the IT route. At intervals, mice were killed and then BAL was performed using eight 0.5-ml aliquots of sterile PBS. The BAL cell and supernatant fractions were separated by centrifugation and the BALF samples were frozen to −80°C. RBCs were removed from the cell fraction by hypotonic lysis, and the cell-free supernatants from this step were harvested and hemoglobin levels quantified using a chromogenic kit. Total and differential WBC counts were performed on the BAL cells, or myeloperoxidase activity was quantified in BAL cell lysates [2]. Lung water content was measured by the wet/dry lung weight ratio method.
Bleomycin-mediated ALI in mice
Age- and gender-matched adult mice were anesthetized as above and then were given either 75 mU bleomycin in 30 μl of endotoxin-free saline, or 30 μl of saline alone by the IT route. Three to ten days later, mice were sacrificed, BAL was performed and total and differential WBC counts were performed on the BAL cells as outlined above.
Hyperoxia-mediated ALI in mice
WT or MMP-8−/− mice both in the pure C57BL/6J strain were exposed to room air or hyperoxia (95% O2, 5% N2) for up to 96 h. For analysis of lung injury, mice were sacrificed after 72 h of exposure to hyperoxia, and wet to dry lung weight ratios were measured as outlined above. For survival experiments, animal mortality was evaluated twice-a day for up to 96 h of continuous exposure to hyperoxia.
Lung physiology
Preliminary studies demonstrated that two IT instillations of 20 μg of LPS 24 h apart were necessary to induce significant increases in lung elastance in WT mice. Thus, WT, MMP-8−/−, MIP-1α−/−, and MMP-8−/− X MIP-1α−/− mice were given two doses of LPS (20 μg) 24 h apart and 24 h after the second dose of LPS, mice were anesthetized with pentobarbital (100 mg/Kg) and a tracheostomy was performed. Lung elastance (H), as a measure of lung stiffness, was measured in LPS-treated mice versus unchallenged mice in each genotype using a computer-controlled small animal ventilator (Flexivent, SCIREQ Inc., Montreal, PQ, Canada) set at a tidal volume of 10 ml/Kg, 120 breaths per min, and 0 cm H2O positive end expiratory pressure, as described previously [26,27].
Chemotaxis assays on BALF samples
BALF harvested from WT or MMP-8−/− mice 4 h and 24 h after IT LPS or IT PBS was diluted 1:2 in RPMI containing 10 mM HEPES and 1% albumin (pH 7.4), and then tested in triplicate for chemotactic activity for human PMNs using Boyden microchemotaxis assays chambers [Neuroprobe, Cabin John, MD [28]].
Cytokines and chemokine levels in BALF
Cell-free BALF harvested from WT or MMP-8−/− mice 4 h and 24 h after IT LPS or IT PBS was analyzed in triplicate for KC, MIP-2α, LIX, LTB4, MIP-1 α, TNF- α, G-CSF, GM-CSF, IL-1β, IL-6, and IL-10 using commercial ELISA kits. The results were expressed as pg/BAL after correcting for the volume of BALF that was recovered from each mouse (≥ 3.7 ml of BALF was recovered from the total of 4 ml of PBS delivered to each mouse).
Real-time PCR
Two hours after IT LPS or PBS lungs were removed from mice, RNA was isolated, and the samples were reverse transcribed [29]. Real-time PCR analysis was performed using a GeneAmp 570 Sequence Detection System (Applied Biosystems, Foster City, CA). The comparative cycle threshold method was used using GAPDH as an endogenous reference housekeeping gene and SYBR green as the fluorophore. The primer sequences for GAPDH and MIP-1α have been described elsewhere [29].
Human PMN isolation and activation
Approval for all studies involving human subjects was obtained from the Brigham and Women’s Hospital Institutional Review Board. PMNs (>95% pure) were isolated from peripheral blood of healthy donors [30]. Cells were activated at 37°C with PAF (10−7M) for 15 min followed by fMLP (10−7M) for 30 min to induce optimal surface MMP-8 expression [2]. PMNs were fixed for 3 min at 4°C in PBS (pH 7.4) containing 3% (w/v) paraformaldehyde and 0.5% (v/v) glutaraldehyde, which prevents release of soluble MMP-8 and other proteinases from cells but has minimal effect on surface MMP-8 catalytic activity [2]. PMNs were resuspended in Tris-buffered saline (0.05M Tris containing 0.15 M NaCl and 0.02M CaCl2; pH 7.4) for MIP-1α proteolysis assays.
Murine PMN apoptosis assays
PMNs isolated from the lungs of WT and MMP-8−/− mice 24 h after IT LPS by BAL were immediately stained with annexin V-Alexa 488 and propidium iodide using a commercial kit, and the percentage of positive cells were determined with MetaMorph® image analysis software (Molecular Devices, Sunnyvale, CA). PMN lysates (5 × 106/ml) were also prepared and assayed in triplicate for active caspase-3 with a commercial fluorogenic assay kit and assay standards of recombinant active caspase-3 using fluorimetry (Hitachi F2500 spectrofluorimeter, Hitachi Spectroscopy, Tokyo, Japan; Ex λ 400 nm Em λ 490 nm).
To assess PMN apoptosis in vitro, we isolated PMNs from the bone marrow of WT and MMP-8−/− mice by positive selection for Ly6G using immunomagnetic beads [31]. PMNs were incubated at 37°C in a humidified atmosphere of 5% CO2 in HBSS containing 10 mM HEPES and 1% albumin for up to 18 h. PMNs were also incubated at 37°C in a humidified atmosphere of 5% CO2 in RPMI containing 10% fetal bovine serum, 100 U/ml penicillin, 10 μg/ml streptomycin, and 250 ng/ml amphotericin B both in the presence and absence of 5 μg/ml hamster anti-CD95 or non-immune hamster IgG. At intervals, PMN cell death was quantified by staining cells with annexin V-Alexa 488 and propidium iodide as above, and the percentage positive cells analyzed using flow cytometry (Canto II, BD, San Jose, CA). Caspase 3 activity was measured in PMN lysates (all prepared at 5 × 106/ml) as outlined above.
MIP-1α degradation assays
Human and murine proMMP-8 and proMMP-9 were activated with p-aminophenylmercuric acetate as described previously [2,31]. Purified rmMIP-1α or rhMIP-1α (2.5 μM) was incubated for up to 18 h at 37°C in Tris-buffered saline with and without 500 nM MMP-8 or 500 nM MMP-9. Reduced cell-free reaction products were separated by electrophoresis on 16.5% Tris-tricine gels, and proteins were visualized by staining the gels with silver using a commercial kit. Human PMNs or PMNs isolated from the bone marrow of mice [31] were activated at 37°C with PAF (10−7M) for 15 min followed by fMLP (10−7M) for 30 min which induces optimal surface expression of MMP-8 on human and WT PMNs [2]. Cells were then fixed as described to prevent free release of soluble serine proteinases and MMPs PMNs [2,31–34] without significantly reducing the activity of membrane-bound MMP-8 [2]. Equal numbers of human or murine PMNs (5 × 106/assay) were then incubated with rmMIP-1α or rhMIP-1α (2.5 μM) in Tris-buffered saline containing 1 mM phenylmethylsulphonyl fluoride for 18 h at 37°C. Cell-free supernatant samples were analyzed on silver-stained Tris-tricine gels. Cell-free supernatant samples from the PMN-based rhMIP-1α proteolysis assays (or rhMIP-1α incubated without PMN) were also diluted 1:25 in chemotaxis assay buffer to dilute the rMIP-1α aliquot which was incubated without PMNs to 10−7M (the optimal concentration for inducing PMN migration in vitro). Samples were then tested for residual PMN chemotactic activity using Boyden microchemotaxis assay chambers (vide supra).
Detection of soluble active MMP-8 and membrane-bound MMP-8 in BALF samples
WT and MMP-8−/− mice were given 10 μg of LPS by the IT route. After 6, 24, and 72 h, BAL was performed using a single 1 ml aliquot of Tris-buffered saline. PMNs were separated from BALF as described above, fixed, and then immunostained with Alexa 488 for surface MMP-8 using rabbit anti-MMP-8 IgG or non-immune rabbit IgG as a control primary antibody [2]. Soluble, active MMP-8 was also measured in BALF cell-free samples from LPS-treated WT mice (or LPS-treated MMP-8−/− mice as a control) by immunoblotting [2] or by quantifying collagenase activity in these samples using quenched DQ FITC-conjugated type I collagen as a substrate [2]. BALF or assay standards of soluble active MMP-8 (10–500 nM) were incubated in triplicate with 50μg/ml quenched FITC-conjugated type I collagen in 0.25 ml of Tris-buffered saline in the presence and absence of 1 mM 1, 10-phenanthroline for 18 h at 37°C and MMP-mediated cleavage of the substrate was quantified as 1, 10-phenanthroline-sensitive cleavage of the substrate by fluorimetry (Ex λ 495; Em λ 520 nm).
Statistics
Data are expressed as mean ± SEM or mean ± SD. The results for paired and unpaired data were compared by the Student’s t-test for parametric data and the Mann-Whitney rank sum test for non-parametric data; p values less than 0.05 were considered significant.
Results
MMP-8 dampens neutrophilic inflammation during LPS-mediated ALI in mice
We compared neutrophilic lung inflammation in WT and MMP-8−/− mice that were generated initially in the mixed SvEv129 X C57BL/6J strain 4 h to 7 d after IT LPS or IT PBS. SvEv129 X C57BL/6J strain WT and MMP-8−/− mice given IT PBS had no PMNs in their BAL samples (not shown). However, PMN counts in BAL samples were significantly higher in MMP-8−/− mice than WT control mice 24 h after IT LPS, and remained higher for 72 h after IT LPS (Fig. 1A). Neutrophilic lung inflammation resolved in both genotypes ~7 d after IT LPS. To provide assurance that the increased PMN counts in the lungs of LPS-treated MMP-8−/− mice were due to the lack of MMP-8 in these mice rather than to minor differences in the genetic background between MMP-8−/− vs. WT mice in this mixed genetic background, we backcrossed the mice into the pure C57BL/6J background (to the F10 generation) and compared lung inflammatory responses to LPS of C57BL/6J MMP-8−/− mice versus C57BL/6J WT littermate control mice. PMN counts were also significantly higher in BAL samples (Fig. 1B) and in lung sections (Figs. 1C & 1D) from C57BL/6J MMP-8−/− mice compared to C57BL/6J WT littermate control mice 24–72 h after IT LPS. These data confirm that MMP-8 has anti-inflammatory activities during LPS-mediated ALI in mice. In subsequent experiments, we studied WT and MMP-8−/− mice only in the pure C57BL/6J background.
Figure 1. MMP-8−/− mice have increased lung PMNs during LPS-mediated ALI.

