

Abstract
Background and purpose:
RhoA kinase (ROCK) participates in K+ depolarization (KCl)-induced Ca2+ sensitization of contraction. Whether constitutive, depolarization- or Ca2+-activated ROCK plays the major role in this signalling system remains to be determined. Here, we determined whether Bay K 8644, a dihydropyridine that promotes Ca2+ channel clusters to operate in a persistent Ca2+ influx mode, could cause ROCK-dependent Ca2+ sensitization.
Experimental approach:
Renal and femoral artery rings from New Zealand white rabbits were contracted with Bay K 8644. Tissues were frozen and processed to measure active RhoA and ROCK substrate (myosin phosphatase targeting subunit, MYPT1) and myosin light chain (MLC) phosphorylation, or loaded with fura-2 to measure intracellular free Ca2+ ([Ca2+]i). Effects of selective inhibitors of contraction were assessed in resting (basal) tissues and those contracted with Bay K 8644.
Key results:
Bay K 8644 produced strong increases in [Ca2+]i, MLC phosphorylation and tension, but not in MYPT1 phosphorylation. ROCK inhibition by H-1152 abolished basal MYPT1-pT853, diminished basal MLC phosphorylation and inhibited Bay K 8644-induced increases in MLC phosphorylation and tension. MLC kinase inhibition by wortmannin abolished Bay K 8644-induced contraction and increase in MLC phosphorylation but did not inhibit basal MYPT1-pT853. H-1152 and wortmannin had no effect on MYPT1-pT696, but 1 µM staurosporine inhibited basal MYPT1-pT853, MYPT1-pT696 and MLC phosphorylation.
Conclusions and implications:
These data suggest that the constitutive activities of ROCK and a staurosporine-sensitive kinase regulate basal phosphorylation of MYPT1, which participates along with activation of MLC kinase in determining the strength of contraction induced by the Ca2+ agonist, Bay K 8644.
Keywords: cardiovascular pharmacology, Ca-channels, vascular smooth muscle, calcium sensitization, active RhoA, RhoA kinase, ROCK, MYPT1, myosin light chain phosphorylation, H-1152
Introduction
The level of tension generated upon activation of smooth muscle by a contractile stimulus is largely dependent on the degree of phosphorylation of myosin light chain (MLC) induced by the stimulus. Stimuli regulate the degree of MLC phosphorylation by activation of kinases (MLCK) that recognize the 20 kDa MLC as a substrate, and by kinases that regulate MLC phosphatase (MLCP) activity through phosphorylation of the large molecular weight subunit, myosin phosphatase targeting subunit (MYPT1) and the 17 kDa protein kinase C (PKC) inhibitory protein, CPI-17 (Somlyo and Somlyo, 2003). The primary cell signalling system activated by contractile stimuli is Ca2+, and elevations of intracellular free Ca2+ ([Ca2+]i) activate Ca2+ calmodulin-dependent MLCK, causing increases in MLC phosphorylation, cross-bridge cycling and tension development (Kamm and Stull, 1985). Interestingly, several studies support the hypothesis that elevations in [Ca2+]i also can activate RhoA kinase (ROCK) to cause increased levels of MYPT1-pT853 responsible for MLCP inhibition (Ratz et al., 2005). Thus, increases in [Ca2+]i are proposed to elevate MLC phosphorylation above the resting, basal level both by activation of MLCK and inhibition of MLCP (Brozovich, 2003). However, in smooth muscle, basal MLC phosphorylation is above zero, and contraction does not occur until MLC phosphorylation is elevated above a threshold level of ∼15% (Rembold et al., 2004).
One tool used by smooth muscle biologists to examine the regulation of contraction in intact smooth muscle is to activate tissues with high concentrations of KCl, a stimulus that clamps cells at a particular depolarized membrane potential, activating voltage-operated Ca2+ channels and elevating intracellular Ca2+ ([Ca2+]i) levels (Casteels et al., 1977; Somlyo and Himpens, 1989). KCl has been used for decades as a stimulus to bypass activation of G protein-coupled receptors (GPCR). By comparison with KCl, GPCR agonists can cause stronger increases in muscle tension for a given increase in [Ca2+]i (Karaki et al., 1988). Such Ca2+ sensitization of actomyosin cross-bridges is attributed to activation of mechanisms causing MLCP inhibition (Somlyo et al., 1999). Although GPCR agonists often induce greater increases in tension for a given increase in [Ca2+]i compared with KCl, KCl can cause Ca2+ sensitization (Yanagisawa and Okada, 1994; Ratz, 1999). Early in this decade, several laboratories independently determined that ROCK appears to participate in KCl-induced Ca2+ sensitization. The ROCK inhibitor Y-27632 reduces the tonic phase of a KCl-induced contraction and the tonic increase in MLC phosphorylation of rat caudal artery (Mita et al., 2002) and rabbit femoral artery (Urban et al., 2003) without causing a concomitant reduction in [Ca2+]i. In rat mesenteric artery, Y-27632 relaxes the steady-state tension induced by 100 mM KCl without reducing [Ca2+]i (Ghisdal et al., 2003). In rabbit thoracic aorta, 60 mM KCl and 3 µM noradrenaline produce nearly identical increases in active RhoA and tension (Sakurada et al., 2003). Notably, Y-27632 produces a greater relaxation of KCl-stimulated mesenteric artery isolated from spontaneously hypertensive rat compared with normotensive Wistar Kyoto rat (Asano and Nomura, 2003). KCl increases MYPT1-p696 in rabbit thoracic aorta (Sakurada et al., 2003), and MYPT1-p853 but not MYPT1-p696 in rabbit femoral artery (Porter et al., 2006). Although KCl can activate periarterial release of noradrenaline, all of these studies employed agents to prevent potential vascular smooth muscle α-adrenoceptor activation. KCl-induced contraction is attenuated by ROCK inhibition in smooth muscle tissues other than arterial, including airway (Gosens et al., 2004; Janssen et al., 2004) and myometrial (Kupittayanant et al., 2001) smooth muscles. The significance of these studies is that any agent that elevates [Ca2+]i or causes membrane depolarization may cause ROCK-dependent Ca2+ sensitization. Such a mechanism could participate in diseases linked to arterial hyper-contraction because Ca2+ entry and ROCK activity are associated with hypertension and coronary artery vasospasm in animals and humans (Uehata et al., 1997; Masumoto et al., 2002; Seasholtz et al., 2006; Sonkusare et al., 2006).
Based on studies using inhibitors of voltage-operated Ca2+ channels (Urban et al., 2003; Janssen et al., 2004) and calmodulin (Sakurada et al., 2003), calcium entry and calmodulin have been proposed to play a role in KCl-induced Ca2+ sensitization. KCl-induced Ca2+ sensitization may also involve phosphoinositide 3-kinase (Wang et al., 2006). We recently showed that ROCK appears to be constitutively active, and that KCl-induced Ca2+ entry correlates with PKCζ-dependent increases in MYPT1 phosphorylation (Ratz and Miner, 2009). On the other hand, Takuwa's group showed, using ionomycin as the stimulus in cultured rat aortic vascular smooth muscle cells, that phosphoinositide 3-kinase-C2α is necessary for Ca2+-induced ROCK-dependent negative regulation of MLCP (Yoshioka et al., 2007). Thus, although some progress has been made, the precise cell signalling cascade leading from membrane depolarization and increases in [Ca2+]i to activation of ROCK causing Ca2+ sensitization remains to be determined.
In the present study, we used Bay K 8644 rather than K+ depolarization (KCl) as a stimulus to elevate Ca2+ entry and cause contraction. Bay K 8644 is a dihydropyridine Ca2+ channel agonist (Schramm et al., 1983) that elevates Ca2+ entry not by causing strong membrane depolarization, but instead, by increasing the probability that individual voltage-operated Ca2+ channels open and by promoting clusters of voltage-operated Ca2+ channels to operate in a persistent calcium influx mode (Brown et al., 1984; Navedo et al., 2005). Thus, Bay K 8644 serves as a more specific stimulus than KCl because it causes contraction that is dependent solely on an increase in [Ca2+]i. This study was designed to determine whether ROCK-dependent Ca2+ sensitization plays a critical role in the Bay K 8644-induced increase in sustained tension. Our results support a hypothesis contrary to that formulated from studies that used KCl as a stimulus, namely, that ROCK is constitutively active and possibly not stimulated by increases in [Ca2+]i.
