

Abstract
Background and purpose:
We showed previously that a new Pt(II) complex ([Pt(O,O′-acac)(γ-acac)(DMS)]) exerted high and fast apoptotic processes in MCF-7 cells. The objective of this study was to investigate the hypothesis that [Pt(O,O′-acac)(γ-acac)(DMS)] is also able to exert anoikis and alter the migration ability of MCF-7 cells, and to show some of the signalling events leading to these alterations.
Experimental approach:
Cells were treated with sublethal doses of [Pt(O,O′-acac)(γ-acac)(DMS)], and the efficiency of colony initiation and anchorage-independent growth was assayed; cell migration was examined by in vitro culture wounding assay. Gelatin zymography for MMP-2 and -9 activities, Western blottings of MMPs, MAPKs, Src, PKC-ε and FAK, after [Pt(O,O′-acac)(γ-acac)(DMS)] treatment, were also performed.
Key results:
Sub-cytotoxic drug concentrations decreased the: (i) anchorage-dependent and -independent growth; (ii) migration ability; and (iii) expression and activity of MMP-2 and MMP-9. [Pt(O,O′-acac)(γ-acac)(DMS)] provoked the generation of reactive oxygen species (ROS), and the activation of p38MAPK, Src and PKC-ε. p38MAPK phosphorylation, cell anoikis and migration due to [Pt(O,O′-acac)(γ-acac)(DMS)] were blocked by PKC-ε inhibition. Furthermore, Src inhibition blocked the [Pt(O,O′-acac)(γ-acac)(DMS)]-provoked activation of PKC-ε, while ROS generation blockage inhibited the activation of Src, and also the decrement of phosphorylated FAK observed in detached [Pt(O,O′-acac)(γ-acac)(DMS)]-treated cells.
Conclusions and implications:
Sublethal concentrations of [Pt(O,O′-acac)(γ-acac)(DMS)] induced anoikis and prevented events leading to metastasis via alterations in cell migration, anchorage independency, stromal interactions and MMP activity. Hence, [Pt(O,O′-acac)(γ-acac)(DMS)] may be a promising therapeutic agent for preventing growth and metastasis of breast cancer.
Keywords: [Pt(O,O′-acac)(γ-acac)(DMS)]; motility; anoikis; p38MAPK; PKC-ε; ROS; MCF-7
Introduction
Breast cancer is the most common cancer in women and a frequent cause of cancer deaths in women (Ferlay et al., 2004). Most deaths from breast cancer are due to metastases that are resistant to conventional therapies. During the progress of a neoplasm, tumour cells can detach themselves from the primary tumour at an early stage, travel through the blood or lymph pathways and acquire further alterations in secondary organs under the selective influence of their new microenvironment. Cell migration is a complex dynamic process that involves multiple biological features. In order for malignant cells to leave the epithelium, after overcoming normal cell adhesion, migrating cells must break through the basement membrane by secreting enzymes called metalloproteinases (Wolf and Friedl, 2009). MMPs are collagenases (e.g. MMP-1, MMP-13), stromelysins (e.g. MMP-10, MMP-12), gelatinases (e.g. MMP-2, MMP-9) or membrane-type enzymes (e.g. MMP-14, MMP-16), and have been recognized as important mediators of extracellular matrix (ECM) degradation. In almost all human cancers, the increased expression of certain MMPs correlates with tumour expansion, increased invasiveness and poor prognosis (Wolf and Friedl, 2009). MMP-2 and MMP-9 appear to have clinical value as diagnostic factors for breast cancer or predictive factors for metastases. In addition, proportions between the different forms or between MMPs and their tissue inhibitors (TIMPs), in terms of concentration or activity, could provide useful clinical information on breast cancer disease and classification (Somiari et al., 2006).
Not only proteolytic enzymes, but also other molecular factors, such as angiogenic factors and cell surface adhesion proteins, are involved in adhesion and migration, and contribute to the formation of metastases. Furthermore, changes in the organization and distribution of cytoskeletal proteins are necessary for focal adhesion formation, cell motility and cell invasion (Ridley, 2001). Integrin–ECM interaction leads to activation of signalling modules such as focal adhesion kinase (FAK) and Src family tyrosine kinases. Hence, detachment from the cell substrate that activates FAK potentially stimulates maximal activity of both FAK and Src kinases, which are responsible for the anchorage-independent survival of the tumour cells and its signalling transduction. It is known that Src is one of the key protein tyrosine kinases involved in cell survival and the metastasis potential of malignant tumours (Jones et al., 2000; Schlessinger, 2000; Mitra and Schlaepfer, 2006). Reactive oxygen species (ROS) act as signalling molecules in the activation of several pathways, including FAK and mitogen-activated protein kinases (MAPKs) (Finkel, 2003), in mediating a wide range of cellular responses, such as proliferation, apoptosis, adhesion and migration. MAPKs including the extracellular signal-regulated protein kinase (ERK), c-jun N-terminal kinase (JNK) and p38MAPK, appear to be the key downstream effectors of FAK in mediating cell survival. Whereas ERK predominantly confers survival advantage to cells during most stress conditions in various cell types, JNK and p38MAPK are preferentially activated by stress-inducing stimuli, such as UV light and heat shock, and are associated with cytokine-induced cell migration in several cell types cell death (Blaschke et al., 2002). In addition, many studies have demonstrated that MAPKs have been correlated with the activation and expression of MMPs, and play a crucial role in cell migration (Mendes et al., 2007).
Recently, this group has synthesized and studied a platinum complex containing two acetylacetonate (acac) ligands, one O,O′-chelate and the other σ-linked by methine in the γ position, and dimethylsulphide (DMS) in the metal coordination sphere, that has shown interesting biological activities (De Pascali et al., 2005). This compound ([Pt(O,O′-acac)(γ-acac)(DMS)]) not only was able to induce apoptosis in endometrial cancer cells (HeLa), with activity up to about 100 times higher than that of cisplatin, but also showed high cytotoxicity in cisplatin-resistant MCF-7 breast cancer cells. Differently from cisplatin, for which the activity appears to be associated both with its intracellular accumulation and with the formation of DNA adducts (Muscella et al., 2007; 2008;), the cytotoxicity of this new compound is related to the intracellular accumulation only, showing a low reactivity with nucleobases and a specific reactivity with sulphur ligands, suggesting that the cellular targets could be amino acid residues of proteins. The different mechanism of action of the new complex, having biological targets, different from those of cisplatin, may render it intrinsically able to evoke less chemo-resistance. Theoretically, this compound could produce, in addition, a broader spectrum of application, and here we have assessed its ability to exert specific anti-metastatic responses in vitro. We provide evidence that low, sublethal doses of [Pt(O,O′-acac)(γ-acac)(DMS)] altered some basic biological features of the MCF-7 cells, such as anchorage-independent growth, migration and MMP production, and show some of the signalling events leading to these alterations.
Methods
Cell lines
MCF-7 cell line was cultured in Dulbecco's modified Eagle's medium (DMEM) (Celbio, Pero, Milan, Italy) supplemented with 10% fetal bovine serum, penicillin (100 U·mL−1) and streptomycin (100 mg·mL−1). Cells were grown to 70% confluence and then treated with [Pt(O,O′-acac)(γ-acac)(DMS)] at various concentrations and for different incubation periods.