In A, WT and MMP-8−/− mice both in a mixed SvEv129 X C57BL/6J strain were given IT LPS (10 μg) or IT PBS. BAL was performed 4 h to 7 d later and PMN counts were determined. Data are mean ± SEM; n = 5–9 mice/group. Asterisk indicates p =0.015 and ** indicates p = 0.003. In B, to provide assurance that MMP-8 reduces acute lung inflammation in mice, PMN counts in BAL samples were quantified 24 h and 72 h after IT LPS (n = 8–14 mice/group) or IT PBS (4–8 mice/group) in MMP-8−/− mice backcrossed to the pure C57BL/6J background (F10 generation) or pure C57BL/6J WT littermate control mice. Data are mean ± SEM. Asterisk indicates p < 0.006 compared to mice of the same genotype given IT PBS at the same time point and ** indicates p ≤ 0.034. C shows representative lung sections from C57BL/6J WT littermate control mice (left panels) and C57BL/6J MMP-8−/− mice (right panels) 24 h after IT PBS (top panels) or IT LPS (bottom panels) immunostained for a PMN marker (Ly-6G). Arrows indicate Ly-6G-positive PMN. Magnification X 200. In D, Ly-6G-positive PMN were counted in eight consecutive 1000 X magnification fields in lung sections harvested from C57BL/6J WT littermate control mice or C57BL/6J MMP-8−/− mice 24 h after IT PBS (n = 4 mice/group) or IT LPS (n = 6–7 mice/group). Data are mean ± SEM. Asterisk indicates p ≤ 0.01 and ** indicates p = 0.047. No staining was observed in lung sections from WT or MMP-8−/− mice immunostained with an isotope-matched non-immune control antibody (not shown).
MMP-8 reduces lung injury after IT instillation of LPS in mice
LPS-induced lung injury was greater in MMP-8−/− than in WT mice, since lungs from LPS-treated MMP-8−/− mice had more extensive and severe lung inflammation and hemorrhage (Fig. 2A), and higher hemoglobin levels in BAL samples (Fig. 2B) than LPS-treated WT mice confirming greater leakage of erythrocytes from the vasculature into the alveolar space in the absence of MMP-8. MMP-8−/− mice also had higher wet/dry lung weight ratios than WT mice 24 h after IT LPS (Fig. 2C) which persisted for 72 h after IT LPS (wet/dry lung weight ratios after 72 h were 5.248 ± SEM 0.062 versus 5.024 ± 0.068 for LPS-treated MMP-8−/− mice versus LPS-treated WT mice; n = 7–8 mice/group; p = 0.032). IT instillation of LPS significantly increased lung elastance (H, a measure of lung stiffness) in both genotypes when compared to untreated mice, but LPS-treated MMP-8−/− mice had greater increases in lung elastance than LPS-treated WT mice (Fig. 2D). Thus, MMP-8 also reduces physiologically significant lung injury after IT instillation of LPS in mice.
Figure 2. MMP-8−/− mice have greater LPS-induced lung injury than WT mice.

In A, C57BL6J WT mice and C57BL/6J MMP-8−/− mice were given 10 μg of LPS by the IT route and 24 h later lungs were removed. Note the more extensive inflammation and hemorrhage in the lungs of the MMP-8−/− mouse (right) given IT LPS than in those from the LPS-treated WT mouse (left). Lungs from WT and MMP-8−/− mice given IT PBS had no obvious lung inflammation or hemorrhage (not shown). In B, hemoglobin levels were measured in BAL samples from WT and MMP-8−/− mice 24 h after IT PBS or LPS. Data are mean ± SEM; n = 3 for groups given IT PBS and 13 for groups given IT LPS. In C, wet/dry lung weight ratios were measured in WT C57BL/6J littermate control mice, C56BL/6J MIP-1α−/−, C57BL/6J MMP-8−/−, and C56BL/6J MMP-8−/− X MIP-1α−/− mice 24 h after IT LPS (n = 11–22 mice/group) or IT PBS (n = 5–9 mice/group). Data are mean ± SEM. Asterisk indicates p <0.001; ** indicates p < 0.03; and *** indicates p < 0.05. NS indicates not significantly different. In D, WT or MMP-8−/− mice (C57BL/6J strain) were given two doses of LPS (20 μg) by the IT route 24 h apart, and 24 h after the last dose of LPS, lung elastance (H which is a measure of lung stiffness) was measured in LPS-treated mice vs. unchallenged mice using a Flexivent™ apparatus. Data are mean ± SEM; n = 6–9 mice per group. Asterisk indicates p < 0.05 compared with unchallenged mice of the same genotype and ** indicates p = 0.017.
MMP-8 reduces lung inflammation and injury in other models of ALI in mice
To determine whether MMP-8 regulates acute lung inflammation and injury in other models of acute lung injury, we assessed WT vs. MMP-8−/− mice in two other models of ALI including one that induces robust lung inflammation in WT mice (IT instillation of bleomycin) and one that induces robust injury to the alveolar capillary barrier and is associated with mortality in WT mice (hyperoxia). IT instillation of bleomycin induced an inflammatory response (consisting of PMNs, macrophages, and lymphocytes) in the lungs of WT and MMP-8−/− mice, and this inflammatory response peaked in both genotypes 7 days after IT bleomycin was delivered. However, total BAL WBC counts were significantly greater in MMP-8−/− mice than WT mice 7 days after IT bleomycin (Fig. 3A), but differential WBC counts did not differ between the genotypes (not shown). There was also a trend towards higher BAL leukocytes in MMP-8−/− mice 10 days after IT bleomycin (not shown).
Figure 3. MMP-8−/− mice have greater lung inflammation and injury in other models of ALI.

In A, WT, MMP-8−/−, MIP-1α−/−, and MMP-8−/− X MIP-1α−/− mice (all in the pure C57BL/6 strain) were given 75 mU bleomycin (n = 11–18 mice/group) or saline (n = 4–6 mice/group) by the IT route. After 7 days, WBC were counted in BAL samples. Data are mean ± SEM. Asterisk indicates p ≤ 0.027 and **, p < 0.001. In B, WT (n =15), MMP-8−/− (n = 13), MIP-1α−/− (n =9), and MMP-8−/− X MIP-1α−/− (n= 13) mice (all in the pure C57BL/6 strain) were exposed to 95% 02 for up to 96 h and survival was recorded. Asterisk indicates p < 0.001 compared to WT mice at 96 h. In C, the same genotypes described in B were exposed for 72 h to 95% 02 (n = 7–13 mice/group) or 21% 02 (n = 4–9 mice/group) and wet to dry lung weight ratios were measured. Data are mean ± SEM. Asterisk indicates p< 0.001 compared to the same genotype exposed to 21% O2 and ** indicates p ≤ 0.007.
We also subjected WT and MMP-8−/− mice to hyperoxia (95% O2 for up to 96 h) and found that mortality was significantly greater in MMP-8−/− mice with hyperoxia-induced ALI than WT mice exposed to hyperoxia (Fig. 3B). After being exposed to hyperoxia for 96 h, 100% of MMP-8−/− mice died compared to only 13% of WT mice (p< 0.001). In addition, lung injury (as assessed by wet to dry lung weight ratios) was also significantly greater in MMP-8−/− mice compared to WT mice with hyperoxic lung injury (Fig. 3C). These data confirm that MMP-8 limits lung inflammation and injury (and mortality) in several murine models of ALI.
MMP-8 reduces lung PMN chemotactic activity during LPS-mediated ALI in mice, but has no effect on peripheral blood PMN counts, PMN migration, or PMN apoptosis in the lung
Total WBC counts and PMN, lymphocyte, and monocyte counts in peripheral blood were similar in unchallenged WT and MMP-8−/− mice (Table 1). In addition, 6, 24 and 72 h after IT instillation of LPS there were no significant differences in total WBC counts or in absolute numbers of PMNs, lymphocytes, or monocytes in peripheral blood between WT and MMP-8−/− mice (Table 2). These data suggest that MMP-8 does not regulate bone marrow production or release of PMNs or other leukocytes.
Table 1.
WT and MMP-8−/− mice have similar total WBC counts in peripheral blood at baseline and after IT LPS.
| Treatment | WT Total WBC (millions/ml) | MMP-8−/− Total WBC (millions/ml) | WT vs. MMP-8−/− Mice |
|---|---|---|---|
| unchallanged | 5.89 (0.25) | 5.55 (0.29) | p = 0.416 |
| IT LPS, 6 h | 6.11 (1.40) | 3.74 (0.51) | p = 0.138 |
| IT LPS, 24 h | 2.51 (0.59) | 2.42 (0.46) | p = 0.912 |
| IT LPS 72 h | 4.84 (0.58) | 3.36 (0.60) | p = 0.103 |
Blood was drawn from the right ventricles of unchallenged WT and MMP-8−/− mice (n = 4 mice per genotype) and from WT and MMP-8−/− mice 6 h, 24, and 72h after IT LPS (n = 7–12). Total leukocyte counts were determined on the blood samples.
Data are mean (SEM).
WT and MMP-8−/− mice did not differ significantly in total WBC counts in blood either at baseline or at any time point studied after IT LPS.
Table 2.
WT and MMP-8−/− mice do not differ in PMN, lymphocyte, or monocytes counts in peripheral blood
| WT Millions/ml | MMP-8−/− Millions/ml | |||||
|---|---|---|---|---|---|---|
| PMN | Lymph | Mono | PMN | Lymph | Mono | |
| unchallenged | 1.32 (0.15) | 4.51 (0.18) | 0.22 (0.04) | 1.41 (0.05) | 3.98 (0.23) | 0.22 (0.06) |
| IT LPS, 6 h | 3.50 (0.76) | 1.66 (0.54) | 0.09 (0.04) | 2.11 (0.39) | 1.44 (0.32) | 0.173 (0.04) |
| IT LPS, 24 h | 1.32 (0.23) | 1.13 (0.44) | 0.06 (0.03) | 1.41 (0.37) | 0.76 (0.16) | 0.02 (0.01) |
| IT LPS 72 h | 1.88 (0.38) | 2.59 (0.24) | 0.20 (0.04) | 1.29 (0.28) | 2.13 (0.46) | 0.15 (0.03) |
Differential leukocyte counts were determined on the blood samples from unchallenged (n=4 mice/group) vs. LPS-treated WT and MMP-8−/− mice (n = 7–12 mice/group) studied in Table 1 and absolute leukocyte subset counts in blood were determined from the total and differential leukocyte counts.
Data are mean values (SEM).
There were no significant differences in PMN, lymphocyte, or monocytes counts in peripheral blood from WT or MMP-8−/− mice at any time point tested.