Methods
Tissue preparation and isometric tension
All animal care and experimental protocols complied with the appropriate animal welfare regulations and guidelines of the US Public Health Service and were approved by the Virginia Commonwealth University Institutional Animal Care and Use Committee. Tissues were prepared and contraction measured as described previously (Ratz, 1993). In brief, renal and femoral arteries from New Zealand white rabbits were cleaned of adhering tissue, denuded of endothelium, cut into 3–4 mm wide rings and stored in physiological salt solution [PSS; composition in mM, NaCl 140, KCl 4.7, MgSO4 1.2, CaCl2 1.6, NaHPO4 1.2, morpholino-propanesulfonic acid (MOPS) 2.0 (adjusted to pH 7.4), Na2EDTA (to chelate heavy metals) 0.02 and D-glucose 5.6] at 4°C for later analyses or immediately secured between wires in the myograph tissue chamber containing PSS maintained at 37°C. To produce K+ depolarization, KCl (110 mM) was substituted isosmotically for NaCl. In some experiments, tissues were incubated in a nominally Ca2+-free solution (0 Ca2+) in which CaCl2 was excluded. The Ca2+ chelator EGTA was not added because the reduction in [Ca2+]i caused by extracellular EGTA activates phospholipase A2 in arterial muscle cells (Smani et al., 2004), and arachidonic acid produced by phospholipase A2 activation can strongly activate ROCK (Feng et al., 1999). Contractile tension was measured as previously described (Ratz, 1993). For each tissue preloaded to the optimum length for muscle contraction (Lo), the degree of KCl-induced steady-state tension was equal to the optimum tension for muscle contraction (To). Subsequent contractions were calculated as T/To, as normalized to the steady state of a control contraction induced by Bay K 8644 (fold-control BayK), or as normalized to the tension produced by Bay K 8644 prior to addition of a drug (T2/T1). Arteries were incubated with 1 µM phentolamine to block potential α-adrenoceptor activation caused by release of noradrenaline from periarterial nerves.
[Ca2+]i
[Ca2+]i was measured in artery rings at Lo as previously described (Ratz, 1993) with minor modifications. Arterial rings were secured in a temperature-controlled myograph suitable for simultaneous measurement of tension and fluorescence (DMT-USA, Atlanta, GA, USA) that was positioned on the stage of an Olympus IX71 inverted microscope with attachments to a DeltaRam V fluorometer and photomultiplier tube (Photon Technology International, Lawrenceville, NJ, USA). Tissues were loaded with 7.5 µM fura 2-PE3 (AM) and 0.01% (w/v) Pluronic F-127(TefLabs, Austin, TX, USA) in PSS. Fluorescence emission intensities at 510 nm collected by a photomultiplier tube were expressed as excitation ratios (340 nm/380 nm) using Felix software (Photon Technology International). Background fluorescence, determined by incubating tissues in 4 mM MnCl2 plus 20 µM ionomycin, was subtracted prior to calculating the fluorescence ratios. For each tissue preloaded at its optimum length for muscle contraction (Lo), the degree of KCl-induced steady-state tension was equal to the optimum tension for muscle contraction (To). Subsequent contractions were calculated as T/To or as normalized to the tension produced by Bay K 8644 prior to addition of a drug (T2/T1). At the end of each experiment, tissues were exposed to 20 µM ionomycin in the presence of Ca2+ and then a Ca2+-free solution plus EGTA to chelate calcium, allowing measurements of, respectively, the maximum and minimum [Ca2+]i responses. Ca2+ responses were then normalized to these maximum and minimum responses (Ca/Cao), or normalized to the tension produced by Bay K 8644 prior to addition of a drug (Ca2/Ca1).
General protocol for protein phosphorylation assays
For each replicate (n value) of the assay, we used four artery rings cut from a renal (or femoral) artery dissected from each rabbit. Tissues were preloaded by stretching the muscle to Lo as described above. For Figures 2, 3, 5 and 8, two of the four tissues were stimulated to contract until steady-state tension was achieved (∼15–30 min) at which time the designated inhibitor (or vehicle for control) was added. Tension tracings were recorded for approximately an additional 15 min at which time tissues were frozen as described below (Western blots). To provide the ‘basal’ tissue values, the other two of the four tissues were not contracted but were otherwise treated identically to those that were contracted. For Figures 6 and 7, one tissue was frozen at time = 0 to provide the basal values, the other three tissues were then contracted and one each was frozen at 5, 15 and 60 min. Usually, but not always, there was sufficient sample from each tissue to assay for both MLC phosphorylation and MYPT phosphorylation.
Figure 2.
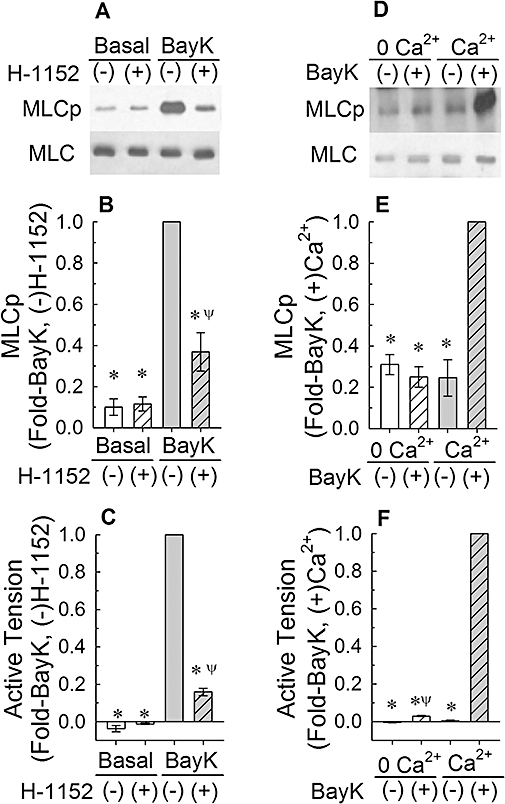
Effect of 30 nM Bay K 8644 (BayK) on myosin light chain phosphorylation (MLCp, A,B) and tension (C), and the effects of 1 µM H-1152 and a Ca2+-free solution (0 Ca2+) on basal and BayK-induced increases in MLCp (A,B,D,E) and tension (C,F). (A) and (D) are representative Western blots. (−): tissues were not exposed to drug; (+): tissues were exposed to drug. Bars are means ± SE, n = 5–11. *P < 0.05, compared with the BayK (−)H-1152, and BayK (+)Ca2+ responses. ψP < 0.05, compared with the basal values (B and C) and compared with BayK (−) 0 Ca2+ (F); anova /Newman-Keuls
Figure 3.
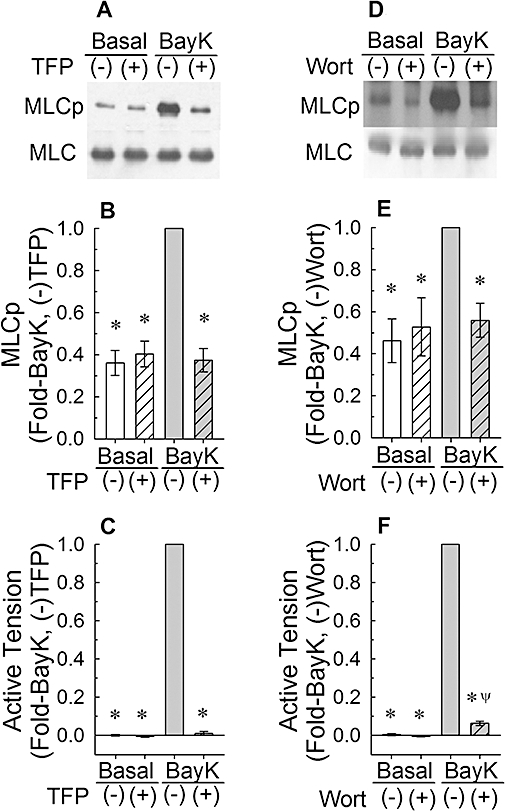
Effects of 50 µM trifluoperazine (TFP, A–C) and 1 µM wortmannin (Wort, D–F) on myosin light chain phosphorylation (MLCp, A,B,D,E) and tension (C,F) in unstimulated muscle (basal) and in tissues stimulated to contraction by addition of 30 nM Bay K 8644 (BayK). (A) and (B) are representative Western blots. (−): tissues were not exposed to drug; (+): tissues were exposed to drug. Bars are means ± SE, n = 7–8. *P < 0.05, compared with BayK (−)TFP, and BayK (−)Wort responses. ψP < 0.05 compared with the basal values; anova /Newman-Keuls.
Figure 5.
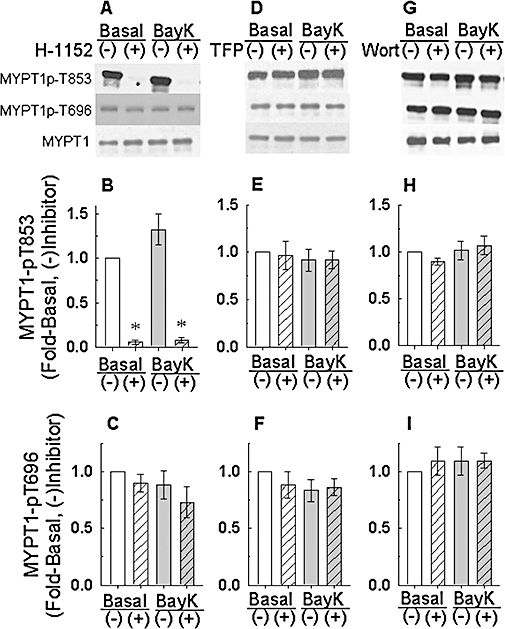
Effect of 1 µM H-1152 (A–C), 50 µM trifluoperazine (TFP, D–F) and 1 µM Wortmannin (Wort, G–I) on basal and Bay K 8644 (BayK)-induced myosin phosphatase targeting subunit (MYPT1) phosphorylation at T853 and T696 and total MYPT1. (A), (D) and (G) are representative Western blots. (−): tissues were not exposed to drug; (+): tissues were exposed to drug. Bars are mean ± SE, n = 5–9. *P < 0.05, compared with basal (−), and BayK (−) values; anova /Dunnett.