Cytotoxicity assay
Cells at 70–80% confluency were treated with trypsin (0.25% trypsin with 1 mmol·L−1 EDTA), washed and resuspended in growth medium; 100 µL of a cell suspension (105 cells·mL−1) was added to each well of a 96-well plate. After overnight incubation, the cells were treated with specific reagents for different incubation periods. The conversion of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenol tetrazolium bromide (MTT) by cells was used as an indicator of cell number as previously described (Muscella et al., 2002). This method measures the reduction of MTT by active mitochondria, which results in a colour change measured at 550 nm wavelength. Experiments were performed to define the linear range of the assay. A good correlation was observed up to 5 × 104 cells per well (data not shown). Increasing the concentration of heat-killed cells per well (killed by incubating at 70°C for 15 min) caused no significant change in the absorbance; thus, this spectrophotometric method was a valid technique for measuring the number of viable cells. All subsequent experiments performed were within the linear range of the assay. The percentage cell survival was calculated as the absorbance ratio of treated-to-untreated cells. The data presented are means ± SD from eight replicate wells per microtitre plate, repeated four times.
Colony-forming assay
Cells were seeded in 100 mm culture dishes at low density (3 × 104 cells per dish) and left to adhere for 24 h in a standard medium. Increasing concentrations of [Pt(O,O′-acac)(γ-acac)(DMS)] were added; after 2 h, the cells were washed, immediately treated with trypsin, resuspended in single-cell suspension and plated for the determination of macroscopic colony formation. After 15 days of growth, colonies were fixed with a 3:1 mixture of methanol/acetic acid, and stained with crystal violet. Only colonies consisting of more than 50 cells were scored. Four separate experiments were performed using duplicate samples.
Soft agar assay
The soft agar assay was performed as previously described with slight modification (McPherson, 1973). Cells were seeded in 100 mm cell culture dishes at low density (3 × 104 per dish), and left to adhere for 24 h in a standard medium. Increasing concentrations of [Pt(O,O′-acac)(γ-acac)(DMS)] were added; after 2 h, the cells were washed, immediately treated with trypsin, resuspended in a medium containing 0.3% agar (Sigma, Milan, Italy) and allowed to grow for 15 days before counting viable colonies fixed with a 3:1 mixture of methanol/acetic acid and stained with crystal violet. Four separate experiments were performed using duplicate samples.
Cell wounding and migration assay
The cells were seeded in 12-well culture dishes and cultured until they reached confluence. The cells were then scraped with a 200 µL micro-pipette tip, denuding a strip of the monolayer approximately 500 µm in diameter. Variation in the wound diameter within experiments was approximately 5%. Cultures were washed twice with phosphate-buffered saline (PBS) to remove cell debris, and incubated for additional 6, 12 and 24 h. When specific inhibitors were used, an appropriate concentration of each inhibitor was added to the culture medium 30 min before [Pt(O,O′-acac)(γ-acac)(DMS)] treatment. After incubation, the cells were photographed with a digital camera, and the migrated area was measured using NIH Image (v1.63) software (National Institutes of Health, Bethesda, MD, USA). To ensure that the same wounds were compared, we used a permanent marker to make positioning marks at the bottom of the culture dishes. The migration area in the wound was calculated according to the following formula: cell free area at 0 h – cell free area at 16 h. At least 10 fields per dish were analysed, and the migrated area was expressed as a percentage of that in untreated control cells.
Collagen type I invasion assay
The assay was performed in 96-well plates as previously described (Bracke et al., 2001). The wells were coated with 24 µg·mL−1 type I collagen in PBS, incubated overnight at 4°C and then treated with 1% BSA (Fisher Scientific, Fair lawn, NJ, USA) for 1 h at 37°C. Untreated cells or cells treated for 2 h with [Pt(O,O′-acac)(γ-acac)(DMS)] were plated into coated wells (5 × 104 cells per well) and incubated at 37°C (5% CO2) for 60 min. Adherent cells, fixed with 3% paraformaldehyde for 10 min, washed with 2% methanol for 10 min, were stained with 0.5% crystal violet in 20% methanol for 10 min. The stain was eluted, and the absorbance at 540 nm was measured by a 96-well plate reader. The data presented are means ± SD from eight replicate wells per microtitre plate, repeated four times.
PolyHEMA assay
In order to completely prevent cell adhesion, cell culture dishes were coated with a film of polyHEMA (Sigma) following the protocol reported by Folkman and Moscona (1978). Briefly, a solution of 120 mg·mL−1 of polyHEMA in 95% ethanol was mixed overnight, centrifuged at 800×g for 30 min to remove undissolved particles and diluted 1:10 with 95% ethanol. Then, 0.95 µL·mm−2 was pipetted in dishes of 35 mm or multi-well plates, and was left to dry at room temperature. Before use, polyHEMA-coated dishes were washed twice in PBS.
Preparation of subcellular fraction
To obtain protein cell extracts, cells were washed twice in ice-cold PBS and harvested in 1 mL of PBS. The samples were centrifuged for 30 s at 10 000×g, and cell pellets were resuspended in the following buffer (mmol·L−1): 20 Tris–HCl (pH 8) containing 100 NaCl, 2 EDTA, 2 Na3VO4, 0.2% Nonidet P-40 and 10% glycerol, supplemented with a cocktail of protease inhibitors (1 µg·mL−1 of each of the proteinase inhibitors aprotinin, leupeptin, soybean trypsin inhibitor, and 1mmol·L−1 phenylmethylsulphonyl fluoride, all from Sigma, Milan, Italy). After a 10 min incubation on ice, the cells were passed several times through a 20-gauge needle, and then centrifuged at 13 000×g for 10 min at 4°C. For preparation of subcellular fractions, we used a procedure described by Homma et al. (1986) with some modifications. The cells were ruptured in homogenization buffer (mmol·L−1): 20 Tris–HCl (pH 7.5) containing 250 sucrose, 2 EDTA, 0.5 EGTA, 0.2 phenylmethylsulphonyl fluoride and a cocktail of protease inhibitors, by Dounce homogenization, and centrifuged immediately at 2000×g for 10 min. The supernatant was collected and centrifuged at 100 000×g for 1 h to separate cytosolic and membrane fractions. The membrane fraction was subsequently resuspended in extraction buffer (mmol·L−1): 20 Tris–HCl (pH 7.5) containing 150 NaCl, 1 EDTA, 1 EGTA, 0.2 phenylmethylsulphonyl fluoride and a cocktail of protease inhibitors with 1% (v/v) Nonidet P-40. Nuclei were pelleted by centrifugation at 2000×g for 15 min at 4°C, and resuspended in high-salt buffer (mmol·L−1): 20 Tris–HCl (pH 7.9), 420 NaCl, 10 KCl, 0.1 Na3VO4, 1 EDTA, 1 EGTA, 20% glycerol, supplemented with a cocktail of protease inhibitors, and sonicated until no nuclei remained intact. The samples were then centrifuged at 13 000×g for 10 min at 4°C, and the resultant supernatant was used as the nuclear extract. The purity of fractions was tested by immunoblotting with antibodies specific to NucP62 (nuclear protein) and β-actin (cytoplasmic protein). Proteins in the homogenates and cellular fraction were determined using the Bio-Rad (Milan, Italy) protein assay kit 1. Lyophilized BSA was used as a standard.