To test whether MMP-8 regulates PMN migration in response to chemoattractants, we tested the capacity of WT vs. MMP-8−/− PMNs to migrate using Boyden chamber assays. WT and MMP-8−/− PMNs had similar capacity to undergo random migration when buffer alone was tested in the lower chambers (Table 3). Keratinocyte-derived chemokine (KC), fMLP, and zymosan-activated serum [ZAS, as a source of C5a [35]] induced similar 2–3 fold increased in the migration of WT and MMP-8−/− PMNs indicating that MMP-8 is not required for PMNs to migrate in response to chemoattractants. Next, we assessed whether MMP-8 increases PMN apoptosis or necrosis during ALI in mice. LPS-treated MMP-8−/− mice have markedly increased PMN counts in lung sections compared to WT mice (Fig. 1C and 1D) which would hinder direct quantitative comparisons of rates of PMN apoptosis and necrosis in lung sections from the mice. To quantify rates of PMN apoptosis and necrosis in the lungs of LPS-treated WT vs. LPS-treated MMP-8−/− mice, we used BAL to harvest lung inflammatory cells 24 h after instilling LPS. Without further purifying or culturing the lung inflammatory cells (which are 90–95% PMNs in both genotype), we stained equal numbers of cells ex vivo with annexin V-Alexa 488 and propidium iodide and also measured intracellular levels of active caspase 3. Lung PMNs from LPS-treated WT and MMP-8−/− mice had similar low percentages of BAL PMNs staining positively with annexin-V-Alexa 488 and propidium iodide (Table 4), and contained similar low levels of active caspase-3 (Table 5). The low percentages of apoptotic and necrotic cells in the lungs of mice with LPS-mediated ALI likely reflects the fact that PMNs undergoing apoptosis in the lung are rapidly cleared by macrophages present in the alveolar spaces.
Table 3.
MMP-8 does not regulate PMN migration in vitro
| Buffer | fMLP | KC | ZAS | |
|---|---|---|---|---|
| WT PMNG | 25.6 (2.1)H | 70.4 (3.4) | 64.6 (6.9) | 65.5 (5.1) |
| MMP-8−/− PMN | 29.0 (4.7) | 65.4 (5.7) | 72.2 (16.3) | 70.9 (15.9) |
PMNs isolated from the bone marrow of WT and MMP-8−/− mice were resuspended at 4 × 106/ml in RPMI-HEPES containing 1% albumin and incubated for 4 h at 37°C through in Boyden microchemotaxis assay chambers [28] using buffer alone, 10−6M fMLP, 10−7M KC, or 10% zymosan-activated serum [ZAS as a source of C5a [35]] in 6 replicate lower wells for each experimental condition. PMN that migrated through the pores in the membranes were counted in 6–8 high magnification fields for each well on Diff Quik™-stained membranes.
Data are mean ± SEM number of cells migrating (n = 3 separate experiments). There were no significant differences in the migratory responses of WT vs. MMP-8−/− PMNs to buffer or any of the chemoattractants tested.
Table 4.
PMNs from the lungs of LPS-treated WT and MMP-8−/− mice have similar rates of apoptosis and necrosis
| Condition | Annexin-V % Positive | Propidium Iodide % Positive |
|---|---|---|
| WT, IT LPSA | 1.8 (0.5)B | 4.1 (0.8) |
| MMP-8−/−, IT LPS | 4.2 (0.5) | 3.2 (0.5) |
WBC (~95% PMNs) were isolated from the lungs of WT and MMP-8−/− mice (both in the C57BL/6 strain) by BAL 24 h after IT LPS. Cells were stained with annexin-V and propodium iodide and the percentage of positive cells was determined.
Data are mean ± SD; n = 4–8 mice per group.
Table 5.
Lung PMNs from LPS-treated WT and MMP-8−/− mice have similar levels of intracellular active caspase-3.
| Condition | Intracellular active caspase-3 (pg) |
|---|---|
| WT BAL PMN, IT LPSC | 444.7 (50.7)E |
| MMP-8−/− BAL PMN, IT LPS | 407.7 (58.6) |
| Human PMN, T= 0 hD | N.D.F |
| Human PMN, T=18 h | 17,095 (1690) |
PMNs were isolated from the lungs of WT and MMP-8−/− mice, both in the C57BL/6 strain, by BAL 24 h after IT instillation of LPS (n=7 mice/group). Extracts of BAL PMNs were prepared at 5 × 106/ml and assayed for levels of active caspase-3 using a fluorogenic substrate specific for caspase-3 and assay standards of purified active caspase-3.
As controls, human PMNs were induced to undergo apoptosis by culturing them in serum-free medium for 18 h at 37°C (Human PMNs T= 18 h), and active caspase 3 levels were quantified in extracts of these PMNs versus extracts of freshly isolated human PMN (Human PMN, T= 0 h) prepared at 5 × 106/ml.
Data are mean ± SD.
N.D. = not detected.
To confirm that MMP-8 does not regulate PMN survival, we harvested PMN from the bone marrow of WT and MMP-8−/− mice and induced them to undergo cell death in vitro by either activating: 1) the intrinsic apoptosis pathway (by incubating the cells at 37°C in the absence of agonists and serum for up to 18 h); or 2) the extrinsic apoptosis pathway by incubating cells with an anti-FAS (anti-CD95) antibody which ligates and thereby activates FAS death domain-containing receptors on the PMN surface. At interval thereafter, we quantified apoptosis and necrosis by measuring annexin V and propidium iodide staining and intracellular levels of active caspase 3. As expected, there was minimal apoptosis or necrosis in freshly isolated PMNs from both genotypes (Fig. 4A and 4B; no serum starvation). When WT PMNs were induced to undergo apoptosis by triggering either the intrinsic (Fig. 4) or extrinsic (Fig. 5) apoptosis pathways, we observed robust increases in rates of apoptosis and necrosis of WT PMNs as expected. However, WT and MMP-8−/− PMNs did not differ in the rates at which they underwent apoptosis or necrosis in vitro (Figs. 4 and 5). Together, these results indicate that it is unlikely that MMP-8 decreases lung PMN counts during LPS-mediated ALI by increasing rates of PMN apoptosis or necrosis in the murine lung.
Figure 4. MMP-8 does not regulate survival of PMNs induced to undergo apoptosis in vitro by serum starvation.

In A-C, PMNs were isolated from the bone marrow of WT and MMP-8−/− mice and aliquots of the cells were induced to undergo apoptosis by incubating the cells in the absence of serum or agonists to trigger the intrinsic apoptosis pathway. Freshly isolated cells (no serum starvation) or cells serum starved for 18 h were stained with annexin-V conjugated to Alexa 488 or propidium iodide (A and B) or active caspase 3 was measured in cell lysates as described in Methods (C). Representative FACS plots are shown in A and quantitative data for annexin V and propodium iodide staining in B. In B and C, data are mean ± SEM (n = 4 mice/group).
Figure 5. MMP-8 does not regulate survival of PMNs induced to undergo apoptosis in vitro by FAS (CD95) activation.

In A-C, PMNs were isolated from the bone marrow of WT and MMP-8−/− mice and aliquots of the cells were induced to undergo apoptosis by incubating the cells for 4 h at 37°C with equal amounts of anti-FAS (anti-CD95) to ligate and activate FAS death domain-containing receptors on PMNs. Cells were then stained with annexin-V conjugated to Alexa 488 or propidium iodide (A and B) or active caspase 3 was measured in cell lysates as described in Methods (C). Representative FACS plots are shown in A and quantitative data for annexin V and propodium iodide staining in B. In B and C, data are mean ± SEM (n = 4 mice/group).
BALF harvested from MMP-8−/− mice 4–24 h after IT LPS had more PMN chemotactic activity than BALF from WT mice (Fig. 6A) indicating that MMP-8 reduces lung PMN chemotactic activity during LPS-mediated ALI. Surprisingly, BALF from WT and MMP-8−/− mice did not differ in levels of traditional PMN chemokines including KC, MIP-2α, LIX, or leukotriene B4 (LTB4) either 4 h or 24 h after IT LPS (Supplemental Tables 1 and 2, respectively). BALF from LPS-treated WT and LPS-treated MMP-8−/− mice also did not differ in levels of other pro-inflammatory mediators that regulate endothelial cell activation, PMN transendothelial migration, or PMN survival (TNF-α, IL-1β, IL-6, G-CSF, and GM-CSF), or in levels of the anti-inflammatory cytokine, IL-10 (\Supplemental Tables 1 and 2). Surprisingly, BALF from MMP-8−/− mice contained 2-fold more MIP-1α than BALF from WT mice 4 h and 24 h after IT LPS (Fig. 6B), indicating that MMP-8 reduces lung MIP-1α levels during LPS-mediated ALI.
Figure 6.

MMP-8 reduces lung PMN chemokine activity and lung MIP-1α levels during LPS-mediated ALI in mice. In A, cell-free BALF samples from WT or MMP-8−/− mice 4 h or 24 h after IT PBS or IT LPS were tested for PMN chemotactic activity in triplicate using Boyden microchemotaxis assay chambers, along with buffer and 10−7M fMLP as controls (white bars). PMNs migrating through the membranes were counted in 6–10 consecutive 1000 X magnification fields for each well. Data are mean ± SEM. Asterisk indicates p ≤ 0.036 compared with BALF from the same genotype of mice given IT PBS at the same time point and ** indicates p ≤ 0.035. B. MIP-1α levels were measured in BALF samples from WT and MMP-8−/− mice 4–72 h after IT PBS (n = 5–11 mice/group) or IT LPS (9–23 mice/group) by ELISA. Data are mean ± SEM. Asterisk indicates p < 0.04 and ** indicates p = 0.032.
MIP-1α promotes the accumulation of PMN and acute lung injury after IT instillation of LPS in mice
MIP-1α (CCL3) is a CC chemokine that potently stimulates mononuclear cell migration. However, human PMNs constitutively express CCR1, a key receptor for MIP-1α and migrate in response to MIP-1α in vitro [36–39]. Murine PMN do not constitutively express CCR-1. However, during endotoxemia, the expression of CCRs including CCR-1 increases in peripheral blood PMNs which permits murine PMNs to become responsive to MIP-1α and other CC chemokines in vitro [40]. We first confirmed that 10−7M MIP-1α increases human PMN migration in vitro using Boyden chamber assays (Fig. 8D) and that a neutralizing antibody to MIP-1α blocks MIP-1α-mediated increases in PMN migration (Supplemental Table 3). Next, we assessed the role of MIP-1α in promoting lung inflammation and injury in mice by comparing lung PMN counts in WT versus MIP-1α−/− mice 24 h after IT LPS. LPS-treated MIP-1α−/− mice had ~40% fewer lung PMNs (Fig. 7A) and were protected from developing increases in wet-dry lung weight ratios (Fig. 2C) and increases in lung elastance (Fig. 7B) compared to LPS-treated WT mice. These data indicate that MIP-1α significantly increases LPS-mediated neutrophilic lung inflammation and lung injury in mice.
Figure 8. MMP-8 cleaves and inactivates MIP-1α in vitro.

Equal amounts of recombinant MIP-1α (2.5 μM in each lane) were incubated at 37°C for 18 h with and without 500 nM MMP-8 or 500 nM MMP-9 in A, or in B and C with or without (lane 1; No PMN) or with equal numbers (5 × 106 cells/assay) of activated and fixed human PMNs (human PMN), activated and fixed WT murine PMNs (WT PMN), or activated and fixed MMP-8−/− murine PMNs (MMP-8−/− PMN). In A-C, reduced cell-free reaction products were analyzed on silver-stained 16.5% Tris-tricine gels. Arrow indicates intact MIP-1α and arrowhead indicates a cleavage product of MIP-1α. In D, rMIP-1α (2.5 μM) was incubated at 37°C for 18 h with and without fixed, activated human PMNs, WT PMNs or MMP-8−/− PMNs as described above. Residual MIP-1α-mediated PMN chemotactic activity was tested in cell-free supernatants samples in triplicate using Boyden microchemotaxis assay chambers (along with buffer alone or 10−7M fMLP as assay controls). Data are mean ± SEM; n = 3–5 separate samples/group. Asterisk indicates p = 0.029 compared to buffer alone and ** indicates p < 0.037.