Figure 8.
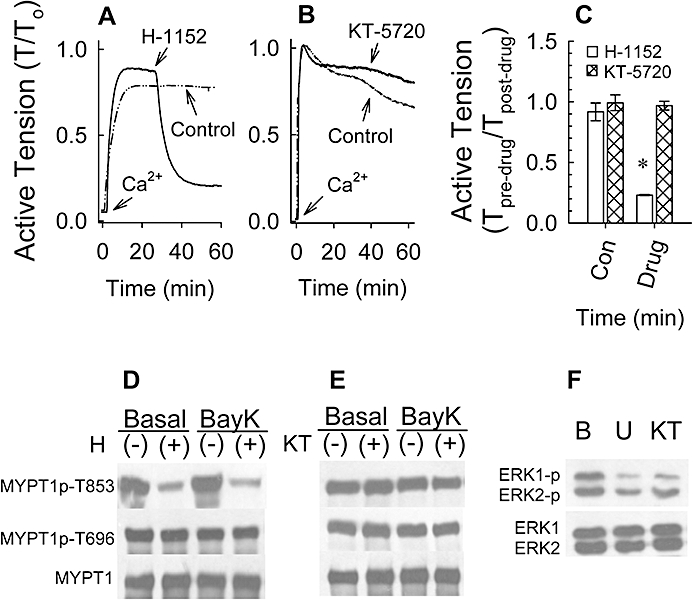
Examples (A,B) and summary data (C) of 100 nM Bay K 8644 (BayK)-induced tension versus time tracings produced in the absence of an inhibitor and after addition of an inhibitor in rabbit femoral artery. Inhibitors H-1152 and KT-5720 were added to tissues once contractions achieved a pseudo-steady state. Effect of 1 µM H-1152 (D) and 1 µM KT-5720 (E) on BayK-induced myosin phosphatase targeting subunit (MYPT1) phosphorylation at T853 and T696 and total MYPT1. Effect of 1 µM U-0126 and 1 µM KT-5720 on basal ERK1 and ERK2 phosphorylation (F). (D), (E) and (F) are representative Western blots. (−): tissues were not exposed to drug; (+): tissues were exposed to drug. B, basal; U, U-126 and KT, KT-5720. Bars in (C) are means ± SE, n = 4. *P < 0.05, compared with control (Con) values.
Figure 6.
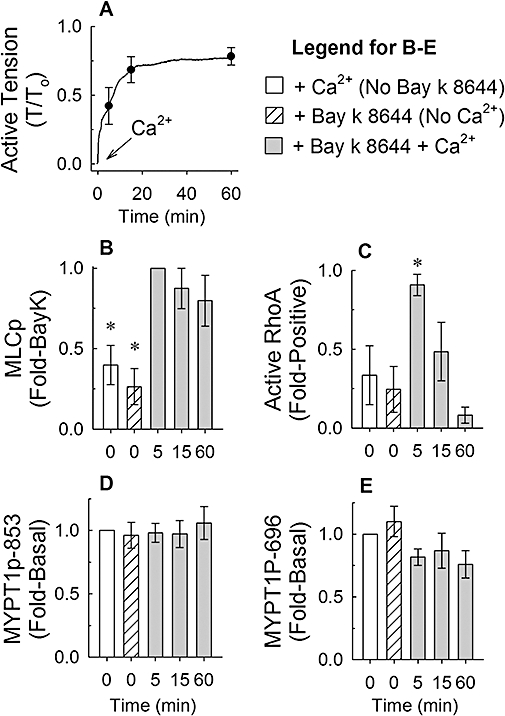
Effects of Ca2+-free solutions. There was considerably less variability in the rate of tension development during a Bay K 8644-induced contraction, when tissues were incubated for 30 min in a Ca2+-free solution in the presence of 100 nM Bay K 8644 plus 10 mM KCl, then activated by adding 2 mM CaCl2, compared with results obtained from tissues activated by adding Bay K 8644 plus 7.5 or 10 mM KCl directly to a Ca2+-containing solution (A, Ca2+, tracing and symbols are average values, symbols reveal ±SE at 5, 15 and 60 min, n = 7). Tissues quick-frozen at 0 (basal, either with Bay K 8644 in 0 Ca2+ or with 2 mM Ca2+ and no Bay K 8644), 5, 15 and 60 min were processed to measure the time course of changes in myosin light chain phosphorylation (MLCp, B), active RhoA (C), myosin phosphatase targeting subunit (MYPT1)-pT853 (D) and MYPT1-pT696 (E). Bars are mean ± SE, n = 5. *P < 0.05, compared with Bay K 8644 + Ca2+ (B) and compared with basal values (C); anova /Newman-Keuls.
Figure 7.
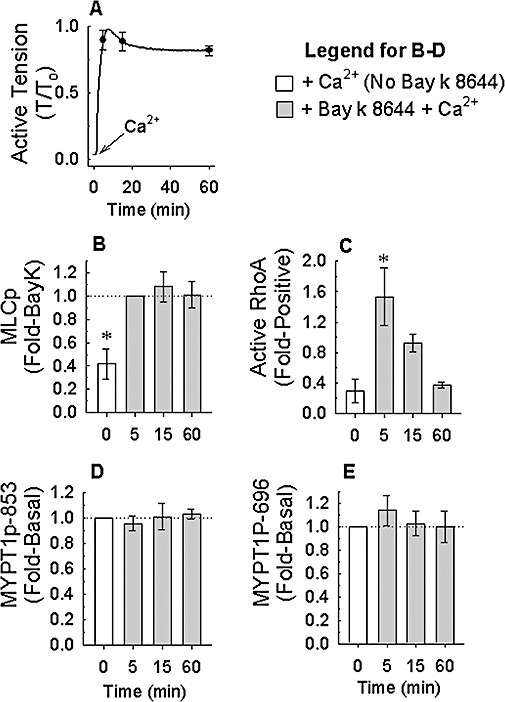
Example of 100 nM Bay K 8644 (BayK)-induced tension (A) versus time tracings produced in rabbit femoral artery. Tracing and symbols are average values; symbols reveal ±SE at 5, 15 and 60 min (n = 4). Tissues quick-frozen at 0 (basal), 5, 15 and 60 min were processed to measure the time course of changes in myosin light chain phosphorylation (MLCp, B), active RhoA (C), myosin phosphatase targeting subunit (MYPT1)-pT853 (D) and MYPT1-pT696 (E). Bars are mean ± SE, n = 4. *P < 0.05, compared with Bay K 8644 + Ca2+ (B) and compared with basal values (C); anova /Newman-Keuls.
Western blots
Phosphorylation of MYPT1 was measured by Western blot analysis of artery ring homogenates using phospho-specific antibodies as described previously (Porter et al., 2006). Tissues were stimulated with Bay K 8644 in the presence or absence of the inhibitors and quick-frozen in an acetone-dry ice slurry in order to stop chemical reactions. Tissues were homogenized in 1% SDS, 10% glycerol, 20 mM dithiothreitol, 25 mM Tris-HCl (pH 6.8), 5 mM EGTA, 1 mM EDTA, 50 mM NaF, 1 mM sodium orthovanadate, 20 mg·mL−1 leupeptin, 2 mg·mL−1 aprotinin and 20 mg·mL−1 (4-amidino-phenyl)-methanesulfonyl fluoride. Samples were boiled at 100°C for 10 min, clarified and stored at −70°C. Thawed homogenates were assayed for protein concentration as follows: samples (5 µL), and BSA for the generation of a standard curve, were loaded in a 12% acrylamide gel. The gel was stained with Coomassie Blue R-250 and destained with a mix containing 25% ethanol and 10% acetic acid in deionized water. The intensity of actin, the most abundant smooth muscle protein representing ∼50 mg·g−1 wet wt in arterial samples (Cohen and Murphy, 1979), was compared with the BSA standard curve and used to quantify the total protein content for each sample. Homogenates were loaded into gel wells at 60 µg for MLC phosphorylation, 20 µg for total MLC, 35 µg for MYPT1-p853, 18 µg for MYPT1-p 696, 5 µg for total MYPT1 and 12 µg for ERKp and total ERK. Proteins were separated by 1-dimensional SDS-PAGE on 13%, 7.5% or 10% gels followed by Western blotting. Phosphorylated MYPT1 was identified using anti-MYPT1-p853 antibody (Millipore, Billerica, MA, USA) and anti-MYPT1-p696 antibody (Millipore). Total MYPT1 (BD Biosciences, San Jose, CA, USA) was assessed to quantify loading accuracy. Phosphorylated MLC was identified using anti-phospho-MLC-[pSer19] antibody (Sigma-Aldrich, St. Louis, MO, USA). Total MLC (Sigma-Aldrich) was assayed for loading accuracy. Phosphorylated ERK was identified using anti-ERK1p and 2p (Sigma-Aldrich). Total ERK (Sigma-Aldrich) was assayed for loading accuracy. Antibodies were detected using horseradish peroxidase (HRP)-labelled secondary antibody (Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA) and enhanced chemiluminescence and ECL films (GE Healthcare Biosciences, Pittsburgh, PA, USA). Quantification of bands was obtained by digital image analysis (Scion Image software, ScionCorp, Frederick, MD, USA).