Western blot analysis
Proteins in homogenates and cellular fraction were determined using the Bio-Rad protein assay kit 1. Lyophilized BSA was used as a standard. Total cell proteins or proteins of the distinct subcellular fractions were dissolved in sodium dodecyl sulphate (SDS) sample buffer and separated on 10 or 15% SDS gels. Separated proteins were transferred electrophoretically onto polyvinylidene difluoride membrane (PVDF) (Amersham International, Piscataway, NJ, USA). Equal protein loading was confirmed by Ponceau S staining. Blots were incubated with specific primary antibodies, and the immune complexes were detected using appropriate peroxidase-conjugated secondary antibodies and enhanced chemiluminescent detection reagent ECL (Amersham International). The blots were stripped and used for several sequential incubations with control antibodies. Densitometric analysis was carried out on the Western blots using the NIH Image (v1.63) software. The pixel intensity for each region was analysed, the background was subtracted and the protein expressions were normalized to β-actin loading control for each lane.
Actin content
MCF-7 cells were grown either to confluence or 40% confluence on 100 mm cell culture dishes. Cells were washed with PBS and lysed with Triton lysis buffer (mmol·L−1): 1 EDTA, 150 NaCl, 50 Tris, 200 PMSF, 1% Triton X-100, 0.1 mg·mL−1 aprotinin, leupeptin and benzamidine (pH 7.4). Cell lysates were collected by scraping the dishes with a rubber scraper. Protein remaining adherent to dishes was solubilized with a small volume of lysis buffer containing 2% SDS, and assayed for completeness of protein recovery. Triton-soluble and -insoluble cell fractions were separated by centrifugation at 100 000×g for 30 min at 37°C. The total protein loaded for the two phenotypes was matched on SDS–PAGE gels, then transferred to PVDF membranes; membranes blocked with 5% non-fat dry milk were then incubated with monoclonal anti-actin antibody, followed by incubation with the appropriate labelled secondary antibody. Blots were developed using ECL Western blot kit, and bands were quantified using the NIH Image (v1.63) software.
Immunofluorescence and F-actin staining
For confocal fluorescence imaging, cells harvested from the subculture were seeded on sterilized 12 mm coverslips and kept in a six-well microplate in the incubator for 2–3 days, prior to experimentation. The filamentous actin (F-actin) was labelled with 0.1 µg·mL−1 TRITC-phalloidin (Sigma-Aldrich, St Louis, MO, USA). Fluorescence images were taken using a confocal microscope (Nikon TE2000, Tokyo, Japan).
Design and preparation of small interfering RNA (siRNA)
siRNAs were prepared by an in vitro transcription method, according to the manufacturer's protocol (Promega, Madison, WI, USA). Initially, four siRNA target sites specific to human protein kinase C (PKC)-ε mRNA, four siRNA target sites specific to human PKC-δ mRNA and five siRNA-p38α as determined by blast analysis were chosen. For each siRNA, sense and antisense templates were designed based on each target sequence and partial T7 promoter sequence. All template oligonucleotides were chemically synthesized and PAGE purified. In vitro transcription, annealing and purification of siRNA duplexes were performed using the protocol supplied with the T7 RiboMAX Express RNAi System (Promega Corporation). Briefly, approximately 2 µg of each single-stranded transcription template was first annealed with the T7 promoter and filled in by Klenow DNA polymerase to form double-stranded transcription templates. For preparation of each siRNA duplex, transcription reactions were first performed with separated antisense and sense templates using the T7 RNA polymerase provided with the kit, and then annealed to form siRNA duplexes. Then, the siRNA duplex was treated with DNase and RNase to remove the extra nucleotides of transcribed siRNA to meet the structural 3′UUU overhang and 5′ phosphate requirement. Immunoblottings were performed 24 and 48 h post-transfection to determine the efficiency of siRNA incorporation in MCF-7 cells and to measure P PKC-ε expression. Quantitative analysis of PKC-ε expression, as measured by intensity of immunoreactivity in siRNA-transfected MCF-7 cells, revealed a higher reduction in PKC-ε and PKC-δ expression, with the following sense RNA sequences: 5′-GCCCCUAAAGACAAUGAAGTT-3′ and 5′-AACUGUUUGUGAAUUUGCCUU-3′ respectively. A 90% reduction in p38MAPK expression was revealed with the following siRNA-p38α sequences: 5′-CAGTCCATCATTCATAGCGAAA-3′. Negative control siRNA (Qiagen S.p.A., Milan, Italy) was used to assess non-specific gene silencing effects.
siRNA transfection
The cells (50–70% confluence) were transfected with siRNA duplexes using the protocol supplied with the CodeBreaker siRNA transfection reagent (Promega Corporation). Briefly, the transfection reagent was first diluted into DMEM medium without serum and antibiotics for about 15 min, and then the sense and non-sense siRNA (siRNA-NS) duplex were added to the medium to form a lipid–siRNA complex. Following additional 15 min incubation, transfection was initiated by adding the lipid–siRNA complex to six-well plates. The final concentrations of siRNAs were 10 nmol·L−1.
Gelatin zymography
The effect of [Pt(O,O′-acac)(γ-acac)(DMS)] on gelatinolytic activity of MMPs was examined by gelatin zymography. MCF-7 cells (1 × 105) were treated in six-well plates with a medium containing increasing concentrations of [Pt(O,O′-acac)(γ-acac)(DMS)]. The conditioned medium was removed 24 h after treatment and centrifuged at 25 000×g for 5 min at 4°C to pellet cellular debris. Each sample (40 µL) was mixed with equal amounts of SDS sample buffer, and analysed by electrophoresis on 10% polyacrylamide gels containing 1 mg·mL−1 gelatin as the protease substrate. Following electrophoresis, the gels were placed in Triton X-100 solution (2.5% Triton X-100 and 50 mmol·L−1 Tris–HCl, pH 7.4) for 1 h to remove SDS, and then incubated for 16–18 h at 37°C in developing buffer (in mmol·L−1): 50 Tris base, 200 NaCl, 5 CaCl2 and 1% Triton X-100 (pH 7.4) on a rotary shaker. After incubation, the gels were stained in 30% methanol, 10% acetic acid and 0.5% (w/v) Coomassie brilliant blue for 1 h followed by destaining. Gelatinolytic activity was manifested as horizontal white bands on a blue background.
Intracellular ROS formation
ROS generation was detected by nitroblue tetrazolium (NBT) assay (Oliveira et al., 2003). NBT (1 mg·mL−1) was added to the medium of treated MCF-7, and incubations were carried out at 37°C for 15–60 min. NBT is reduced by ROS to a dark blue insoluble form of NBT called formazan. Cells were then carefully washed and lysed in a 90% dimethylsulphoxide solution containing 0.01 N NaOH and 0.1% SDS. Absorbance of formazan was measured at 550 nm against lysis buffer blank. Data are expressed as % of control untreated MCF-7 cells.