Figure 7. MIP-1α promotes LPS-mediated acute lung inflammation and injury and neutralization or genetic deletion of MIP-1α in MMP-8−/− mice reduces lung PMN counts in LPS-treated MMP-8−/− mice.

In A, C57BL6J WT mice and C57BL/6J MIP-1α−/− mice were given PBS (n = 4 mice/group) or 10 μg LPS (n = 12–15 mice/group) by the IT route and 24 h later PMN were counted in BAL samples. Data are mean ± SEM. Asterisk indicates p < 0.003 compared to mice of the same genotype given IT PBS and ** indicates p = 0.038. In B, WT, MIP-1α−/−, and MMP-8−/− X MIP-1α−/− mice were not treated or given two doses of LPS (20 μg) by the IT route 24 h apart and 24 h after the last dose of LPS lung elastance (H) was measured using a Flexivent™ apparatus. Data are mean ± SEM; n = 7–17 mice/group. Asterisk indicates p < 0.001. There was no statistically significant difference in H between unchallenged MIP-1α−/− mice and LPS-treated MIP-1α−/− mice, or between unchallenged MMP-8−/− X MIP-1α−/− mice and LPS-treated MMP-8−/− X MIP-1α−/− mice, or between LPS-treated MMP-8−/− X MIP-1α−/− mice and LPS-treated MIP-1α−/− mice. In C, WT, MIP-1α−/−, MMP-8−/−, and MMP-8−/− X MIP-1α−/− mice were given 10 μg of LPS or PBS by the IT route, and 24 h later PMNs were counted in BAL samples. Data are mean ± SEM. Asterisk indicates p = 0.047; ** indicates p = 0.04; and *** indicates p = 0.002; n = 12–26 mice/group. PMNs were not detected in BAL samples from any mice given IT PBS (not shown).
MIP-1α is the key substrate in the lung for MMP-8 during LPS-mediated ALI in mice
Genetic deletion of MIP-1α in MMP-8−/− mice decreased the lung inflammatory response to IT LPS (Fig. 7C) and IT bleomycin (Fig. 3A) when compared to measures of lung inflammation in MMP-8−/−. Genetic deletion of MIP-1α in MMP-8−/− mice reduced the increases in wet/dry lung weight ratios observed in MMP-8−/− mice with LPS-mediated ALI (Fig. 2C) and hyperoxia-induced ALI (Fig. 3C) and also reduced the increases in lung elastance in LPS-treated MMP-8−/− mice to similar levels to those observed in LPS-treated MIP-1α−/− mice in Fig. 7B. In addition, genetic deletion of MIP-1α in MMP-8−/− mice also impressively reduced the mortality that was associated with hyperoxia in MMP-8−/− mice (Fig. 3B). These data indicate that MMP-8 limits lung acute inflammation and injury in mice in a MIP-1α-dependent mechanism.
MMP-8 cleaves and inactivates MIP-1α in vitro
Steady state MIP-1α mRNA levels measured by real-time RT-PCR in lung homogenates 2 h after IT LPS increased to a similar extent in WT and MMP-8−/− mice compared with PBS-treated control mice of the same genotype (154-fold vs. 210 average fold increases in steady state MIP-1α mRNA levels relative to GAPDH levels for LPS-treated WT vs. LPS-treated MMP-8−/− mice, respectively; n = 5–6 mice/group). Thus, it is unlikely that MMP-8 regulates lung MIP-1α levels at the steady state mRNA level.
Next, we tested whether soluble MMP-8 and membrane-bound MMP-8 on activated PMNs can cleave and inactivate recombinant MIP-1α (rMIP-1α) in vitro. Soluble MMP-8 cleaved rhMIP-1α in vitro, generating a lower Mr cleavage product (arrowhead in Fig. 8A). Surprisingly, MMP-9 [the other MMP family member expressed by PMNs which has broader substrate specificity than MMP-8 [1]] had no activity against rhMIP-1α. Neutrophil elastase and cathepsin G, two serine proteinases expressed by PMN having a very broad spectrum of catalytic activity, also had no activity against MIP-1α (data not shown). The cleavage of rMIP-1α that was mediated by soluble MMP-8 was always incomplete even when high concentrations (500 nM) of soluble MMP-8 were incubated for up to 24 h with rMIP-1α. Purified murine MMP-8 (500 nM) also had only weak activity against rmMIP-1α (not shown).
Since activated human and murine PMNs express an active form of MMP-8 on their surface [2], next we tested the activity of membrane-bound MMP-8 against rhMIP-1α. We incubated rhMIP-1α with PMNs from humans and WT mice which had been optimally activated to induce surface MMP-8 expression, and then fixed to completely prevent PMN release of soluble MMP-8, MMP-9, and serine proteinases which can contribute to PMN extracellular proteolytic activity [2,31–34]. We have confirmed previously that our fixation process does not significantly reduce the activity of membrane-bound MMP-8 on PMNs [2]]. Membrane-bound MMP-8 on activated and fixed human and WT murine PMNs substantially or completely degraded rhMIP-1α in vitro (Figs. 8B and 8C), but an equal number of activated and fixed MMP-8−/− PMNs did not (Fig. 8C) despite the fact that MMP-8−/− PMN express similar amounts of MMP-9 [4] and serine proteinases (not shown) as WT PMN. Membrane-bound murine MMP-8 on PMNs also efficiently cleaved rmMIP-1α (not shown).
Recombinant MIP-1α incubated with membrane-bound MMP-8 on activated human or WT murine PMNs lost its activity to stimulate PMN migration in vitro, but rMIP-1α incubated with an equal number of activated MMP-8−/− PMNs fully retained its biologic activity (Fig. 8D). Thus, among proteinases expressed on the surface of activated PMNs, MMP-8 is the key enzyme contributing to the PMN surface-associated MIP-1α-degrading activity.
MMP-8 also reduces macrophage accumulation during LPS-mediated ALI
Since MIP-1-α is known to regulate mononuclear cell migration, we also quantified lung lymphocytes and macrophages in PBS-treated and LPS-treated WT vs. MMP-8−/− vs. MIP-1α−/− mice. BAL lymphocyte counts were low (< 104 lymphocytes per mouse in all three genotypes) and did not differ between WT, MMP-8−/−, and MIP-1α−/− mice at any time point assessed (from 4 h to 7 days) after IT LPS (data not shown). BAL macrophage counts increased modestly but significantly in MMP-8−/− mice 24 h after IT LPS compared to MMP-8−/− mice given IT PBS and WT mice given IT LPS (Fig. 9). However, BAL macrophage counts did not increase in LPS-treated WT mice or LPS-treated MIP-1α−/− mice compared to their PBS-treated controls at the 24 h time point. Seventy two hours after IT LPS, BAL macrophage counts increased impressively in both WT and MMP-8−/− mice compared to their PBS-treated controls but BAL macrophage counts did not increase in LPS-treated MIP-1α−/− mice (Fig. 9). Macrophage counts were also significantly higher in MMP-8−/− mice than in WT mice 72 h after IT LPS, but MMP-8−/− X MIP-1α−/− mice had lower BAL macrophage counts than MMP-8−/− mice 72 h after IT LPS (Fig. 9). Together, these data indicate that MMP-8-mediated cleavage of MIP-1α reduces macrophage as well as PMN accumulation during LPS-mediated ALI in mice.
Figure 9. MMP-8 reduces macrophage accumulation during LPS-mediated ALI.

WT, MIP-1α−/−, MMP-8−/−, and MMP-8−/− X MIP-1α−/− mice were given 10 μg of LPS (n = 7–25 mice) or PBS (n = 5–10 mice) by the IT route and after 24–72 h, macrophages were counted in BAL samples. Data are mean ± SEM. Asterisk indicates p < 0.016; ** indicates p ≤ 0.05; and *** indicates p = 0.012.
Membrane-bound MMP-8 is a major form of MMP-8 during ALI in mice
Since membrane-bound MMP-8 on activated PMN is a more potent MIP-1α degrading enzyme than soluble MMP-8, next we compared the levels of soluble, active MMP-8 in BALF versus membrane-bound MMP-8 on lung PMNs harvested from WT mice with LPS-mediated ALI. We performed BAL on WT mice (and MMP-8−/− mice as a control) 24 h after IT instillation of LPS and quantified soluble MMP-8 activity in BALF cell-free supernatant samples by its capacity to cleave FITC-conjugated type I collagen substrate (a good substrate for MMP-8) and by Western blotting to assess the forms of soluble MMP-8 present in the lung. We also immunostained BAL PMNs to assess whether they express membrane-bound MMP-8.
Soluble active MMP-8 was not detected in any of the cell-free BALF samples harvested 6 h, 24 h, or 72 h after IT LPS was delivered to WT mice as assessed by their complete lack of ability to cleave FITC-conjugated type I collagen ( n = 5–6 mice per time point; data not shown). Western blot analysis of BALF cell-free supernatant samples harvested 24 h after IT LPS from WT mice demonstrated latent proMMP-8 (~80 kDa; arrowhead) in all of the samples and a low Mr fragment of MMP-8 (~27 kDa; arrow) in three of the samples, but no forms of MMP-8 corresponding to the intact active enzyme (~60 kDa) in any of the BALF samples from WT mice (Supplemental Fig. 1A). Soluble active MMP-8 was also not detected by western blot analysis in BALF from WT mice 6 h or 72 h after IT LPS (n =5 mice per time point). The 27 kDa MMP-8 fragment that we detected in WT BALF samples 24 h after IT LPS has been identified previously when purified human proMMP-8 is activated in vitro [2,41,42]. It corresponds to the COOH terminal hemopexin domain of MMP-8 (which lacks enzyme activity) and is generated when the active site of MMP-8 cleaves MMP-8 at a locus between the catalytic domain and the hemopexin domain. Neither proMMP-8 nor the 27 kDa MMP-8 fragment was detected in BALF samples harvested 24 h after IT LPS from two MMP-8−/− mice as expected (Supplemental Fig. 1B; lanes 1 and 2).
Lung PMNs isolated from WT mice 6 h after IT LPS had minimal surface MMP-8 expression (Supplemental Fig. 2). However, 24 h after IT LPS lung PMNs expressed high levels of membrane-bound MMP-8, but MMP-8−/− lung PMNs did not, as expected (Fig. 10). High level expression of MMP-8 on the surface of lung PMNs persisted to 72 h after IT LPS (Supplemental Fig. 2). Since membrane-bound MMP-8 on activated PMNs is an active enzyme [Figs. 8B and 8C and [2]], these data suggest that membrane-bound MMP-8 on PMNs (rather than soluble MMP-8 freely released by activated PMNs) is likely to be the critically important active form of the proteinase mediating its anti-inflammatory activities in the murine lung.