G-LISA RhoA activation assay biochemistry kit for expression analysis of active RhoA (GTP bound RhoA)
Active RhoA was determined using a RhoA G-LISA™ assay as recommended by the manufacturer (Cytoskeleton Inc., Denver, CO, USA). Tissues were quick-frozen in liquid nitrogen and homogenized in 250 µL of ice-cold lysis buffer containing protease inhibitor provided by the kit. The lysates were clarified by centrifugation at 25 000×g for 10 min at 4°C. The cell lysates were adjusted to a final concentration of 1 mg·mL−1 protein. An equal volume of the samples was added to the wells of the Rho G-LISA plate coated with Rho-GTP-binding protein. The plate was placed on a cold microplate shaker set at 400 rpm at 4°C for 30 min. After three washes at room temperature using a ‘wash buffer’ supplied by the manufacturer, 50 µL of anti-RhoA primary antibody (diluted 1:250) was added to each well and left on the shaker for 45 min. The plate was washed three times, and 50 µL of diluted HRP-labelled secondary antibody (1:250) was added to the wells and placed on the shaker at room temperature for 45 min. After three washes, 50 µL of HRP detection reagent was added to the wells, and the luminescence signal was detected using a microplate luminescence reader (PerkinElmer 2030 Multilabel Reader, PerkinElmer Life and Analytical Science, Turku, Finland).
Statistics
The null hypothesis was examined using Students’t-test (when 2 groups were compared) or using a one-way analysis of variance (anova) followed by Dunnett's post hoc test to assess whether each test group was different than a control group, or by Newman-Keuls's post hoc test. In all cases, the null hypothesis was rejected at P < 0.05. For each study described, the n value was equal to the number of rabbits from which arteries were taken.
Materials
Phentolamine, Bay K 8644 and trifluoperazine were from Sigma-Aldrich. H-1152 and Y-27632 were from EMD Biosciences (formerly Calbiochem), San Diego, CA, USA. Wortmannin and ML-7 were from Alexis Biochemicals (San Diego, CA, USA). Staurosporine and U0126 were from Calbiochem (San Diego, CA, USA). Ionomycin was from VWR (Bridgeport, NJ, USA). Fura 2-PE3 (AM) and Pluronic F-127 were from Tef Laboratories. KT-5720 was from Tocris Bioscience (Ellisville, MO, USA). Ionomycin and Bay K 8644 were dissolved in ethanol; Wortmannin, ML-7, staurosporine, KT-5720 and U-0126 were dissolved in dimethylsulfoxide; all other inhibitors were dissolved in distilled water. Ethanol and dimethylsulfoxide were added at a final concentration no greater than 0.1%, a concentration that had no effect on Bay K 8644-induced increases in tension or [Ca2+]i.
Results
Effect of inhibitors of ROCK, Ca2+ entry, calmodulin and calmodulin-dependent MLCK on Bay K 8644-induced contraction and increase in MLC phosphorylation
Quiescent rabbit femoral arteries do not contract when Bay K 8644 alone is added to tissues (Ratz et al., 1989). In general, what permits Bay K 8644 to produce a strong contraction in quiescent arteries is to raise the extracellular [KCl] to about 10–15 mM, a concentration that alone also does not induce contraction. In the present study, we found that 30 nM Bay K 8644 often could produce a strong contraction in the presence of 7.5 mM added extracellular KCl (∼12.5 mM total) in rabbit renal arteries (Figure 1A,D). However, in some tissues, addition of 10 mM extracellular [KCl] (∼ 15 mM total) was required to permit Bay K 8644 to induce a strong contraction (Figure 1B,C,E,F). Thus, to induce contraction, we routinely started at 7.5 mM extracellular [KCl] and 30 nM Bay K 8644, and if contraction rate was slow, added 2.5 mM more KCl to the tissue bath to speed the rate and enhance the strength of contraction.
Figure 1.
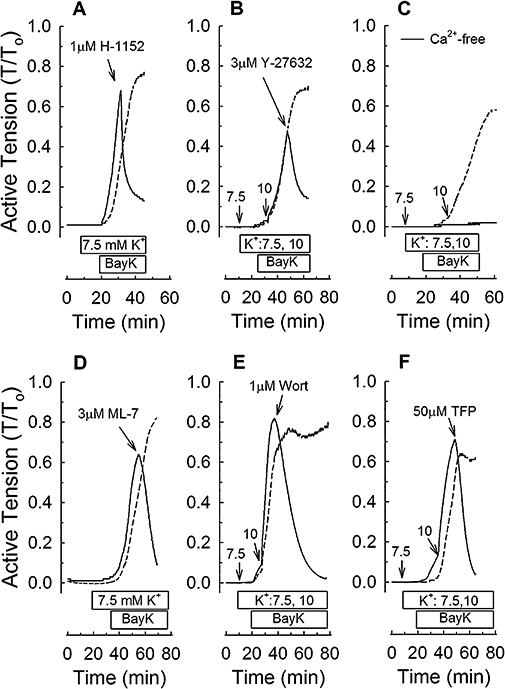
Examples of tension versus time tracings showing control 30 nM Bay K 8644 (BayK)-induced contractions (dashed lines) and the effect of selective inhibitors (A,B,D–F) and a Ca2+-free solution (C) on contractions induced by BayK (solid lines). RhoA kinase inhibitors H-1152 (1 µM, A) and Y-27632 (3 µM, B), myosin light chain kinase inhibitors ML-7 (3 µM, D) and wortmannin (1 µM, E), and the calmodulin inhibitor trifluoperazine (50 µM, F) were added at the times indicated by arrows. 7.5 and 10 refer to the amount (in mM) of KCl (K+) added. Horizontal bars indicate the duration of time tissues were exposed to K+ and BayK.
Compared with control tissues, the ROCK inhibitors, H-1152 (Figure 1A) and Y-27632 (Figure 1B) produced a rapid and strong inhibition of Bay K 8644-induced contraction. As expected because Bay K 8644 increases Ca2+ entry, the contraction induced by Bay K 8644 was entirely dependent on the presence of extracellular Ca2+ (Figure 1C). Moreover, inhibitors of MLCK (ML-7 and wortmannin) and calmodulin (trifluoperazine) abolished Bay K 8644-induced contractions (Figure 1D–F). These data confirmed that Bay K 8644 induced a Ca2+ entry-dependent contraction in rabbit renal artery and suggested that ROCK, MLCK and calmodulin participate in this contraction.
Effect of H-1152, trifluoperazine and wortmannin on basal and Bay K 8644-induced increases in MLC phosphorylation and tension
As expected from our previous work with Bay K 8644, a strong and sustained increase in tension induced by Bay K 8644 (Figure 2C) correlated with an increase above the basal level in MLC phosphorylation in rabbit renal artery (Figure 2A,B and see Figure 3). Normalization of the data to the relatively weak basal MLC phosphorylation signal introduced some variability in the apparent degree of Bay K 8644-induced increase in MLC phosphorylation. When all of the data from Figure 2B (n = 11), Figure 2E (n = 5), Figure 3B (n = 8) and E (n = 7) were averaged, we found that Bay K 8644 produced an approximately fourfold increase in MLC phosphorylation above the basal level (n = 31). The ROCK inhibitor, H-1152 (1 µM), strongly inhibited the increases in steady-state tension (Figure 2C) and MLC phosphorylation induced by Bay K 8644, and did not appear to reduce the basal level of MLC phosphorylation (Figure 2A,B, but see Figure 9). The same results were obtained with Y-27632, a ROCK inhibitor that is structurally different than H-1152 (data not shown). Incubation of tissues in a Ca2+-free solution (0 Ca2+) abolished the ability of Bay K 8644 to increase MLC phosphorylation (Figure 2D,E) and inhibited Bay K 8644-induced increase in tension by 98% (Figure 2F). A Ca2+-free solution did not reduce the basal MLC phosphorylation level (Figure 2D,E).
Figure 9.