Statistical analysis
Experimental points represent means ± SD of three to six replicates. Statistical analysis was carried out using anova. When indicated, post hoc tests (Bonferroni/Dunn) were also performed. P < 0.05 was taken to denote statistical significance.
Materials
[Pt(O,O′-acac)(γ-acac)(DMS)] was prepared according to previously reported procedures (De Pascali et al., 2005). The PKC inhibitors GF109203X, rottlerin and Gö6976; the PKC-ε-selective translocation inhibitor εV1; the MAPK pharmacological inhibitors PD98059, SP600125 and SB203580; and the MMP-2/MMP-9 inhibitor II were obtained from Calbiochem (Darmstadt, Germany). Phospho-specific PKC-ε, PKC-δ, ERK1/2, JNK and polyclonal unphosphorylated antibodies, goat anti-rabbit IgG conjugated with peroxidase, as well as control antibodies, were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Phospho-specific p38MAPK (Thr180/Tyr182) and total (phosphorylated and unphosphorylated) p38MAPK antibodies, phospho-specific pFAK (Tyr397) and total (phosphorylated and unphosphorylated) FAK antibodies, phospho-specific p-Src (Tyr416) and total (phosphorylated and unphosphorylated) Src antibodies were obtained from Cell Signaling Technology (Celbio). The inhibitors of NADPH oxidase, diphenyleneiodonium (DPI) and apocynin, were obtained from Sigma.
Results
Effects of sublethal doses of [Pt(O,O′-acac)(γ-acac)(DMS)] on activation of MAPKs in MCF-7 mammary tumour cells
We showed previously that exposure of the MCF-7 cells to [Pt(O,O′-acac)(γ-acac)(DMS)] at concentrations ranging from 1 to 200 µmol·L−1 resulted in a dose-dependent inhibition of cell survival assessed by MTT assay (Muscella et al., 2008). In order to determine whether [Pt(O,O′-acac)(γ-acac)(DMS)] had effects on MCF-7 cell adhesion and migration, we here used low drug concentrations (0.10, 0.25 and 0.50 µmol·L−1) which were not able to induce apoptosis nor assayable cytotoxicity (Figure 1A–C). We evaluated the effects of such concentrations of [Pt(O,O′-acac)(γ-acac)(DMS)] on the activation state of MAPKs such as ERK, JNK and p38MAPK, in as much as they all play a crucial role in cell migration and adhesion (Huang et al., 2004). Western blot analysis using specific antibodies to phosphorylated ERK1/2, p38MAPK and JNK revealed that sublethal doses of [Pt(O,O′-acac)(γ-acac)(DMS)] induced a clear phosphorylation of p38MAPK, while the effects on the phosphorylation state of JNK and ERK1/2 were barely detectable (Figure 1D). [Pt(O,O′-acac)(γ-acac)(DMS)] provoked p38MAPK phosphorylation in a time-dependent manner, without affecting the overall level, as detected with an antibody recognizing both phosphorylated and unphosphorylated p38MAPK (Figure 1D).
Figure 1.
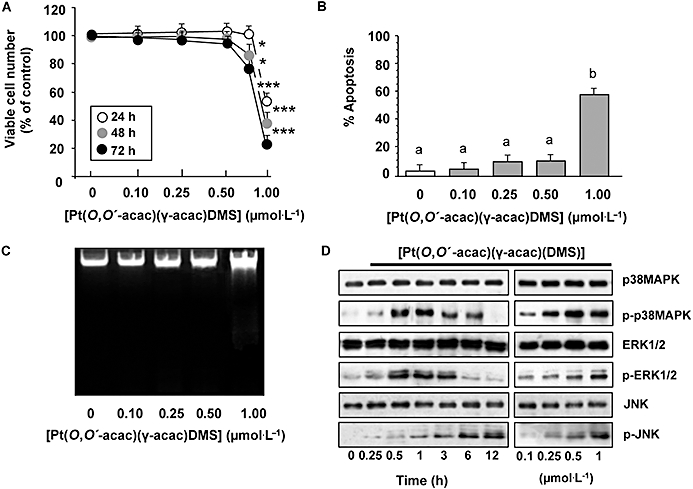
MCF-7 cell responses to low concentrations of [Pt(O,O′-acac)(γ-acac)(DMS)]. Cells were treated or not with increasing concentrations of [Pt(O,O′-acac)(γ-acac)(DMS)] or with 1 µmol·L−1 for the indicated time intervals. (A) Viable cell numbers were assessed by an MTT assay as described in Methods. The data are means ± SD of four different experiments run in eight replicate, and are presented as per cent of control. The results are statistically different from control at *P < 0.05; **P < 0.01; ***P < 0.001, n = 4. (B) Quantification of the percentage of apoptotic nuclei obtained from cells stained with DAPI (means ± SD; n = 4). Values with shared letters are not significantly different according to Bonferroni/Dunn post hoc tests. (C) Visualization of DNA fragmentation in [Pt(O,O′-acac)(γ-acac)(DMS)]-treated MCF-7 cells. Total DNA was isolated and separated on a 1% agarose gel. A representative example of three independent experiments is shown. (D) Cell lysates were analysed by Western blotting with anti- unphosphorylated and phosphorylated p38MAPK, ERK1/2 and JNK antibodies. Representative immunoblots of three experiments are depicted.
[Pt(O,O′-acac)(γ-acac)(DMS)] inhibits both anchorage-dependent and -independent growth of MCF-7 mammary tumour cells at sublethal doses
The clonogenicity of MCF-7 cells was examined using colony-forming assays. There was a significant reduction in the efficiency of colony initiation (Figure 2A) of MCF-7 cells after [Pt(O,O′-acac)(γ-acac)(DMS)] treatment, together with a significant decrease in colony sizes (Figure 2A, inset). A phenotypic hallmark of cancer cells is their ability to survive and grow under non-adhesive or anchorage-independent conditions. The ability of tumour cells to grow in soft agar correlates with the tumourigenic potential of cells through anchorage-independent growth (Rittling and Denhardt, 1999). When cultured on soft agar (anchorage-independent growth), MCF-7 cells formed more than 150 colonies greater than 50 µm in size per well at day 10. As shown in Figure 2B, 0.10 and 0.25 µmol·L−1[Pt(O,O′-acac)(γ-acac)(DMS)] decreased the ability of MCF-7 cells to grow in soft agar by 42 and 73%, respectively (P = 0.048 and 0.016), while 0.50 µmol·L−1[Pt(O,O′-acac)(γ-acac)(DMS)] completely inhibited it (Figure 2B). Thus, because [Pt(O,O′-acac)(γ-acac)(DMS)] inhibited the anchorage-independent growth of MCF-7 cells, we determined whether such growth inhibition was due to anoikis. To this end, cells were cultured on polyHEMA-coated plates which completely prevent cell adhesion (Zhu et al., 2001), and analysed for apoptosis. Indeed, when MCF-7 cells were seeded on polyHEMA-coated plates and treated with 0.25 µmol·L−1[Pt(O,O′-acac)(γ-acac)(DMS)], they showed some signs of apoptosis, as indicated by DAPI analysis, caspase-9 and degradation of poly(ADP-ribose) polymerase (PARP) (Figure 2C,D). In contrast, cells which were not incubated with [Pt(O,O′-acac)(γ-acac)(DMS)] had only basal (<5%) apoptosis. Noteworthy, when MCF-7 cells were seeded on normal culture dishes, they survived when stimulated with 0.10–0.50 µmol·L−1[Pt(O,O′-acac)(γ-acac)(DMS)] (Figure 1A), and, consistently, no apoptosis was observed (Figure 1B,C). These results strongly suggest that [Pt(O,O′-acac)(γ-acac)(DMS)] induces anoikis in MCF-7 cells grown under anchorage-independent conditions.