Figure 10. Lung PMNs from WT mice given IT LPS express membrane-bound MMP-8.
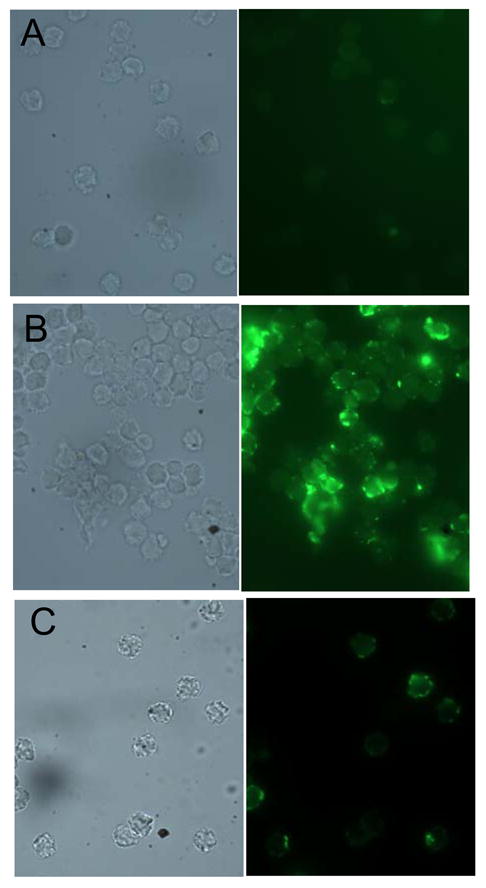
A WT mouse (A and B) and an MMP-8−/− mouse (C) were given 10 μg of LPS by the IT route, and 24 h later PMNs were isolated from their lungs by BAL. Cells were fixed and non-permeabilized cells immunostained with a rabbit anti-MMP-8 IgG (B and C) or a non-immune rabbit IgG (A) followed by goat anti-rabbit IgG conjugated to Alexa-488. Note the intense staining for MMP-8 on the surface of WT BAL PMNs stained with anti-MMP-8 (B) but minimal staining associated with WT PMNs stained with the control antibody (A) or with MMP-8−/− PMNs incubated with the anti-MMP-8 antibody (C).
Discussion
Acute lung injury is characterized by the rapid influx of PMNs into the lung and alveolar capillary barrier dysfunction. PMN-derived proteinases have enormous potential to regulate both of these key pathologic features of ALI because they proteolytically regulate the activities of pro-inflammatory mediators, degrade lung extracellular matrix proteins, and injure the cellular components of the alveolar capillary barrier in vitro [43]. Surprisingly, until now, PMN-derived proteinases (including neutrophil elastase and MMP-9) have not been shown to contribute to these processes in vivo [44,45]. Herein, we used the aseptic models of ALI (IT LPS, IT bleomycin, and hyperoxia) rather than in a sepsis model system to assess the roles of MMP-8 in these pathologies since: 1) the aseptic models that we chose causes robust acute lung inflammation and/or alveolar capillary barrier injury; and 2) other leukocyte-derived proteinases promote bacterial killing [46–49] but the roles of MMP-8 in host defense have not yet been assessed. Thus, the aseptic IT LPS model of ALI enabled us to quantify the contributions of MMP-8 to both of the key pathologic components of ALI in the absence of any potentially confounding effects of MMP-8 on bacterial killing.
Our results identify MMP-8 as a PMN-derived proteinase that plays a significant role in regulating lung inflammation, alveolar capillary barrier injury, and mortality during ALI in mice by cleaving and inactivating a CC chemokine. Moreover, we show that MIP-1α plays a surprisingly important role in increasing PMN (as well as macrophage) accumulation in the lung during ALI and in promoting injury to the alveolar capillary barrier. This is also the first report that membrane-bound MMP-8 is abundantly expressed on the surface of activated PMN in vivo. Our data indicate that membrane-bound MMP-8 contributes in important ways to its activities in the lung.
Roles for MMP-8 in regulating inflammatory processes
Our earlier study showed that MMP-8−/− mice in the mixed SVeV129 X C57Bl/6J strain had higher PMN counts in BAL samples than WT mice 24 h after IT LPS [2]. Herein, we back-crossed MMP-8−/− mice into the pure C57BL/6J strain and confirmed that pure C57BL/6J MMP-8−/− mice have more severe and prolonged lung inflammation and injury after IT LPS than C57BL/6J WT mice. We also confirmed that MMP-8 limits bleomycin-mediated ALI and hyperoxia-induced lung injury and mortality. The current study is the first report that MMP-8 cleaves a CC chemokine in vivo not only to reduce PMN accumulation in the lung, but also to decrease lung macrophage accumulation, and to limit injury during ALI in mice.
Previous studies have shown that MMP-8 regulates neutrophilic inflammation in different organ systems in mice by various mechanisms. MMP-8 promotes PMN accumulation in TNF-α-induced acute hepatitis in mice by releasing LIX from hepatic ECM stores in the liver [13]. However, we found no role for MMP-8 in releasing of LIX from ECM stores in the lung since LIX levels were similar in lung samples from LPS-challenged WT and MMP-8−/− mice. MMP-8 promotes early increases in PMNs in the skin and dorsal air pouches of mice injected with chemical carcinogens or LPS due to MMP-8 cleaving LIX to generate a form with increased biologic activity [4,12]. However, in our study, MMP-8 reduced rather than increased PMN chemotactic activity in lung samples 4–24 h after IT LPS. It is noteworthy that the levels of LIX in the lung after IT LPS are 5- to 20-fold lower than those of KC, MIP-2α, and MIP-1α, suggesting that, in contrast to acute inflammatory reactions in murine skin and liver, LIX likely plays only a minor role in regulating PMN accumulation in the murine lung. Interestingly, Balbin et al. also showed that, once skin inflammation is established in male MMP-8−/− mice in response to chemical carcinogens, it is abnormally increased, creating an environment that promotes skin tumor development [4]. Our data showing that MMP-8−/− mice have greater neutrophilic lung inflammation in the IT LPS model 24–72 h after IT LPS are consistent with the latter results of Balbin et al, but the mechanism by which MMP-8 reduces skin inflammation was not identified in the study of Balbin and coworkers.
MMP-8 reduces the number of airway PMNs and eosinophils in allergen-induced airway inflammation in mice by promoting granulocyte apoptosis by unknown mechanisms [17], but we found no role for MMP-8 in regulating PMN apoptosis in the lung in our model of ALI. The percentage of PMNs that were undergoing apoptosis in the lungs of LPS-treated WT and MMP-8−/− mice was very low. One possible explanation for the lack of an effect of MMP-8 in regulating PMN apoptosis in our study is that during LPS-mediated ALI in mice very high levels of G-CSF and GM-CSF are generated in the lung, and both of these mediators strongly promote PMN survival in the lung [50]. It is also noteworthy that MIP-1α levels were not measured in MMP-8−/− mice in either the allergen-induced airway inflammation or the skin chemical carcinogenesis models.
MIP-1α and PMN and macrophage accumulation in the lung
Our studies of MMP-8−/− X MIP-1α−/− mice during LPS-, bleomycin- and hyperoxia-mediated ALI identified MIP-1α as the key substrate in the lung that is degraded by MMP-8. However, genetic deletion of MIP-1α in MMP-8−/− mice did not eliminate PMN or macrophage accumulation in the lungs of LPS- or bleomycin-treated mice indicating that other chemokines also promote the accumulation of PMNs and macrophages in the lung during ALI, and that MMP-8 does not play a significant role in degrading these mediators. It is also noteworthy that MMP-8 is not required for resolution of LPS-induced neutrophilic lung inflammation or removal of MIP-1α from the lung since lung PMN counts and MIP-1α levels in LPS-treated MMP-8−/− mice returned to baseline after 7 days. Rather, MMP-8 reduces the intensity and shortens the duration of the lung inflammatory response to LPS. Anti-inflammatory pathways and molecules other than MMP-8 clearly promote clearance of MIP-1α from the lung and resolution of acute lung inflammation in MMP-8−/− mice. For example, after a single dose of LPS is delivered to the lungs of mice, LPS is cleared from the lung leading to reduced LPS-mediated induction of chemokine expression by lung macrophages and epithelial cells. In addition, PMNs undergo apoptosis and are then cleared by lung macrophages. Lipid mediators (including lipoxins, resolvins, and protectins) are also generated in the inflamed lung that actively promote resolution of inflammation [51–54]. MIP-1α is likely removed from the lung by mechanisms other than MMP-8-mediated cleavage. For example, unidentified proteinase(s) other than MMP-8 produced by cells other than PMNs (macrophages and epithelial cells) may degrade MIP-1α in MMP-8−/− mice since macrophage-derived MMPs (MMPs-12 -2, and -3) have been shown to degrade CC chemokines [55]. Pro-inflammatory mediators can be removed from the extracellular space by binding to functional decoy receptors on myeloid cells [56] or by undergoing receptor- or clathrin-mediated endocytosis by PMNs followed by intracellular degradation [57,58]. Also, RBCs function as a sink for chemokines, binding them and removing them from the extracellular space and RBCs are cleared themselves by phagocytes in the liver and spleen [59]. Exudate fluid generated in the injured lung (containing chemokines) is cleared from the lung by passive diffusional pathways including the pulmonary lymphatics [60–62].