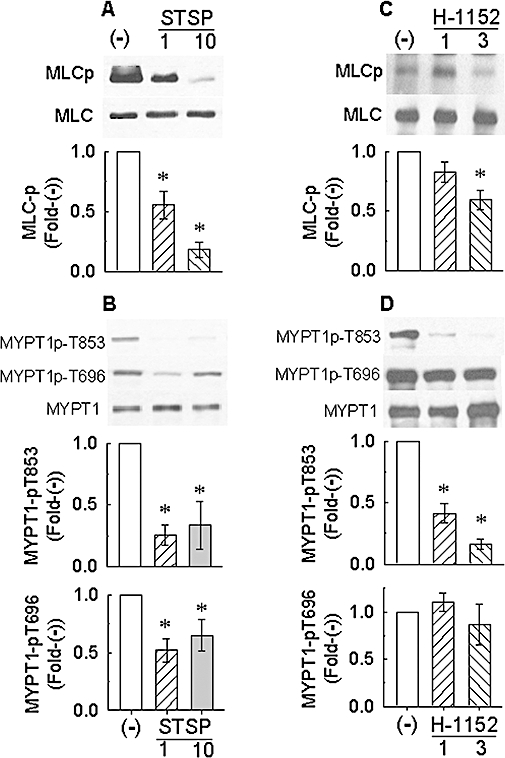
Effect of staurosporine (STSP) and H-1152 on basal myosin light chain phosphorylation (MLCp, A and C respectively) and basal myosin phosphatase targeting subunit (MYPT1) phosphorylation at T853 and T696 and total MYPT1 (B and D respectively). Protein bands are representative Western blots. (−): tissues were not exposed to drug. Bars are means ± SE, n = 4–9. *P < 0.05, compared with (−) values; anova /Dunnett.
Like H-1152, trifluoperazine (50 µM) and wortmannin (1 µM) inhibited Bay K 8644-induced steady-state increases in tension and MLC phosphorylation and did not inhibit basal MLC phosphorylation (Figure 3). At the concentration of H-1152 used, neither MLC phosphorylation nor tension induced by Bay K 8644 were inhibited completely (i.e. they were not reduced all the way to the basal levels, see Figure 2B,C). By contrast, trifluoperazine and wortmannin abolished the Bay K 8644-induced increase in MLC phosphorylation and caused complete (trifluoperazine) and nearly complete (wortmannin) relaxation (Figure 3). These data together support the hypothesis that ROCK, MLCK and calmodulin participate in Bay K 8644-induced contraction, but that these proteins may not participate in maintaining the basal level of MLC phosphorylation.
Effect of H-1152, trifluoperazine, wortmannin and ML-7 on Bay K 8644-induced increase in [Ca2+]i
To determine whether H-1152 and other inhibitors caused relaxation by reducing [Ca2+]i, tissues were loaded with the Ca2+ indicator, fura-2, and tension and calcium were simultaneously measured over time. Bay K 8644 induced a strong and sustained increase in [Ca2+]i (Figure 4B) concomitantly with an increase in tension (Figure 4A). When added during the sustained phase of contraction, 1 µM H-1152 produced an immediate relaxation (Figure 4C,E) without causing a reduction in [Ca2+]i (Figure 4D,F). Trifluoperazine, wortmannin and ML-7 also caused a strong relaxation of Bay K 8644-induced tension (Figure 4E). Like H-1152, 1 µM wortmannin did not affect the Bay K 8644-induced increase in [Ca2+]i (Figure 4F). However, 50 µM trifluoperazine and 3 µM ML-7 reduced the Bay K 8644-induced increase in [Ca2+]i nearly to the basal level (Figure 4F). Thus, the relaxation and reduction in MLC phosphorylation induced by H-1152 and wortmannin were not caused by reductions in [Ca2+]i, whereas inhibition of Ca2+ entry participated in causing relaxation induced by trifluoperazine and ML-7.
Figure 4.
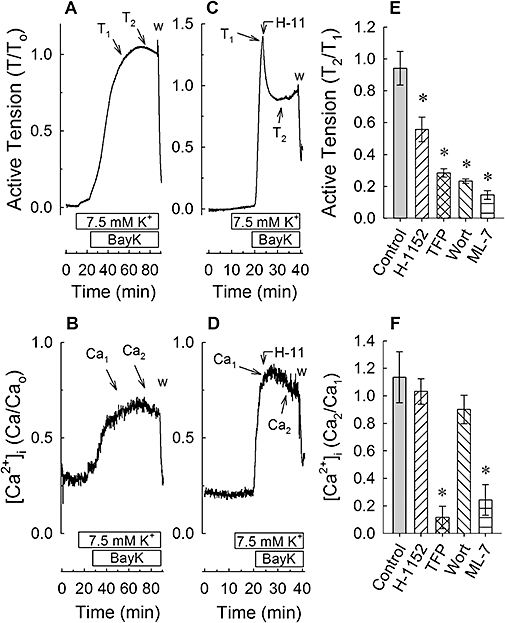
Examples of 30 nM Bay K 8644 (BayK)-induced tension (A,C) and [Ca2+]i (B,D) versus time tracings, and summary tension (E) and [Ca2+]i (F) data produced in the absence of an inhibitor (A and B and ‘Control’ in E and F) and after addition of an inhibitor (C,D and inhibitors in E,F). Inhibitors H-1152, trifluoperazine (TFP), wortmannin (Wort) and ML-7 were added to tissues once contractions achieved a pseudo-steady state [arrows linking H-11 (H-1152) to the tension (C) and [Ca2+]i (D) tracings]. T1 and Ca1 indicate the times at which, respectively, pre-drug tensions and [Ca2+]i values were recorded. T2 and Ca2 indicate the times at which, respectively, post-drug tensions and [Ca2+]i values were recorded. Data in (E) and (F) are means ± SE, n = 3–4. *P < 0.05 compared with control; anova /Dunnett.
Effect of H-1152, trifluoperazine and wortmannin on MYPT1 phosphorylation
Myosin light chain phosphatase inhibition in smooth muscle involves phosphorylation of MYPT1 at both T696 and T853 (human numbering scheme). To determine whether Bay K 8644 produces an increase in MLC phosphorylation by causing MYPT1 phosphorylation, tissues were stimulated with Bay K 8644 and frozen at steady-state contraction to quantify by Western blot the levels of MYPT1p-T696 and MYPT1-pT853. Unlike KCl, which produces at least a twofold increase in MYPT1-pT853 in tonic arteries (Porter et al., 2006; Dimopoulos et al., 2007), Bay K 8644 did not increase MYPT1p-T853 above the basal level of ‘1’[Figure 5B,E,H, ‘Basal (−)’ vs. ‘BayK (−)’; note that when pooling the data from these 3 experiments, the ‘BayK (−)’ value was 1.14 ± 0.04, n = 19, and this was not significantly greater than the basal level of ‘1’, P = 0.17]. Moreover, Bay K 8644 did not induce an increase in MYPT1-p696 (Figure 5). Thus, the increase in MLC phosphorylation induced by Bay K 8644 was not associated with significant increases in MYPT1 phosphorylation. Notably however, H-1152 (1 µM) abolished basal MYPT1-pT853 and did not affect basal MYPT1-T696 (Figure 5). Moreover, in the presence of H-1152, Bay K 8644 did not increase MYPT1-pT853 above the H-1152-inhibited basal level (Figure 5A,B).
Neither trifluoperazine nor wortmannin altered MYPT1 phosphorylation in the presence or absence of Bay K 8644 (Figure 5). These data support the hypotheses that MLCK and calmodulin do not play a role in MYPT1 phosphorylation, that ROCK is constitutively active and that an increase in Ca2+ induced by Bay K 8644 does not increase the activity of ROCK.
The rate of contraction induced by Bay K 8644 often displayed tissue-to-tissue variability. Thus, we may have missed a potential transient increase in MYPT1 phosphorylation by freezing tissues at a single time point once tissues had achieved a stable steady-state contraction. We therefore adopted a protocol where tissues generated more rapid contractions that reproducibly reached a steady-state value within 15 min. Tissues were incubated for 30 min in a nominally Ca2+-free solution (0 Ca2+; CaCl2 was omitted from the PSS) in the presence of 100 nM Bay K 8644 plus 10 mM KCl, then contracted by addition of 2 mM CaCl2 (Figure 6A). Tissues were frozen at 5, 15 and 60 min and processed to measure active RhoA, MLC phosphorylation and MYPT1 phosphorylation. Control tissues were either exposed to 2 mM CaCl2 and not Bay K 8644, or to Bay K 8644 and not 2 mM CaCl2. Addition of 2 mM CaCl2 to tissues bathed in 0 Ca2+ plus 100 nM Bay K 8644 and 10 mM KCl produced a monotonic contraction that reached steady state within ∼15 min and maintained strong tension ∼75% of that produced by 110 mM KCl for at least 60 min (Figure 6A). MLC phosphorylation was increased by 5 min and remained at an elevated level for the duration of contraction (Figure 6B). Interestingly, RhoA was elevated only transiently (at 5 min) despite maintenance of tension for 60 min (Figure 6C). MYPT1p-T853 (Figure 6D) and MYPT1p-T696 (Figure 6E) were not elevated at any of the time points measured. These experiments were also performed in femoral arteries and, except for a more rapid and stronger contraction produced in femoral artery compared with renal artery, the results were the same (Figure 7), suggesting that the conclusion of this study is not confined to one specific tissue.