Figure 2.
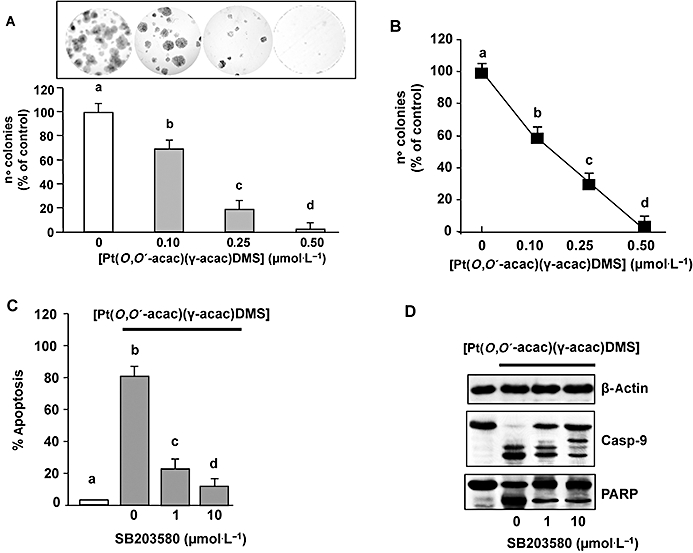
[Pt(O,O′-acac)(γ-acac)(DMS)] inhibition of MCF-7 cell invasion. (A and B). Cells were treated or not with increasing concentrations of [Pt(O,O′-acac)(γ-acac)(DMS)] for 2 h, or cells were transfected with siRNA-p38 [or control siRNA (NS) and then were incubated with 0.50 µmol·L−1[Pt(O,O′-acac)(γ-acac)(DMS)]. (A) After 15 days of growth, colonies consisting of more than 50 cells were scored. (B) The independent growth was determined by the soft agar assays. Data presented are the mean ± SD of colony number counted of four different experiments, and expressed as per cent of control. (C and D) Cells, pretreated with SB203580 (1–10 µmol·L−1) or transfected with siRNA–p38 [or control siRNA (NS)] were incubated or not with 0.50 µmol·L−1[Pt(O,O′-acac)(ã-acac)(DMS)], and then plated in polyHEMA plates. (C) Quantification of the percentage of apoptotic nuclei obtained from cells stained with DAPI (means ± SD; n = 4). Values with shared letters are not significantly different according to Bonferroni/Dunn post hoc tests. (D) On total cell lysates, Western blotting was performed with anti-unphosphorylated (p38MAPK), phosphorylated-p38MAPK (p-p38MAPK) (inset) and anti-caspase-9 antibodies. Nuclear fractions were analysed by Western blotting with anti-PARP polyclonal antibody. The same blots were stripped and reprobed with an anti-β-actin monoclonal antibody. Representative immunoblots of three experiments are depicted.
We next examined the effect of specific inhibitors of MAPKs on [Pt(O,O′-acac)(γ-acac)(DMS)]-induced anoikis. Thus, MCF-7 cells were pretreated with 10 µmol·L−1 PD98059 (MEK1/2 inhibitor), SP600125 (JNK inhibitor) and SB203580 (p38MAPK inhibitor) prior to treatment with 0.25 µmol·L−1[Pt(O,O′-acac)(γ-acac)(DMS)] for 30 min, and then further incubated for 24 h. Our results showed that [Pt(O,O′-acac)(γ-acac)(DMS)] effects were reversed by the use of SB203580 (Figure 2C,D), but not by JNK or MEK1/2 inhibitors (data not shown).
To further evaluate the role of p38MAPK in regulating the response to [Pt(O,O′-acac)(γ-acac)(DMS)], p38α–siRNA experiments were also performed, as p38α is the most abundant isoform in MCF-7 cells (Chen et al., 2009). Western blotting experiments demonstrated that p38α–siRNA was able to reduce the p38 total protein expression by nearly 90% (Figure 2D, inset), and completely inhibited the phosphorylation of p38MAPK provoked by [Pt(O,O′-acac)(γ-acac)(DMS)]. Cells transfected with siRNA–p38α or control siRNA (NS) were incubated or not with 0.50 µmol·L−1[Pt(O,O′-acac)(γ-acac)(DMS)], and then colony-forming and soft agar assays were performed; these cells were also seeded in polyHEMA plates. The p38α–siRNA, but not siRNA-NS, inhibited the effects of [Pt(O,O′-acac)(γ-acac)(DMS)] on anchorage-dependent and -independent growth and anoikis of MCF-7 cells (Figure 2).
A type collagen invasion assay was performed to evaluate the [Pt(O,O′-acac)(γ-acac)(DMS)]-anti-invasive effect on MCF-7 cells. The cells showed limited growth in plates coated with type I collagen, the most prevalent component of the basement membrane, after 2 h treatment with [Pt(O,O′-acac)(γ-acac)(DMS)]. A significant dose-dependent anti-invasive effect of [Pt(O,O′-acac)(γ-acac)(DMS)] could also be observed (Figure 3A). This result suggests that the ability of MCF-7 cells to attach and grow at sites distant from the primary tumour may be inhibited by [Pt(O,O′-acac)(γ-acac)(DMS)].
Figure 3.