MIP-1α is expressed by macrophages and also by PMNs activated with LPS and other pro-inflammatory mediators [63], and it binds to CCR-1 and CCR-5 [64,65] with CCR-1 serving as the most important receptor during inflammatory responses in mice [66]. While MIP-1α is a well-established chemokine for mononuclear cells, there is increasing evidence that it also regulates PMN function in humans and mice. MIP-1α binds to CCR-1 that is constitutively expressed by human PMNs to stimulate PMN degranulation and migration [37,64,65]. Pro-inflammatory mediators generated during LPS-mediated ALI up-regulate human PMN levels of CCR-1 and increase PMN migratory responses to MIP-1α [38,39]. While murine PMN do not constitutively express CCR-1, during endotoxemia in mice, the expression of CCR-1 and -5 increases in peripheral blood PMNs, and this permits PMNs to become more responsive to MIP-1α and other CC chemokines in vitro [40]. Adenovirus-mediated over-expression of MIP-1α in the murine lung also promotes the accumulation of PMNs as well as NK T cells in the lung during Klebsiella pneumoniae pneumonia [36]. Neutralization of MIP-1α in mice with endotoxemia also reduces early neutrophilic inflammation in the murine lung [67]. Our studies of MIP-1α−/− mice confirm that this chemokine plays a key role in promoting PMN accumulation in the lung during LPS-mediated ALI. MIP-1α could promote the accumulation of PMNs and macrophages in the murine lung during LPS-mediated ALI by binding to CCR1 on these cells to stimulate their migration [65] and/or by increasing the expression of adhesion molecules on endothelial cells, PMNs, and monocytes to increase leukocyte adhesion to endothelial cells and transendothelial migration [66–68]. Hsieh et al [69] recently reported that MIP-1α−/− mice also had a reduced systemic inflammatory response and remote organ injury (including lung injury) secondary to trauma and hemorrhage which was associated with lower serum levels of TNF-α, IL-6, IL-10 and MCP-1 compared to WT mice after injury [69] indicating that MIP-1α is a key chemokine driving systemic inflammatory responses to injury. However, it is likely that MIP-1α−/− mice also have a hypo-inflammatory state during LPS- and bleomycin-mediated ALI since we showed that MIP-1α−/− mice are protected from lung inflammation and injury in these models of direct injury to the lung. Detailed studies of MIP-1α−/− mice and signaling pathways leading to their hypo-inflammatory state in models of direct lung injury are beyond the scope of this paper, but will be the focus of future studies in our laboratory. Nevertheless, our data highlight the important (and unexpectedly) critical role of MIP-1α in promoting lung inflammation and injury in response to direct injury to the lung. Whatever the mechanism involved, it is likely that during LPS-mediated ALI, pro-inflammatory mediators generated in the lung increase MIP-1α synthesis by PMNs and macrophages and also increase the expression of MIP-1α receptors on PMNs and monocytes which may initially amplify recruitment of these cells into the lung. However, by stimulating degranulation of PMNs recruited into the lung, pro-inflammatory mediators also increase the release of MMP-8 by PMNs and translocation of MMP-8 to the PMN surface [2]. By degrading MIP-1α in the lung, these forms of MMP-8 may serve as a negative feedback loop that restrains lung inflammation and protects the alveolar capillary barrier from injury. By reducing the accumulation of PMNs in the lung, MMP-8 may have protective effects since PMNs release oxidants and cationic proteins that can injure the alveolar capillary barrier [43,70]. However, in ALI secondary to pneumonia, MMP-8-mediated reductions in lung PMN and macrophage counts may lead to worse outcomes by reducing phagocyte-mediated bacterial killing.
MIP-1α and lung injury
In addition to reducing PMN accumulation, MMP-8 also protects the lung against physiologically significant alveolar capillary barrier injury during LPS-mediated ALI as assessed by measuring leakage of erythrocytes from the pulmonary vasculature into the alveolar space, lung water content, and lung elastance. MMP-8−/− mice also had worse lung injury and higher mortality than WT mice during hyperoxia-induced ALI which leads to more robust alveolar capillary barrier injury than does LPS-mediated ALI [71]. Recently, MMP-8−/− mice were found to be protected from increased lung capillary permeability in a model of ventilator-induced ALI, but the mechanisms involved were not elucidated [72]. MMP-8 may protect the lung from LPS-induce alveolar capillary barrier injury also by cleaving and inactivating MIP-1α since: 1) MIP-1α−/− mice were protected from LPS- and hperoxia-induced ALI (Figs. 2C, 3C and 7B); 2) genetic deletion of MIP-1α in MMP-8−/− mice limited LPS-and hyperoxia-induced lung injury (Figs. 2C, 3C, and 7B); and 3) delivering a neutralizing antibody to MIP-1α reduced lung capillary leak in rats with immune complex-mediated ALI [73] and in mice with endotoxemia [67]. The mechanism by which MIP-1α increases ALI is not clear. Although MIP-1α increases lung levels of TNF-α in rats [73], and TNF-α increases endothelial permeability by destabilizing endothelial microtubules [74], this mechanism is unlikely in our murine model since TNF-α levels were similar in LPS-challenged WT and MMP-8−/− mice. MIP-1α may increase the levels of other mediators that promote ALI in mice or MIP-1α may increase ALI by increasing lung PMN counts, leading to injury to the alveolar capillary barrier by PMNs releasing toxic oxidants and cationic proteins [43,70].
Membrane-bound MMP-8 in ALI in mice
There have been no prior studies of membrane-bound MMP-8 in vivo. In the current study, we found high levels of MMP-8 expressed on the surface of PMNs recruited to the lungs of WT mice which persisted to at least 72 h after IT LPS. However, we did not detect any soluble active MMP-8 in BALF samples from LPS-treated WT mice either by using FITC-conjugated type I collagen as a substrate for active MMP-8 or by Western blot analysis. The lack of soluble active MMP-8 in BALF from LPS-treated WT mice is likely due (in part) to TIMP family members rapidly inactivating MMP-8 freely released by PMNs, followed by rapid clearance of MMP-8-TIMP complexes from the lung especially since TIMP levels are up-regulated in the lung during ALI [75]. Also, soluble MMP-8 has a short half life after it has been activated in vitro [2,41,42]. This is due to the high content of proline residues in the hinge region of the enzyme located between the catalytic domain and the hemopexin domain at the COOH terminus. The proline-rich hinge region of MMP-8 is readily cleaved by the active site of MMP-8 generating two fragments: the catalytic domain (~40 kDa) and the hemopexin domain (~27 kDa) with loss of the enzyme’s activity [2,41,42]. It is noteworthy in this respect that our Western blot analysis of cell-free BALF samples from LPS-treated WT mice detected inactive proMMP-8 along a ~27 kDa fragment which is likely to be the hemopexin domain of MMP-8 which lacks catalytic activity [2,41,42]. We did not detect the ~40 kDa catalytic domain fragment which may be due to its rapid clearance from the lung after binding to TIMPs [2,41,42]. Until now, fragmentation and inactivation of soluble MMP-8 has only been reported in vitro, but our findings indicate that this process also occurs during inflammatory processes in vivo. Not only is membrane-bound MMP-8 the more abundant form of MMP-8 during ALI in mice, but it is more potent than soluble MMP-8 at degrading MIP-1α in vitro even in TIMP-free buffers, which may be due to the greater catalytic stability of membrane-bound MMP-8 compared to soluble MMP-8 [2]. Furthermore, unlike soluble MMP-8, membrane-bound MMP-8 is also resistant to inhibition by TIMPs [2]. Thus, we hypothesize that membrane-bound MMP-8 expressed on activated PMNs is the key bioactive form of the MMP-8 that restrains lung inflammation and limits injury to the alveolar capillary barrier during ALI in mice.
Conclusions
Our results identify MMP-8 as the first PMN-derived proteinase that regulates lung inflammation and injury during ALI in mice. An previous study showed that MMP-8 cleaves CCL2 in vitro with very low catalytic efficiency [55,76], but membrane-bound MMP-8 was not tested against this chemokine in vitro. Herein, we have identified MIP-1α as a CC chemokine substrate that is degraded by MMP-8 with important consequences in vivo. MMP-8-mediated cleavage of MIP-1α surprisingly restrains LPS-induced neutrophilic (as well as macrophage-type) inflammation in the murine lung and reduces physiologically significant alveolar capillary barrier injury during ALI in mice. MMP-8 also significantly limits mortality associated with hyperoxia-induced ALI in mice. Furthermore, we show for the first time that membrane-bound MMP-8 on activated PMNs is the most abundant (and persistent) active form of MMP-8 in the lung during ALI in mice and is likely to be the critical bioactive form of the enzyme mediating its anti-inflammatory activities in the murine lung. Future studies will investigate whether MIP-1α is an important substrate for MMP-8 in other models of inflammation in the lung and other organs. In the future, we will also assess the roles of MMP-8 in murine models of sepsis. However, our data suggest that new treatment strategies designed to increase or prolong MMP-8 activity (especially on the PMN surface) may help to limit injury to the alveolar capillary barrier in patients with ALI, and thereby reduce the morbidity and mortality associated with this syndrome.
Supplementary Material
Acknowledgments
The authors thank Drs. Steven D. Shapiro, M.D. and Carlos Lopez-Otin, Ph.D. for providing the MMP-8−/− mice. We also thank Augustine M. Choi, M.D., Ph.D. for critically reading the manuscript.
Footnotes
Disclosures
None of the authors have conflicts of interest or financial interests to disclose.
Abbreviations used: ALI, acute lung injury; BAL, bronchoalveolar lavage; BALF, bronchoalveolar lavage fluid; IT, intratracheal; MMP, matrix metalloproteinase; TIMP, tissue inhibitor of metalloproteinases.