H-1152 is not entirely selective for inhibition of ROCK. Therefore, based on an extensive study by Cohen's group (Bain et al., 2007) comparing the specificities of 65 inhibitors of protein kinases against a panel of 70–80 protein kinases, we chose KT-5720 to serve as a control for potential non-selective activity of H-1152 in our tissue. Specifically, 1 µM KT-5720 and 1 µM H-1152 inhibit multiple protein kinases in common that may alter smooth muscle contraction, including protein kinase C-related kinase-2 (PRK2) and 3-phosphoinositide-dependent protein kinase (PDK1), and 1 µM KT-5720 is a poor inhibitor of ROCK whereas 1 µM H-1152 is a strong inhibitor of ROCK. Unlike 1 µM H-1152 that produced a rapid and strong inhibition of a Ca2+-induced contraction in the presence of Bay K 8644 (Figure 8A) and inhibited basal MYPT1-pT853 (Figure 8C), 1 µM KT-5720 had no effect on tension (Figure 8B) or MYPT1 phosphorylation (Figure 8D). Unlike H-1152, 1 µM KT-5720 is a good inhibitor of mitogen-activated protein kinase kinase-1 (MEK1) (Bain et al., 2007). Thus, to confirm that KT-5720 was active in our tissue, we determined whether KT-5720 could inhibit basal ERK phosphorylation, and compared this with the effect of the MEK1 inhibitor U-0126. We had previously shown that another MEK1 inhibitor, PD-98059, inhibits basal ERK phosphorylation in rabbit femoral artery (Ratz, 2001). As expected, both 1 µM U-0126 and 1 µM KT-5720 reduced the level of basal ERK phosphorylation (Figure 8E), indicating that KT-5720 was active in rabbit artery.
Effect of staurosporine and H-1152 on basal MLC phosphorylation and MYPT1 phosphorylation
The general protein Ser/Thr kinase inhibitor staurosporine at 1 µM and 10 µM concentration-dependently reduced the basal level of MLC phosphorylation (Figure 9A), indicating that a kinase was responsible for the basal level of MLC phosphorylation. Staurosporine, like H-1152, inhibited basal MYPT1-pT853, but unlike H-1152, inhibited also basal MYPT1-696 (Figure 9B). Inhibition by 10 µM staurosporine was not greater than that induced by 1 µM staurosporine (Figure 9B).
Staurosporine and H-1152 both inhibited basal MYPT1-pT853, but whereas staurosporine inhibited basal MLC phosphorylation, H-1152 did not appear to do so (see Figure 2A,B). However, the Western blot analysis for H-1152 was different than that for staurosporine because the effect of H-1152 was normalized to the level of MLC phosphorylation induced by Bay K 8644 rather than to the basal (pre-stimulus) level. When the effect of H-1152 on basal MLC phosphorylation was re-examined using a protocol similar to that employed for staurosporine, we found that H-1152 appeared to cause a concentration-dependent inhibition of basal MLC phosphorylation (Figure 9C). That is, basal MLC phosphorylation was reduced ∼20% and ∼40% by, respectively, 1 µM and 3 µM H-1152. Although the reduction in the average MLC phosphorylation value induced by 3 µM H-1152 was statistically significant, that induced by 1 µM H-1152 was not. H-1152 also induced a concentration-dependent inhibition of basal MYPT1-pT853 (Figure 9D), but even at 3 µM, H-1152 had no effect on basal MYPT1-p696 (Figure 9D).
Discussion
Results from the present study indicate that increases in tension induced by Bay K 8644 in the presence of extracellular Ca2+ correlated with increases above the basal levels in [Ca2+]i and MLC phosphorylation but not MYPT1 phosphorylation. MYPT1 phosphorylation is a measure of ROCK activity (Kawano et al., 1999; Liu and Liao, 2008). Thus, these data suggest that a strong elevation in [Ca2+]i resulting in tension maintenance at levels comparable to those induced by KCl may not necessarily activate ROCK. These data are the first to present an alternative to the hypothesis proposing that KCl activates ROCK by increasing [Ca2+]i (Sakurada et al., 2003; Urban et al., 2003; Janssen et al., 2004). Despite the absence of an elevation in phosphorylation of the ROCK substrate, MYPT1, ROCK inhibitors quite effectively reduced Bay K 8644-stimulated increases in MLC phosphorylation and tension. Notably, the ROCK inhibitor H-1152 dramatically reduced the basal level of MYPT1-pT853 without affecting basal MYPT1-pT696. Moreover, H-1152 at 3 µM significantly reduced the basal level of MLC phosphorylation by ∼40% (see Figure 9A). Although 1 µM H-1152 did not cause a significant inhibition of basal MLC phosphorylation, the average level was reduced by ∼20%. The general Ser/Thr kinase inhibitor staurosporine likewise reduced basal MLC phosphorylation and MYPT1-pT853, but also reduced basal MYPT1-pT696, even at the relatively low concentration of 1 µM. Smooth muscle, like striated muscle, is generally considered to be ‘off’ until ‘turned on’ by a contractile stimulus. However, a great deal of data now support a model in which key signalling systems that participate in regulation of contraction are partially ‘on’ when smooth muscle cells are in the basal, resting state (Guo et al., 2003; Navedo et al., 2005; Poley et al., 2008). These data together support a novel model in which constitutive MLCP activity is regulated by constitutive ROCK activity, and possibly by other kinases that are also constitutively active and sensitive to inhibition by 1 µM staurosporine (Wilson et al., 2005).
Our working model supports the notion that inhibition of constitutive ROCK activity would result in inhibition of steady-state MLC phosphorylation and tension stimulated by any agent that elevates [Ca2+]i, including Bay K 8644, KCl and GPCR agonists. A recent study by Rembold et al. (2004) shows that there is a threshold level of MLC phosphorylation of ∼15% that must be achieved before muscle tension will increase. This information, combined with results from the present study showing that basal MLC phosphorylation can be dramatically reduced by 1 µM staurosporine and 3 µM H-1152, suggests that contractile stimuli need not elevate ROCK activity to cause strong tension maintenance. What appears to be at least as important as stimulus-induced activation of MLCK is the level of constitutive ROCK activity. In short, agents that increase [Ca2+]i would induce weaker increases in MLC phosphorylation when basal ROCK activity is reduced. This is due to the resulting elevated basal MLCP activity that would shift basal MLC phosphorylation further from the threshold for contraction. This model does not exclude the contribution made by activation of ROCK, PKC and other kinases during stimulation of certain GPCRs, which would add to the level of basal activation and MLCK-induced activation of the MLC phosphorylation.
In cells and tissues, MYPT1-pT696 and MYPT1-pT853 appear to be preferentially phosphorylated by different kinases. In particular, MYPT1-pT853 is the preferred substrate for ROCK, whereas MYPT1-pT696 is the preferred substrate for one or more kinases other than ROCK (Kawano et al., 1999; Muranyi et al., 2005; Yoneda et al., 2005). Moreover, studies using intact smooth muscle tissues support the hypothesis that contractile agonists cause increases in MYPT1-pT853 rather than MYPT1-p696 (Kitazawa et al., 2003; Porter et al., 2006), and quantification of the level of MYPT1-pT853 is used as a measure of ROCK activity in cells and tissues (Liu and Liao, 2008). Although several kinases can phosphorylate MYPT1 at T696 in vitro, including ROCK (Kimura et al., 1996), in the present study, even 3 µM H-1152 did not reduce the basal level of MYPT1-pT696, while abolishing basal MYPT1-pT853 and reducing basal MLC phosphorylation. Wilson et al. (2005) have shown that integrin-linked kinase (ILK) is the kinase responsible for Ca2+-independent MLC phosphorylation and contraction in response to microcystin-mediated phosphatase inhibition. What is most relevant to the present study is that these investigators showed that zipper-interacting kinase was not inhibited by 1 µM staurosporine, but that ILK is. The present study showed that 1 µM staurosporine was sufficient to cause a strong reduction in the basal levels of MLC phosphorylation and MYPT1 phosphorylation at both T853 and T696. Together, these data suggest that zipper-interacting kinase was not responsible for the basal level of MYPT1p-T696. Although still speculative, our data support the notion that the decreases in basal MYPT1-pT853 and MYPT1-T696 were due, respectively, to inhibition of constitutive ROCK and ILK. However, additional studies are required to substantiate this hypothesis.