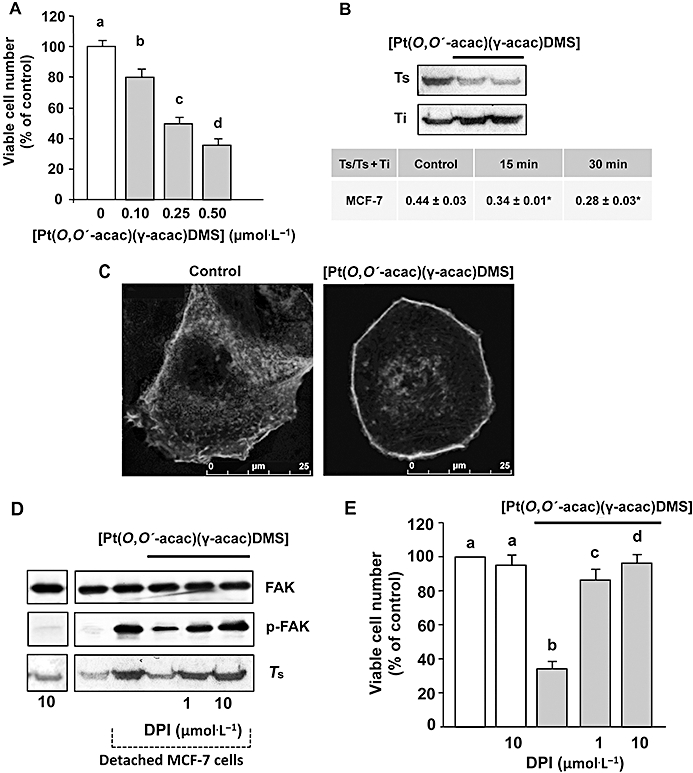
[Pt(O,O′-acac)(γ-acac)(DMS)] induces actin polymerization and reduces the attachment of MCF-7 cells to type I collagen. (A) Cells were treated or not with increasing concentrations of [Pt(O,O′-acac)(γ-acac)(DMS)], and collagen invasion assay was performed. Results are presented as number of penetrating cells relative to control. Data presented are the mean ± SD of two different experiments. Values with shared letters are not significantly different according to Bonferroni/Dunn post hoc tests. (B) Monomeric (Ts) and total actin (Ts+Ti) were measured by quantitative immunoblot analysis after subcellular fractionation of cells treated for different time-points with 0.50 µmol·L−1[Pt(O,O′-acac)(γ-acac)(DMS)]. Upper: a representative immunoblot of three experiments is depicted. Table: Monomeric (Ts) and total actin (Ts+Ti) were measured by quantitative immunoblot analysis. Values are expressed as the mean ± SD of three different Western blots; *P < 0.05, n = 4. (C) Confocal microscopy of F-actin in fixed MCF-7 cells treated or not with 0.50 µmol·L−1 of [Pt(O,O′-acac)(γ-acac)(DMS)] for 20 min. (D) Detached cells, pretreated or not with DPI, were treated with or without 0.50 µmol·L−1[Pt(O,O′-acac)(γ-acac)(DMS)] for 30 min; cell lysates were analysed by Western blotting with anti-p-FAK or anti-total-FAK (unphosphorylated and phosphorylated FAK) antibody. Soluble fractions of cells were analysed by Western blotting with anti-β-actin monoclonal antibody. (E) Cells, pretreated with DPI, were treated or not with increasing concentrations of [Pt(O,O′-acac)(γ-acac)(DMS)], and then collagen invasion assays were performed. Results are presented as number of penetrating cells, relative to control. Data presented are the mean ± SD of two different experiments. Values with shared letters are not significantly different according to Bonferroni/Dunn post hoc tests.
[Pt(O,O′-acac)(γ-acac)(DMS)] induces rapid actin reorganization in MCF-7 cells
Formation of focal adhesion and actin rearrangements are essential events in cell adhesion and spreading (Carragher and Frame, 2004). Thus, we investigated whether [Pt(O,O′-acac)(γ-acac)(DMS)] treatment (for 15 and 30 min) provoked actin cytoskeleton rearrangements. Quantitative immunoblot analysis of Triton-soluble and -insoluble actin cytoskeleton preparations showed a significant decrease in the ratio of globular actin (G) : total (globular and filamentous) actin, evident at 15 min of treatment, indicating rapid actin polymerization (Figure 3B). The proportion of polymerized actin inversely correlates with cell motility (Li et al., 2008). The quantitative data obtained by immunoblot analysis were confirmed by confocal microscopic analysis of actin cytoskeleton in MCF-7 cells incubated or not with [Pt(O,O′-acac)(γ-acac)(DMS)] (Figure 3C). In order to disclose a role of the tyrosine kinase FAK, involved in cell migration and adhesion, Western analyses of phosphorylated and unphosphorylated FAK were performed on MCF-7 cells. In detached MCF-7 cells, high levels of phosphorylated FAK (Tyr397) were detected in a time-dependent manner, with a maximal increase at 2 h (data not shown). In detached [Pt(O,O′-acac)(γ-acac)(DMS)]-treated cells, we observed a significant decrease of phosphorylated FAK (Figure 3C). Because it is known that ROS potently activates Src (Seko et al., 1996; Cheng et al., 2002; Kopetz et al., 2009) and Src then phosphorylates FAK increasing its kinase activity (Mitra and Schlaepfer, 2006), we pre-incubated MCF-7 cells with DPI, a specific inhibitor of NADPH oxidase, and restored FAK phosphorylation, G/total actin ratio (Figure 3C) and the capacity to grow in collagen-coated plates (Figure 3D). Thus, we propose that [Pt(O,O′-acac)(γ-acac)(DMS)] inhibits MCF-7 cell invasion by modulating the phosphorylation of FAK involved in the actin reorganization of detached cells.
[Pt(O,O′-acac)(γ-acac)(DMS)] inhibits the migration of mammary tumour cells
Cell mobility/migration of MCF-7 breast cancer cells was examined by in vitro culture wounding assays. Wounds were created in confluent cell cultures, and repopulation of the wound space was evaluated by measuring the width of injury line. Width of injury line was plotted as percentage of control in order to quantitate the effect of [Pt(O,O′-acac)(γ-acac)(DMS)] on cell migration (Figure 4). Using the wound closure assay, we observed that [Pt(O,O′-acac)(γ-acac)(DMS)] significantly reduced the migration of MCF-7 cells in a concentration- and time-dependent manner (Figure 4); this effect was not due to cytotoxicity as these cells do not exhibit significant growth inhibition under these conditions (Figure 1A). In addition, 0.50 µmol·L−1[Pt(O,O′-acac)(γ-acac)(DMS)] reduced the migration ability of these cells by 53% (P = 0.026) after 11 h. This effect was blocked by SB203580, a specific p38MAPK inhibitor (Figure 5).
Figure 4.
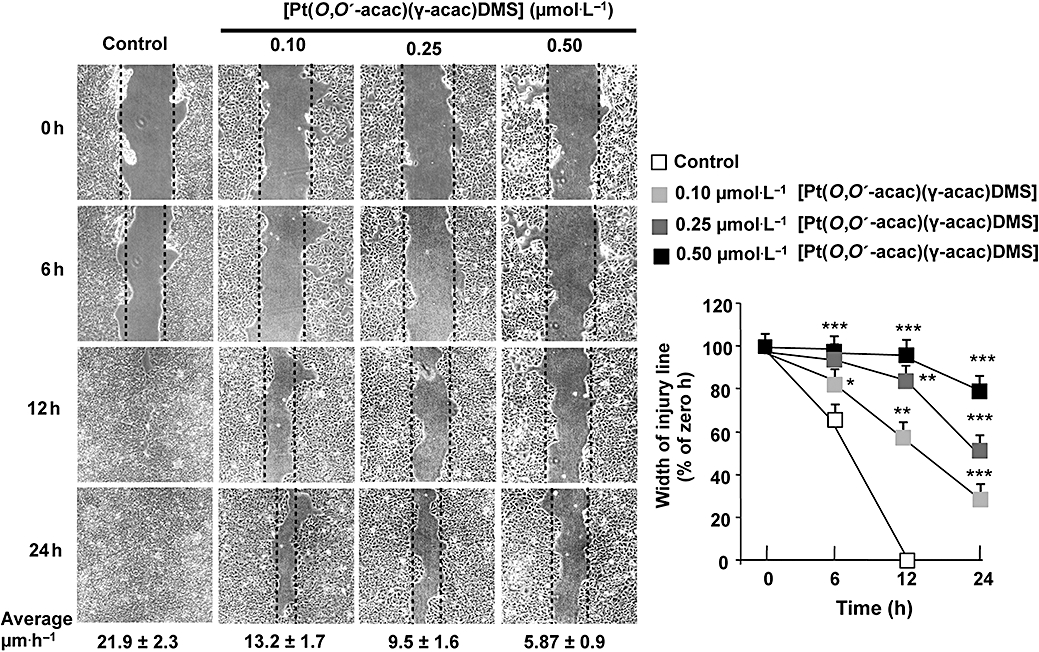
The inhibition by [Pt(O,O′-acac)(γ-acac)(DMS)] of MCF-7 cell migration is concentration and time dependent. Differential cell migration rate was examined using wound closure assay. Cells were treated or not with [Pt(O,O′-acac)(γ-acac)(DMS)] at the indicated concentrations and monitored by microscopy at the indicated times. Migration rate (average ± SD) and degree of wound closure were assessed by measuring the distance between wound edges at indicated time intervals in at last eight randomly chosen regions of three different experiments (means ± SD) normalized to 100% wound closure for control cells. *P < 0.05, **P < 0.01, ***P < 0.001; values different from control. Average velocities for the three different experiments are given under photographs.