References
- 1.Owen CA, Campbell EJ. The cell biology of leukocyte-mediated proteolysis. J Leukoc Biol. 1999;65:137–150. doi: 10.1002/jlb.65.2.137. [DOI] [PubMed] [Google Scholar]
- 2.Owen CA, Hu Z, Lopez-Otin C, Shapiro SD. Membrane-bound matrix metalloproteinase-8 on activated polymorphonuclear cells is a potent, tissue inhibitor of metalloproteinase-resistant collagenase and serpinase. J Immunol. 2004;172:7791–7803. doi: 10.4049/jimmunol.172.12.7791. [DOI] [PubMed] [Google Scholar]
- 3.Balbin M, Fueyo A, Knauper V, Pendas AM, Lopez JM, Jimenez MG, Murphy G, Lopez-Otin C. Collagenase 2 (MMP-8) expression in murine tissue-remodeling processes. Analysis of its potential role in postpartum involution of the uterus. J Biol Chem. 1998;273:23959–23968. doi: 10.1074/jbc.273.37.23959. [DOI] [PubMed] [Google Scholar]
- 4.Balbin M, Fueyo A, Tester AM, Pendas AM, Pitiot AS, Astudillo A, Overall CM, Shapiro SD, Lopez-Otin C. Loss of collagenase-2 confers increased skin tumor susceptibility to male mice. Nat Genet. 2003;35:252–257. doi: 10.1038/ng1249. [DOI] [PubMed] [Google Scholar]
- 5.Cowland JB, Borregaard N. The individual regulation of granule protein mRNA levels during neutrophil maturation explains the heterogeneity of neutrophil granules. J Leukoc Biol. 1999;66:989–995. doi: 10.1002/jlb.66.6.989. [DOI] [PubMed] [Google Scholar]
- 6.Murphy G, Reynolds JJ, Bretz U, Baggiolini M. Collagenase is a component of the specific granules of human neutrophil leukocytes. Biochem J. 1977;162:195–197. doi: 10.1042/bj1620195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hasty KA, Hibbs MS, Kang AH, Mainardi CL. Secreted forms of human neutrophil collagenase. J Biol Chem. 1986;261:5645–5650. [PubMed] [Google Scholar]
- 8.Knauper V, Kramer S, Reinke H, Tschesche H. Characterization and activation of procollagenase from human polymorphonuclear leukocytes. Eur J Biochem. 1990;189:295–300. doi: 10.1111/j.1432-1033.1990.tb15489.x. [DOI] [PubMed] [Google Scholar]
- 9.Gruber BL, Schwartz LB, Ramamurthy NS, Irani AM, Marchese MJ. Activation of latent rheumatoid synovial collagenase by human mast cell tryptase. J Immunol. 1988;140:3936–3942. [PubMed] [Google Scholar]
- 10.Knauper V, Wilhelm SM, Seperack PK, DeClerck YA, Langley KE, Osthues A, Tschesche H. Direct activation of human neutrophil procollagenase by recombinant stromelysin. Biochem J. 1993;295:581–586. doi: 10.1042/bj2950581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Diekmann O, Tschesche H. Degradation of kinins, angiotensins and substance P by polymorphonuclear matrix metalloproteinases MMP 8 and MMP 9. BrazilianJ Med Biol Res. 1994;27:1865–1876. [PubMed] [Google Scholar]
- 12.Tester AM, Cox JH, Connor AR, Starr AE, Dean RA, Puente XS, Lopez-Otin C, Overall CM. LPS responsiveness and neutrophil chemotaxis in vivo require PMN MMP-8 activity. PLoS ONE. 2007;2:e312. doi: 10.1371/journal.pone.0000312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.van Lint P, Wielockx B, Puimege L, Noel A, Lopez-Otin C, Libert C. Resistance of collagenase-2 (matrix metalloproteinase-8)-deficient mice to TNF-induced lethal hepatitis. J Immunol. 2005;175:7642–7649. doi: 10.4049/jimmunol.175.11.7642. [DOI] [PubMed] [Google Scholar]
- 14.Gaggar A, Jackson PL, Noerager BD, O’Reilly PJ, McQuaid DB, Rowe SM, Clancy JP, Blalock JE. A novel proteolytic cascade generates an extracellular matrix-derived chemoattractant in chronic neutrophilic inflammation. J Immunol. 2008;180:5662–5669. doi: 10.4049/jimmunol.180.8.5662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lin M, Jackson P, Tester AM, Diaconu E, Overall CM, Blalock JE, Pearlman E. Matrix metalloproteinase-8 facilitates neutrophil migration through the corneal stromal matrix by collagen degradation and production of the chemotactic peptide Pro-Gly-Pro. Am J Pathol. 2008;173:144–153. doi: 10.2353/ajpath.2008.080081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Weathington NM, van Houwelingen AH, Noerager BD, Jackson PL, Kraneveld AD, Galin FS, Folkerts G, Nijkamp FP, Blalock JE. A novel peptide CXCR ligand derived from extracellular matrix degradation during airway inflammation. Nat Med. 2006;12:317–323. doi: 10.1038/nm1361. [DOI] [PubMed] [Google Scholar]
- 17.Gueders MM, Balbin M, Rocks N, Foidart JM, Gosset P, Louis R, Shapiro S, Lopez-Otin C, Noel A, Cataldo DD. Matrix metalloproteinase-8 deficiency promotes granulocytic allergen-induced airway inflammation. J Immunol. 2005;175:2589–2597. doi: 10.4049/jimmunol.175.4.2589. [DOI] [PubMed] [Google Scholar]
- 18.Gutierrez-Fernandez A, Inada M, Balbin M, Fueyo A, Pitiot AS, Astudillo A, Hirose K, Hirata M, Shapiro SD, Noel A, Werb Z, Krane SM, Lopez-Otin C, Puente XS. Increased inflammation delays wound healing in mice deficient in collagenase-2 (MMP-8) FASEB J. 2007;21:2580–2591. doi: 10.1096/fj.06-7860com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cederqvist K, Sorsa T, Tervahartiala T, Maisi P, Reunanen K, Lassus P, Andersson S. Matrix metalloproteinases-2, -8, and -9 and TIMP-2 in tracheal aspirates from preterm infants with respiratory distress. Pediatrics. 2001;108:686–692. doi: 10.1542/peds.108.3.686. [DOI] [PubMed] [Google Scholar]
- 20.Weiland JE, Davis WB, Holter JF, Mohammed JR, Dorinsky PM, Gadek JE. Lung neutrophils in the adult respiratory distress syndrome. Am Rev Respir Dis. 1986;133:218–225. doi: 10.1164/arrd.1986.133.2.218. [DOI] [PubMed] [Google Scholar]
- 21.Christner P, Fein A, Goldberg S, Lippmann M, Abrams W, Weinbaum G. Collagenase in the lower respiratory-tract of patients with adult respiratory-distress syndrome. Am Rev Respir Dis. 1985;131:690–695. doi: 10.1164/arrd.1985.131.5.690. [DOI] [PubMed] [Google Scholar]
- 22.Gadek JE, Fells GA, Wright DG, Crystal RG. Human neutrophil elastase functions as a type III collagen “collagenase”. Biochem Biophys Res Commun. 1980;95:1815–1822. doi: 10.1016/s0006-291x(80)80110-0. [DOI] [PubMed] [Google Scholar]
- 23.Gadher SJ, Eyre DR, Duance VC, Wotten SF, Heck LW, Schmid TM, Woolley DE. Susceptibility of cartilage collagens type II, IX, X, and XI to human synovial collagenase and neutrophil elastase. Eur J Biochem. 1988;175:1–7. doi: 10.1111/j.1432-1033.1988.tb14158.x. [DOI] [PubMed] [Google Scholar]
- 24.Sabeh F, Ota I, Holmbeck K, Birkedal-Hansen H, Soloway P, Balbin M, Lopez-Otin C, Shapiro S, Inada M, Krane S, Allen E, Chung D, Weiss SJ. Tumor cell traffic through the extracellular matrix is controlled by the membrane-anchored collagenase MT1-MMP. J Cell Biol. 2004;167:769–781. doi: 10.1083/jcb.200408028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kafienah W, Buttle DJ, Burnett D, Hollander AP. Cleavage of native type I collagen by human neutrophil elastase. Biochem J. 1998;330(Pt 2):897–902. doi: 10.1042/bj3300897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lutchen KR, Yang K, Kaczka DW, Suki B. Optimal ventilation waveforms for estimating low-frequency respiratory impedance. J Appl Physiol. 1993;75:478–488. doi: 10.1152/jappl.1993.75.1.478. [DOI] [PubMed] [Google Scholar]
- 27.Ingenito EP, Mora R, Cullivan M, Marzan Y, Haley K, Mark L, Sonna LA. Decreased surfactant protein-B expression and surfactant dysfunction in a murine model of acute lung injury. Am J Respir Cell Mol Biol. 2001;25:35–44. doi: 10.1165/ajrcmb.25.1.4021. [DOI] [PubMed] [Google Scholar]
- 28.Owen CA, Campbell MA, Boukedes SS, Campbell EJ. Monocytes recruited to sites of inflammation express a distinctive pro-inflammatory (P) phenotype. Am J Physiol(Lung Cell Mol Physiol 11) 1994;267:L786–L796. doi: 10.1152/ajplung.1994.267.6.L786. [DOI] [PubMed] [Google Scholar]
- 29.Hautamaki RD, Kobayashi DK, Senior RM, Shapiro SD. Requirement for macrophage elastase for cigarette smoke-induced emphysema in mice. Science. 1997;277:2002–2004. doi: 10.1126/science.277.5334.2002. [DOI] [PubMed] [Google Scholar]
- 30.Boyum A. Isolation of mononuclear cells and granulocytes from human blood: Isolation of mononuclear cells by one centrifugation and of granulocytes by combining centrifugation and sedimentation at 1 g. Scand J Clin Lab Invest. 1963;21(Suppl 97):77–89. [PubMed] [Google Scholar]
- 31.Owen CA, Hu Z, Barrick B, Shapiro SD. Inducible expression of tissue inhibitor of metalloproteinases-resistant matrix metalloproteinase-9 on the cell surface of neutrophils. Am J Resp Cell Mol Biol. 2003;29:283–294. doi: 10.1165/rcmb.2003-0034OC. [DOI] [PubMed] [Google Scholar]
- 32.Owen CA, Campbell MA, Boukedes SS, Campbell EJ. Cytokines regulate membrane-bound leukocyte elastase on neutrophils: A novel mechanism for effector activity. Am J Physiol(Lung Cell Mol Physiol) 1997;272:L385–L393. doi: 10.1152/ajplung.1997.272.3.L385. [DOI] [PubMed] [Google Scholar]
- 33.Owen CA, Campbell EJ. Angiotensin II generation at the cell surface of activated neutrophils: Novel cathepsin G-mediated catalytic activity that is resistant to inhibition. J Immunol. 1998;160:1436–1443. [PubMed] [Google Scholar]
- 34.Campbell EJ, Campbell MA, Owen CA. Bioactive proteinase 3 on the cell surface of human neutrophils: quantification, catalytic activity, and susceptibility to inhibition. J Immunol. 2000;165:3366–3374. doi: 10.4049/jimmunol.165.6.3366. [DOI] [PubMed] [Google Scholar]
- 35.Hill HR, Bohnsack JF, Morris EZ, Augustine NH, Parker CJ, Cleary PP, Wu JT. Group B streptococci inhibit the chemotactic activity of the fifth component of complement. J Immunol. 1988;141:3551–3556. [PubMed] [Google Scholar]
- 36.Zeng X, Moore TA, Newstead MW, Hernandez-Alcoceba R, Tsai WC, Standiford TJ. Intrapulmonary expression of macrophage inflammatory protein 1alpha (CCL3) induces neutrophil and NK cell accumulation and stimulates innate immunity in murine bacterial pneumonia. Infect Immun. 2003;71:1306–1315. doi: 10.1128/IAI.71.3.1306-1315.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Crisman JM, Elder PJ, Wilkie NM, Kolattukudy PE. Identification of amino acids involved in the binding of hMIP-1 alpha to CC-CKR1, a MIP-1 alpha receptor found on neutrophils. Mol Cell Biochem. 1999;195:245–256. doi: 10.1023/a:1006901109902. [DOI] [PubMed] [Google Scholar]