The tonic phase of KCl-induced contraction is attenuated by ROCK inhibitors, which has led to the hypothesis that strongly depolarizing KCl concentrations can activate ROCK (Mita et al., 2002; Sakamoto et al., 2003; Sakurada et al., 2003; Urban et al., 2003). KCl causes both an increase in activated RhoA (Sakurada et al., 2003) and increases in MYPT1-pT853 (Kitazawa et al., 2003; Porter et al., 2006). An increase in [Ca2+]i produced upon membrane depolarization with KCl has been implicated in the activation of ROCK (Sakurada et al., 2003). However, our data using Bay K 8644, a dihydropyridine Ca2+ channel agonist that elevates [Ca2+]i by directly activating voltage-operated Ca2+ channels rather than by causing membrane depolarization, did not elevate the level of either MYPT1-pT853 or MYPT1-T696, but did increase MLC phosphorylation while transiently increasing the level of active RhoA (see Figures 6 and 7). The steady-state level of MLC phosphorylation produced by Bay K 8644 was reduced by 1 µM H-1152 and by 1 µM wortmannin, a MLCK inhibitor. These data, along with the finding that the Bay K 8644-induced contraction was absolutely dependent on extracellular Ca2+, support the hypothesis that Bay K 8644 causes contraction by elevating Ca2+ entry, [Ca2+]i and activation of MLCK. We propose that H-1152 caused a reduction in the levels of MYPT1-pT853 and MLC phosphorylation during stimulation with Bay K 8644 because constitutively active ROCK was inhibited, and not because ROCK was ‘turned on’ by the elevation in [Ca2+]i. Sakurada et al. (2003) showed that KCl causes a sustained increase above the basal level in active RhoA. Thus, our data showing that an elevation in [Ca2+]i induced by Bay K 8644 caused only a transient increase in RhoA suggests that the depolarization rather than increase in [Ca2+]i induced by high depolarizing KCl concentrations may have led to the sustained RhoA activation. Sustained rather than transient RhoA activation may be necessary for activation of ROCK (and thus, MYPT1 phosphorylation) to levels above the resting basal values. Alternatively, KCl may activate signalling systems other than ROCK leading to elevations above the basal level in MYPT1 phosphorylation (Ratz and Miner, 2009; Ratz et al., 2009)
RhoA kinase is a downstream target of phosphoinositide 3-kinase (Wang et al., 2006). Although wortmannin was originally identified as a MLCK inhibitor, it was subsequently shown to also potently inhibit phosphoinositide 3-kinase. In the present study, wortmannin nearly abolished Bay K 8644-induced tension and MLC phosphorylation and had no effect on MYPT1-pT696 or MYPT1-pT853. These data suggest that phosphoinositide 3-kinase does not appear to play a role in constitutive ROCK activity or in Bay K 8644-induced contraction.
In summary, our data support a model in which constitutive ROCK activity rather than Ca2+-induced activation of ROCK can explain the ROCK-dependent contraction induced by Bay K 8644. The implication of these data is that the contraction resulting from any stimulus that increases [Ca2+]i would be expected to be attenuated by ROCK inhibitors because of inhibition of constitutive ROCK activity, and not necessarily because ROCK was activated by the stimulus. These data provide renewed incentive to understand how ROCK may participate in vascular hyper-contractile disorders such as hypertension and coronary artery vasospasm (Uehata et al., 1997; Masumoto et al., 2002; Seasholtz et al., 2006; Sonkusare et al., 2006).
Acknowledgments
This work was supported by a grant from the National Institutes of Health R01-HL61320.
Glossary
Abbreviations:
- [Ca2+]i
intracellular free Ca2+
- CPI-17
17 kDa protein kinase C inhibitory protein
- GPCR
G protein-coupled receptors
- ILK
integrin-linked kinase
- MLC
myosin light chain
- MLCK
myosin light chain kinase
- MLCP
myosin light chain phosphatase
- MLCp
myosin light chain phosphorylation
- MYPT1
myosin phosphatase targeting subunit
- PKC
protein kinase C
- ROCK
RhoA kinase
Statement of conflict of interest
The authors declare no conflict of interest.
References
- Asano M, Nomura Y. Comparison of inhibitory effects of Y-27632, a Rho kinase inhibitor, in strips of small and large mesenteric arteries from spontaneously hypertensive and normotensive Wistar-Kyoto rats. Hypertens Res. 2003;26:97–106. doi: 10.1291/hypres.26.97. [DOI] [PubMed] [Google Scholar]
- Bain J, Plater L, Elliott M, Shpiro N, Hastie CJ, McLauchlan H, et al. The selectivity of protein kinase inhibitors: a further update. Biochem J. 2007;408:297–315. doi: 10.1042/BJ20070797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown AM, Kunze DL, Yatani A. The agonist effect of dihydropyridines on Ca channels. Nature. 1984;311:570–572. doi: 10.1038/311570a0. [DOI] [PubMed] [Google Scholar]
- Brozovich FV. Rho signaling: agonist stimulation and depolarization come together. Circ Res. 2003;93:481–483. doi: 10.1161/01.RES.0000093183.00556.D5. [DOI] [PubMed] [Google Scholar]
- Casteels R, Kitamura K, Kuriyama H, Suzuki H. Excitation-contraction coupling in the smooth muscle cells of the rabbit main pulmonary artery. J Physiol. 1977;271:63–79. doi: 10.1113/jphysiol.1977.sp011990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen DM, Murphy RA. Cellular thin filament protein contents and force generation in porcine arteries and veins. Circ Res. 1979;45:661–665. doi: 10.1161/01.res.45.5.661. [DOI] [PubMed] [Google Scholar]
- Dimopoulos GJ, Semba S, Kitazawa K, Eto M, Kitazawa T. Ca2+-dependent rapid Ca2+ sensitization of contraction in arterial smooth muscle. Circ Res. 2007;100:121–129. doi: 10.1161/01.RES.0000253902.90489.df. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng J, Ito M, Kureishi Y, Ichikawa K, Amano M, Isaka N, et al. Rho-associated kinase of chicken gizzard smooth muscle. J Biol Chem. 1999;274:3744–3752. doi: 10.1074/jbc.274.6.3744. [DOI] [PubMed] [Google Scholar]
- Ghisdal P, Vandenberg G, Morel N. Rho-dependent kinase is involved in agonist-activated calcium entry in rat arteries. J Physiol. 2003;551:855–867. doi: 10.1113/jphysiol.2003.047050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gosens R, Schaafsma D, Meurs H, Zaagsma J, Nelemans SA. Role of Rho-kinase in maintaining airway smooth muscle contractile phenotype. Eur J Pharmacol. 2004;483:71–78. doi: 10.1016/j.ejphar.2003.10.027. [DOI] [PubMed] [Google Scholar]
- Guo Z, Su W, Ma Z, Smith GM, Gong MC. Ca2+-independent phospholipase A2 is required for agonist-induced Ca2+ sensitization of contraction in vascular smooth muscle. J Biol Chem. 2003;278:1856–1863. doi: 10.1074/jbc.M211075200. [DOI] [PubMed] [Google Scholar]
- Janssen LJ, Tazzeo T, Zuo J, Pertens E, Keshavjee S. KCl evokes contraction of airway smooth muscle via activation of RhoA and Rho-kinase. Am J Physiol Lung Cell Mol Physiol. 2004;287:L852–L858. doi: 10.1152/ajplung.00130.2004. [DOI] [PubMed] [Google Scholar]
- Kamm KE, Stull JT. The function of myosin and myosin light chain kinase phosphorylation in smooth muscle. Annu Rev Pharmacol Toxicol. 1985;25:593–620. doi: 10.1146/annurev.pa.25.040185.003113. [DOI] [PubMed] [Google Scholar]
- Karaki H, Sato K, Ozaki H. Different effects of norepinephrine and KCl on the cytosolic Ca2+-tension relationship in vascular smooth muscle of rat aorta. Eur J Pharmacol. 1988;151:325–328. doi: 10.1016/0014-2999(88)90817-5. [DOI] [PubMed] [Google Scholar]
- Kawano Y, Fukata Y, Oshiro N, Amano M, Nakamura T, Ito M, et al. Phosphorylation of myosin-binding subunit (MBS) of myosin phosphatase by Rho-kinase in vivo. J Cell Biol. 1999;147:1023–1038. doi: 10.1083/jcb.147.5.1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura K, Ito M, Amano M, Chihara K, Fukata Y, Nakafuku M, et al. Regulation of myosin phosphatase by Rho and Rho-associated kinase (Rho-kinase) Science. 1996;273:245–248. doi: 10.1126/science.273.5272.245. [DOI] [PubMed] [Google Scholar]
- Kitazawa T, Eto M, Woodsome TP, Khalequzzaman M. Phosphorylation of the myosin phosphatase targeting subunit and CPI-17 during Ca(2+) sensitization in rabbit smooth muscle. J Physiol. 2003;546:879–889. doi: 10.1113/jphysiol.2002.029306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kupittayanant S, Burdyga T, Wray S. The effects of inhibiting Rho-associated kinase with Y-27632 on force and intracellular calcium in human myometrium. Pflugers Arch. 2001;443:112–114. doi: 10.1007/s004240100668. [DOI] [PubMed] [Google Scholar]