Figure 5.
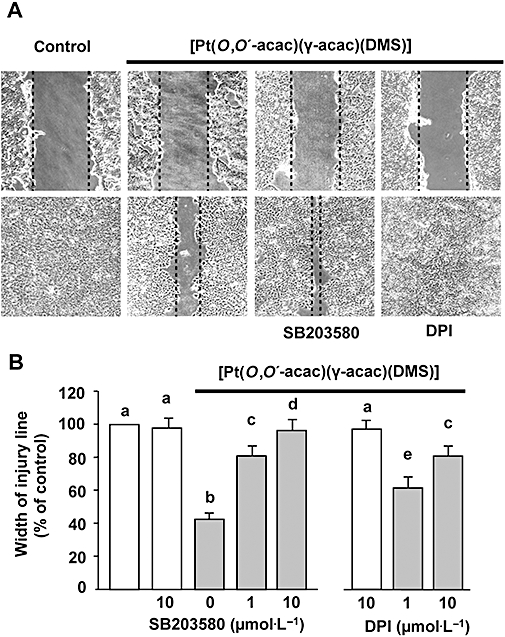
Inhibition of p38MAPK activation or ROS production reversed the effects of [Pt(O,O′-acac)(γ-acac)(DMS)] on MCF-7 cell migration. Cells were wounded with a micro-pipette tip. Next, the cells were treated with the inhibitors SB203580 (1 and 10 µmol·L−1) or DPI (1 and 10 µmol·L−1) for 30 min prior to stimulation with or without [Pt(O,O′-acac)(γ-acac)(DMS)] (0.50 µmol·L−1). After 16 h, migration was evaluated. (A) Raw images of the cells in each condition. (B) Results (means ± SD) from three independent experiments are presented. Migration was expressed as the percentage of unstimulated cells at 16 h. Statistical analysis was carried out using the anova, and values with shared letters are not significantly different according to Bonferroni/Dunn post hoc tests.
[Pt(O,O′-acac)(γ-acac)(DMS)] decreases expression and activity of MMP-2 and -9
It is known that breast tumour cells actively produce MMPs to facilitate tumour cell invasion. We therefore aimed to find out if [Pt(O,O′-acac)(γ-acac)(DMS)] interferes with the expression of MMP-2 and MMP-9, enzymes associated with breast cancer progression (Somiari et al., 2006). By Western blotting analysis with specific antibodies against MMP-2 and −9, we showed that both metalloproteases were drastically reduced in cells treated for 12 h with 0.10–0.50 µmol·L−1[Pt(O,O′-acac)(γ-acac)(DMS)] (Figure 6A). By gelatin zymographic analysis performed on conditioned medium from MCF-7 cells, gelatinolytic activities at around 90 and 72 kDa were observed representing the active forms of MMP-9 and MMP-2, respectively (Figure 6A). Figure 6B shows the time-dependent inhibition of MMP-2 and -9 protein levels obtained in cells treated with 0.50 µmol·L−1[Pt(O,O′-acac)(γ-acac)(DMS)]. When the MCF-7 cells were pretreated for 30 min with SB203580 (1 and 10 µmol·L−1), and then with 0.50 µmol·L−1[Pt(O,O′-acac)(γ-acac)(DMS)] for 12 h, the inhibition of both MMP-2 and MMP-9 expression levels was efficiently blocked (Figure 6C). DPI and apocynin also markedly suppressed [Pt(O,O′-acac)(γ-acac)(DMS)] inhibition secretion of MMP-2 and MMP-9, suggesting that NADPH oxidase was responsible for the decrease in MMPs (data not shown). To examine the role of MMP-2 and MMP-9 in cell migration, MMP activities were blocked using the MMP-2/MMP-9 inhibitor II. Cell migration assays demonstrated that pretreatment with MMP-2/MMP-9 inhibitor II blocked cell migration in a concentration-dependent manner (Figure 6D). The concentration of 50 µmol·L−1 MMP-2/MMP-9 inhibitor II was used in MTT assay performed after 24 h incubation showing no effects on cell viability (data not shown). These data implied that the effect of MMP-2/MMP-9 inhibitor II on cell migration cannot be simply attributed to the inhibition of cell proliferation.
Figure 6.
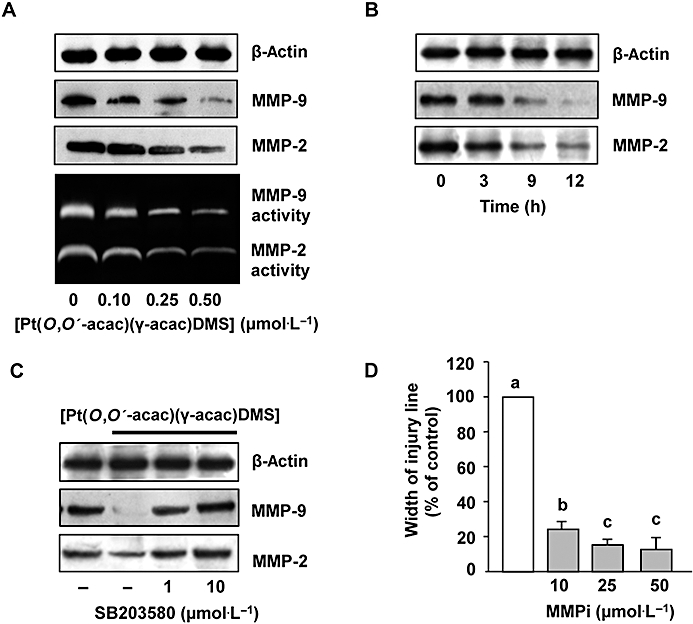
[Pt(O,O′-acac)(γ-acac)(DMS)] inhibited the secretion of MMP-2 and MMP-9. MCF-7 cells were treated or not with [Pt(O,O′-acac)(γ-acac)(DMS)] (A) for the indicated doses or (B) with 0.50 µmol·L−1 for the indicated time intervals. Conditioned media and cell lysates were subjected to gelatin zymography and Western blot analysis with anti-MMP-2 and anti-MMP-9 antibodies respectively. (C) Cells pretreated for 30 min with the p38MAPK inhibitor, SB203580 (1 and 10 µmol·L−1), were treated or not with 0.50 µmol·L−1[Pt(O,O′-acac)(γ-acac)(DMS)] for 12 h. Cell lysates were analysed by Western blotting with anti-MMP-2 and anti-MMP-9 antibodies. For Western blotting, control loadings are shown by β-actin and representative immunoblots of three experiments are depicted. (D) Cells pre-incubated with the MMP-2/MMP-9 inhibitor II (MMPi), for 60 min, were treated with 0.50 µmol·L−1[Pt(O,O′-acac)(γ-acac)(DMS)] for 16 h. Differential cell migration rate was examined using wound closure assay. Data are means ± SD of four different experiments and are presented as per cent of wound closure for control cells. Statistical analysis was carried out using anova, and values with shared letters are not significantly different according to Bonferroni/Dunn post hoc tests.