- 38.Cheng SS, Lai JJ, Lukacs NW, Kunkel SL. Granulocyte-macrophage colony stimulating factor up-regulates CCR1 in human neutrophils. J Immunol. 2001;166:1178–1184. doi: 10.4049/jimmunol.166.2.1178. [DOI] [PubMed] [Google Scholar]
- 39.Bonecchi R, Polentarutti N, Luini W, Borsatti A, Bernasconi S, Locati M, Power C, Proudfoot A, Wells TN, Mackay C, Mantovani A, Sozzani S. Up-regulation of CCR1 and CCR3 and induction of chemotaxis to CC chemokines by IFN-gamma in human neutrophils. J Immunol. 1999;162:474–479. [PubMed] [Google Scholar]
- 40.Speyer CL, Gao H, Rancilio NJ, Neff TA, Huffnagle GB, Sarma JV, Ward PA. Novel chemokine responsiveness and mobilization of neutrophils during sepsis. Am J Pathol. 2004;165:2187–2196. doi: 10.1016/S0002-9440(10)63268-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Knauper V, Osthues A, DeClerck YA, Langley KE, Blaser J, Tschesche H. Fragmentation of human polymorphonuclear-leucocyte collagenase. Biochem J. 1993;291:847–854. doi: 10.1042/bj2910847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Knauper V, Docherty AJP, Smith B, Tschesche H, Murphy G. Analysis of the contribution of the hinge region of human neutrophil collagenase (HNC, MMP-8) to stability and collagenolytic activity by alanine scanning mutagenesis. FEBS Lett. 1997;405:60–64. doi: 10.1016/s0014-5793(97)00158-0. [DOI] [PubMed] [Google Scholar]
- 43.Owen CA, Campbell EJ. Proteinases. In: Haslett C, Evans T, editors. ARDS: Acute Respiratory Distress in Adults. Chapman & Hall Medical; London: 1996. pp. 139–165. [Google Scholar]
- 44.Hirche TO, Atkinson JJ, Bahr S, Belaaouaj A. Deficiency in neutrophil elastase does not impair neutrophil recruitment to inflamed sites. Am J Respir Cell Mol Biol. 2003 doi: 10.1165/rcmb.2003-0253OC. [DOI] [PubMed] [Google Scholar]
- 45.Betsuyaku T, Shipley JM, Liu Z, Senior RM. Neutrophil emigration in the lungs, peritoneum, and skin does not require gelatinase B. Am J Respir Cell Mol Biol. 1999;20:1303–1309. doi: 10.1165/ajrcmb.20.6.3558. [DOI] [PubMed] [Google Scholar]
- 46.Belaaouaj A, McCarthy R, Baumann M, Gao Z, Ley TJ, Abraham SN, Shapiro SD. Mice lacking neutrophil elastase reveal impaired host defense against gram negative bacterial sepsis. Nat Med. 1998;4:615–618. doi: 10.1038/nm0598-615. [DOI] [PubMed] [Google Scholar]
- 47.Belaaouaj A, Kim KS, Shapiro SD. Degradation of outer membrane protein A in Escherichia coli killing by neutrophil elastase. Science. 2000;289:1185–1188. doi: 10.1126/science.289.5482.1185. [DOI] [PubMed] [Google Scholar]
- 48.Hartzell W, Shapiro SD. Macrophage elastase prevents Gemella morbillorum infection and improves outcome following murine bone marrow transplantation. Chest. 1999;116:31S–32S. doi: 10.1378/chest.116.suppl_1.31s-a. [DOI] [PubMed] [Google Scholar]
- 49.Wilson CL, Ouellette AJ, Satchell DP, Ayabe T, Lopez-Boado YS, Stratman JL, Hultgren SJ, Matrisian LM, Parks WC. Regulation of intestinal α-defensin activation by the metalloproteinase matrilysin in innate host defense. Science. 1999;286:113–117. doi: 10.1126/science.286.5437.113. [DOI] [PubMed] [Google Scholar]
- 50.Matute-Bello G, Liles WC, Radella F, Steinberg KP, Ruzinski JT, Hudson LD, Martin TR. Modulation of neutrophil apoptosis by granulocyte colony-stimulating factor and granulocyte/macrophage colony-stimulating factor during the course of acute respiratory distress syndrome. Crit Care Med. 2000;28:1–7. doi: 10.1097/00003246-200001000-00001. [DOI] [PubMed] [Google Scholar]
- 51.Bonnans C, Levy BD. Lipid mediators as agonists for the resolution of acute lung inflammation and injury. Am J Respir Cell Mol Biol. 2007;36:201–205. doi: 10.1165/rcmb.2006-0269TR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bonnans C, Fukunaga K, Levy MA, Levy BD. Lipoxin A(4) regulates bronchial epithelial cell responses to acid injury. Am J Pathol. 2006;168:1064–1072. doi: 10.2353/ajpath.2006.051056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fukunaga K, Kohli P, Bonnans C, Fredenburgh LE, Levy BD. Cyclooxygenase 2 plays a pivotal role in the resolution of acute lung injury. J Immunol. 2005;174:5033–5039. doi: 10.4049/jimmunol.174.8.5033. [DOI] [PubMed] [Google Scholar]
- 54.Schwab JM, Chiang N, Arita M, Serhan CN. Resolvin E1 and protectin D1 activate inflammation-resolution programmes. Nature. 2007;447:869–874. doi: 10.1038/nature05877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.van Lint P, Libert C. Chemokine and cytokine processing by matrix metalloproteinases and its effect on leukocyte migration and inflammation. J Leukoc Biol. 2007;82:1375–1381. doi: 10.1189/jlb.0607338. [DOI] [PubMed] [Google Scholar]
- 56.D’Amico G, Frascaroli G, Bianchi G, Transidico P, Doni A, Vecchi A, Sozzani S, Allavena P, Mantovani A. Uncoupling of inflammatory chemokine receptors by IL-10: generation of functional decoys. Nat Immunol. 2000;1:387–391. doi: 10.1038/80819. [DOI] [PubMed] [Google Scholar]
- 57.Bourke E, Cassetti A, Villa A, Fadlon E, Colotta F, Mantovani A. IL-1 beta scavenging by the type II IL-1 decoy receptor in human neutrophils. J Immunol. 2003;170:5999–6005. doi: 10.4049/jimmunol.170.12.5999. [DOI] [PubMed] [Google Scholar]
- 58.Ray E, Samanta AK. Receptor-mediated endocytosis of IL-8: a fluorescent microscopic evidence and implication of the process in ligand-induced biological response in human neutrophils. Cytokine. 1997;9:587–596. doi: 10.1006/cyto.1997.0206. [DOI] [PubMed] [Google Scholar]
- 59.Darbonne WC, Rice GC, Mohler MA, Apple T, Hebert CA, Valente AJ, Baker JB. Red blood cells are a sink for interleukin 8, a leukocyte chemotaxin. J Clin Invest. 1991;88:1362–1369. doi: 10.1172/JCI115442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Belenko M, Chanana AD, Joel DD, Fagerhol MK, Janoff A. Tracing the leukocyte marker protein in lung fluids and lung-draining lymph nodes during endotoxemia in sheep. Am Rev Respir Dis. 1986;133:866–874. [PubMed] [Google Scholar]
- 61.St John RC, Mizer LA, Weisbrode SE, Dorinsky PM. Increased intestinal protein permeability in a model of lung injury induced by phorbol myristate acetate. Am Rev Respir Dis. 1991;144:1171–1176. doi: 10.1164/ajrccm/144.5.1171. [DOI] [PubMed] [Google Scholar]
- 62.Folkesson HG, Matthay MA, Westrom BR, Kim KJ, Karlsson BW, Hastings RH. Alveolar epithelial clearance of protein. J Appl Physiol. 1996;80:1431–1445. doi: 10.1152/jappl.1996.80.5.1431. [DOI] [PubMed] [Google Scholar]
- 63.Kasama T, Strieter RM, Standiford TJ, Burdick MD, Kunkel SL. Expression and regulation of human neutrophil-derived macrophage inflammatory protein 1 alpha. J Exp Med. 1993;178:63–72. doi: 10.1084/jem.178.1.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jan MS, Huang YH, Shieh B, Teng RH, Yan YP, Lee YT, Liao KK, Li C. CC chemokines induce neutrophils to chemotaxis, degranulation, and alpha-defensin release. J Acquir Immune Defic Syndr. 2006;41:6–16. doi: 10.1097/01.qai.0000188336.94090.14. [DOI] [PubMed] [Google Scholar]
- 65.Zhang S, Youn BS, Gao JL, Murphy PM, Kwon BS. Differential effects of leukotactin-1 and macrophage inflammatory protein-1 alpha on neutrophils mediated by CCR1. J Immunol. 1999;162:4938–4942. [PubMed] [Google Scholar]
- 66.Ramos CD, Canetti C, Souto JT, Silva JS, Hogaboam CM, Ferreira SH, Cunha FQ. MIP-1alpha[CCL3] acting on the CCR1 receptor mediates neutrophil migration in immune inflammation via sequential release of TNF-alpha and LTB4. J Leukoc Biol. 2005;78:167–177. doi: 10.1189/jlb.0404237. [DOI] [PubMed] [Google Scholar]
- 67.Standiford TJ, Kunkel SL, Lukacs NW, Greenberger MJ, Danforth JM, Kunkel RG, Strieter RM. Macrophage inflammatory protein-1 alpha mediates lung leukocyte recruitment, lung capillary leak, and early mortality in murine endotoxemia. J Immunol. 1995;155:1515–1524. [PubMed] [Google Scholar]
- 68.Lee SC, Brummet ME, Shahabuddin S, Woodworth TG, Georas SN, Leiferman KM, Gilman SC, Stellato C, Gladue RP, Schleimer RP, Beck LA. Cutaneous injection of human subjects with macrophage inflammatory protein-1 alpha induces significant recruitment of neutrophils and monocytes. J Immunol. 2000;164:3392–3401. doi: 10.4049/jimmunol.164.6.3392. [DOI] [PubMed] [Google Scholar]
- 69.Hsieh CH, Frink M, Hsieh YC, Kan WH, Hsu JT, Schwacha MG, Choudhry MA, Chaudry IH. The role of MIP-1 alpha in the development of systemic inflammatory response and organ injury following trauma hemorrhage. J Immunol. 2008;181:2806–2812. doi: 10.4049/jimmunol.181.4.2806. [DOI] [PubMed] [Google Scholar]
- 70.Gutteridge MC, Quinlan G. Reactive oxygen species, antioxidant protection and lung injury. In: Evans TW, Haslett C, editors. ARDS: Acute Respiratory Distress in Adults. Chapman & Hall Medical; London: 1996. pp. 167–195. [Google Scholar]
- 71.Matute-Bello G, Frevert CW, Martin TR. Animal models of acute lung injury. Am J Physiol Lung Cell Mol Physiol. 2008;295:L379–L399. doi: 10.1152/ajplung.00010.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Dolinay T, Wu W, Kaminski N, Ifedigbo E, Kaynar AM, Szilasi M, Watkins SC, Ryter SW, Hoetzel A, Choi AM. Mitogen-activated protein kinases regulate susceptibility to ventilator-induced lung injury. PLoS ONE. 2008;3:e1601. doi: 10.1371/journal.pone.0001601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Shanley TP, Schmal H, Friedl HP, Jones ML, Ward PA. Role of macrophage inflammatory protein-1 alpha (MIP-1 alpha) in acute lung injury in rats. J Immunol. 1995;154:4793–4802. [PubMed] [Google Scholar]
- 74.Petrache I, Birukova A, Ramirez SI, Garcia JG, Verin AD. The role of the microtubules in tumor necrosis factor-alpha-induced endothelial cell permeability. Am J Respir Cell Mol Biol. 2003;28:574–581. doi: 10.1165/rcmb.2002-0075OC. [DOI] [PubMed] [Google Scholar]
- 75.Ricou B, Nicod L, Lacraz S, Welgus HG, Suter PM, Dayer JM. Matrix metalloproteinases and TIMP in acute respiratory distress syndrome. Am J Respir Crit Care Med. 1996;154:346–352. doi: 10.1164/ajrccm.154.2.8756805. [DOI] [PubMed] [Google Scholar]
- 76.McQuibban GA, Gong JH, Wong JP, Wallace JL, Clark-Lewis I, Overall CM. Matrix metalloproteinase processing of monocyte chemoattractant proteins generates CC chemokine receptor antagonists with anti-inflammatory properties in vivo. Blood. 2002;100:1160–1167. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.