- Liu PY, Liao JK. A method for measuring Rho kinase activity in tissues and cells. In: Balch WE, Der CJ, Hall A, editors. Small Gtpases in Disease, Pt B. San Diego, CA: Elsevier Academic Press Inc.; 2008. pp. 181–189. Vol. 439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masumoto A, Mohri M, Shimokawa H, Urakami L, Usui M, Takeshita A. Suppression of coronary artery spasm by the Rho-kinase inhibitor fasudil in patients with vasospastic angina. Circulation. 2002;105:1545–1547. doi: 10.1161/hc1002.105938. [DOI] [PubMed] [Google Scholar]
- Mita M, Yanagihara H, Hishinuma S, Saito M, Walsh MP. Membrane depolarization-induced contraction of rat caudal arterial smooth muscle involves Rho-associated kinase. Biochem J. 2002;364:431–440. doi: 10.1042/BJ20020191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muranyi A, Derkach D, Erdodi F, Kiss A, Ito M, Hartshorne DJ. Phosphorylation of Thr695 and Thr850 on the myosin phosphatase target subunit: inhibitory effects and occurrence in A7r5 cells. FEBS Lett. 2005;579:6611–6615. doi: 10.1016/j.febslet.2005.10.055. [DOI] [PubMed] [Google Scholar]
- Navedo MF, Amberg GC, Votaw VS, Santana LF. Constitutively active l-type Ca2+ channels. Proc Natl Acad Sci USA. 2005;102:11112–11117. doi: 10.1073/pnas.0500360102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poley RN, Dosier CR, Speich JE, Miner AS, Ratz PH. Stimulated calcium entry and constitutive RhoA kinase activity cause stretch-induced detrusor contraction. Eur J Pharmacol. 2008;599:137–145. doi: 10.1016/j.ejphar.2008.09.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter M, Evans MC, Miner AS, Berg KM, Ward KR, Ratz PH. Convergence of calcium desensitizing mechanisms activated by forskolin and phenylephrine pretreatment, but not 8-bromo-cGMP. Am J Physiol Cell Physiol. 2006;290:C1552–C1559. doi: 10.1152/ajpcell.00534.2005. [DOI] [PubMed] [Google Scholar]
- Ratz PH. High α1-adrenergic receptor occupancy decreases relaxing potency of nifedipine by increasing myosin light chain phosphorylation. Circ Res. 1993;72:1308–1316. doi: 10.1161/01.res.72.6.1308. [DOI] [PubMed] [Google Scholar]
- Ratz PH. Dependence of Ca2+ sensitivity of arterial contractions on history of receptor activation. Am J Physiol. 1999;277:H1661–H1668. doi: 10.1152/ajpheart.1999.277.5.H1661. [DOI] [PubMed] [Google Scholar]
- Ratz PH. Regulation of ERK phosphorylation in differentiated arterial muscle of the rabbit. Am J Physiol. 2001;281:H114–H123. doi: 10.1152/ajpheart.2001.281.1.H114. [DOI] [PubMed] [Google Scholar]
- Ratz PH, Miner AS. Role of protein kinase Czeta and calcium entry in KCl-induced vascular smooth muscle calcium sensitization and feedback control of cellular calcium levels. J Pharmacol Exp Ther. 2009;328:399–408. doi: 10.1124/jpet.108.142422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratz PH, Hai C-M, Murphy RA. Dependence of stress on cross-bridge phosphorylation in vascular smooth muscle. Am J Physiol. 1989;256:C96–C100. doi: 10.1152/ajpcell.1989.256.1.C96. [DOI] [PubMed] [Google Scholar]
- Ratz PH, Berg KM, Urban NH, Miner AS. Regulation of smooth muscle calcium sensitivity: KCl as a calcium-sensitizing stimulus. Am J Physiol Cell Physiol. 2005;288:C769–C783. doi: 10.1152/ajpcell.00529.2004. [DOI] [PubMed] [Google Scholar]
- Ratz PH, Miner AS, Barbour SE. Calcium-independent phospholipase A2 participates in KCl-induced calcium sensitization of vascular smooth muscle. Cell Calcium. 2009;46:65–72. doi: 10.1016/j.ceca.2009.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rembold CM, Wardle RL, Wingard CJ, Batts TW, Etter EF, Murphy RA. Cooperative attachment of cross bridges predicts regulation of smooth muscle force by myosin phosphorylation. Am J Physiol Cell Physiol. 2004;287:C594–C602. doi: 10.1152/ajpcell.00082.2004. [DOI] [PubMed] [Google Scholar]
- Sakamoto K, Hori M, Izumi M, Oka T, Kohama K, Ozaki H, et al. Inhibition of high K+-induced contraction by the ROCKs inhibitor Y-27632 in vascular smooth muscle: possible involvement of ROCKs in a signal transduction pathway. J Pharmacol Sci. 2003;92:56–69. doi: 10.1254/jphs.92.56. [DOI] [PubMed] [Google Scholar]
- Sakurada S, Takuwa N, Sugimoto N, Wang Y, Seto M, Sasaki Y, et al. Ca2+-dependent activation of Rho and Rho kinase in membrane depolarization-induced and receptor stimulation-induced vascular smooth muscle contraction. Circ Res. 2003;93:548–556. doi: 10.1161/01.RES.0000090998.08629.60. [DOI] [PubMed] [Google Scholar]
- Schramm M, Thomas G, Towart R, Franckowiak G. Novel dihydropyridines with positive inotropic action through activation of Ca2+ channels. Nature. 1983;303:535–537. doi: 10.1038/303535a0. [DOI] [PubMed] [Google Scholar]
- Seasholtz TM, Wessel J, Rao F, Rana BK, Khandrika S, Kennedy BP, et al. Rho kinase polymorphism influences blood pressure and systemic vascular resistance in human twins: role of heredity. Hypertension. 2006;47:937–947. doi: 10.1161/01.HYP.0000217364.45622.f0. [DOI] [PubMed] [Google Scholar]
- Smani T, Zakharov SI, Csutora P, Leno E, Trepakova ES, Bolotina VM. A novel mechanism for the store-operated calcium influx pathway. Nat Cell Biol. 2004;6:113–120. doi: 10.1038/ncb1089. [DOI] [PubMed] [Google Scholar]
- Somlyo AP, Himpens B. Cell calcium and its regulation in smooth muscle. FASEB J. 1989;3:2266–2276. doi: 10.1096/fasebj.3.11.2506092. [DOI] [PubMed] [Google Scholar]
- Somlyo AP, Somlyo AV. Ca2+ sensitivity of smooth muscle and nonmuscle myosin II: modulated by G proteins, kinases, and myosin phosphatase. Physiol Rev. 2003;83:1325–1358. doi: 10.1152/physrev.00023.2003. [DOI] [PubMed] [Google Scholar]
- Somlyo AP, Xuqiong W, Walker L, Somlyo A. Pharmacomechanical coupling: the role of calcium, G-proteins, kinases and phosphatase. Rev Physiol Biochem Pharmacol. 1999;440:183–187. doi: 10.1007/3-540-64753-8_5. [DOI] [PubMed] [Google Scholar]
- Sonkusare S, Palade PT, Marsh JD, Telemaque S, Pesic A, Rusch NJ. Vascular calcium channels and high blood pressure: pathophysiology and therapeutic implications. Vascul Pharmacol. 2006;44:131–142. doi: 10.1016/j.vph.2005.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uehata M, Ishizaki T, Satoh H, Ono T, Kawahara T, Morishita T, et al. Calcium sensitization of smooth muscle mediated by a Rho-associated protein kinase in hypertension. Nature. 1997;389:990–994. doi: 10.1038/40187. [DOI] [PubMed] [Google Scholar]
- Urban NH, Berg KM, Ratz PH. K+ depolarization induces RhoA kinase translocation to caveolae and Ca2+ sensitization of arterial muscle. Am J Physiol Cell Physiol. 2003;285:C1377–C1385. doi: 10.1152/ajpcell.00501.2002. [DOI] [PubMed] [Google Scholar]
- Wang Y, Yoshioka K, Azam MA, Takuwa N, Sakurada S, Kayaba Y, et al. Class II phosphoinositide 3-kinase alpha-isoform regulates Rho, myosin phosphatase and contraction in vascular smooth muscle. Biochem J. 2006;394:581–592. doi: 10.1042/BJ20051471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson DP, Sutherland C, Borman MA, Deng JT, Macdonald JA, Walsh MP. Integrin-linked kinase is responsible for Ca2+-independent myosin diphosphorylation and contraction of vascular smooth muscle. Biochem J. 2005;392:641–648. doi: 10.1042/BJ20051173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanagisawa T, Okada Y. KCl depolarization increases Ca2+ sensitivity of contractile elements in coronary arterial smooth muscle. Am J Physiol. 1994;267:H614–H621. doi: 10.1152/ajpheart.1994.267.2.H614. [DOI] [PubMed] [Google Scholar]
- Yoneda A, Multhaupt HA, Couchman JR. The Rho kinases I and II regulate different aspects of myosin II activity. J Cell Biol. 2005;170:443–453. doi: 10.1083/jcb.200412043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshioka K, Sugimoto N, Takuwa N, Takuwa Y. Essential role for class II phosphoinositide 3-kinase alpha-isoform in Ca2+-induced, Rho- and Rho kinase-dependent regulation of myosin phosphatase and contraction in isolated vascular smooth muscle cells. Mol Pharmacol. 2007;71:912–920. doi: 10.1124/mol.106.032599. [DOI] [PubMed] [Google Scholar]