Role of p38MAPK in [Pt(O,O′-acac)(γ-acac)(DMS)]-mediated inhibition of MCF-7 migration
Recent studies have demonstrated that MAPK activation plays crucial roles in cell migration (Huang et al., 2004). Thus, we investigated the activation of p38MAPK by [Pt(O,O′-acac)(γ-acac)(DMS)] treatment. Using an antibody recognizing the dually phosphorylated (threonine 180 and tyrosine 182) p38MAPK, we found that [Pt(O,O′-acac)(γ-acac)(DMS)] provoked its phosphorylation in a time-dependent manner (Figure 7A), without affecting the overall level, detected with an antibody recognizing both phosphorylated and unphosphorylated p38MAPK (Figure 7A).
Figure 7.
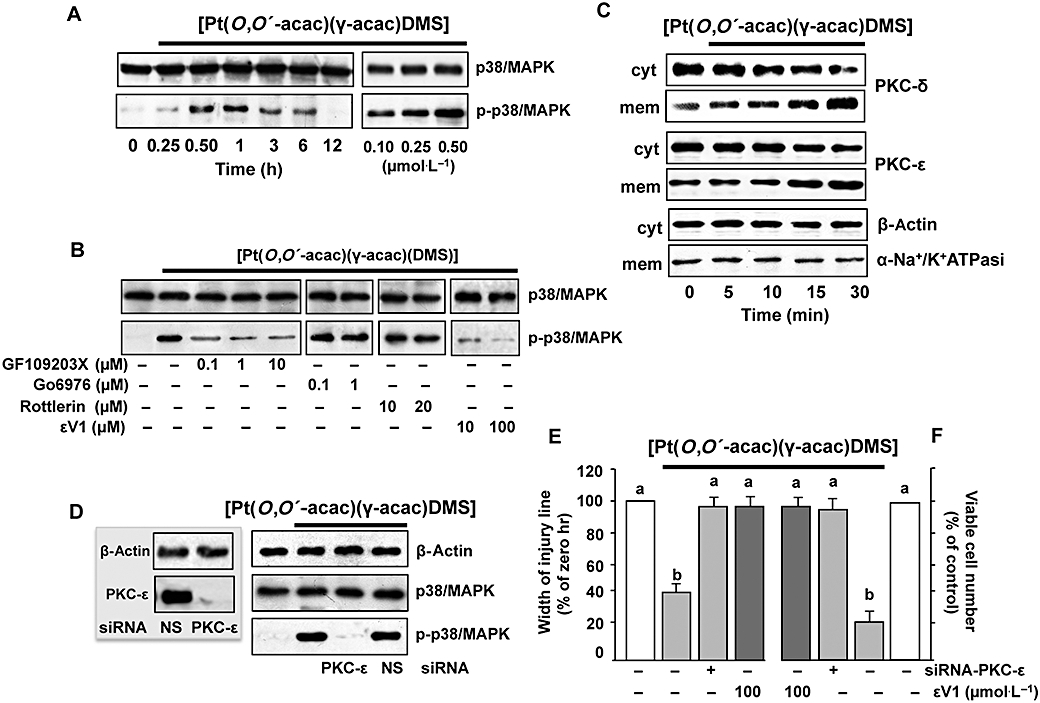
[Pt(O,O′-acac)(γ-acac)(DMS)] induces p38MAPK activation. (A) Cells pretreated or not with the PKC inhibitors, GF109203X, Gö6976, rottlerin or εV1, were treated or not with 0.50 µmol·L−1[Pt(O,O′-acac)(γ-acac)(DMS)] for 1 h. Cell lysates were analysed by Western blotting with anti-total-p38MAPK (unphosphorylated and phosphorylated p38MAPK) and phosphorylated p38MAPK antibodies. Control loading is shown by β-actin. Representative immunoblots of three experiments are depicted. (B) MCF-7 cells were treated without or with [Pt(O,O′-acac)(γ-acac)(DMS)] for the indicated times. Cell fractions (cytosol and membranes for translocation studies) were analysed by Western blotting with specific anti-PKC-ε and anti-PKC-δ antibodies. The purity of fractions was tested by immunoblotting with anti β-actin and anti-α subunit of Na+/K+ATPase monoclonal antibodies. The figures are representative of four independent experiments. (C,D) Cells were transfected with siRNA–PKC-ε or control siRNA (NS) and then were incubated with 0.50 µmol·L−1[Pt(O,O′-acac)(γ-acac)(DMS)]. (C) Western blotting of total lysates was performed with specific anti-PKC-ε (inset) or with anti-unphosphorylated (p38MAPK) and phosphorylated p38MAPK (p-p38MAPK) antibodies. Control loadings are shown by β-actin. Representative immunoblots of three experiments are depicted. (D) Differential cell migration rate was examined using wound closure assay. Data are means ± SD of four different experiments and are presented as per cent of wound closure for control cells. (E) Cells were plated in polyHEMA plates. The number of surviving cells was determined, after 24 h, by an MTT assay and is expressed as per cent change from the initial seeding (time 0). (E,F) Values with shared letters are not significantly different according to Bonferroni/Dunn post hoc tests.
The mechanism of Pt(O,O′-acac)(γ-acac)(DMS)]-induced p38MAPK phosphorylation
Role of PKC-ε
MCF-7 cells express PKC-α, -δ, -ε, -ι and -ζ (Muscella et al., 2005). To investigate whether or not PKCs were involved in Pt(O,O′-acac)(γ-acac)(DMS)]-mediated p38MAPK activation, MCF-7 cells were pre-incubated for 30 min with the PKC inhibitor GF109203X. Figure 7B shows that the effects of Pt(O,O′-acac)(γ-acac)(DMS)] on p38MAPK phosphorylation were inhibited at the lowest GF109203X concentration used, indicating a potential involvement of conventional or novel PKCs; GF109203X is also able to inhibit atypical PKCs, but at higher doses (Martiny-Baron et al., 1993). As the conventional PKC inhibitor Gö6976 had no effect on p38MAPK kinase (Figure 7B), we also investigated the role of PKC-ε and PKC-δ on Pt(O,O′-acac)(γ-acac)(DMS)]-provoked p38MAPK activation. In as much as activated PKCs translocate from the cytosol to the cellular membranes, we analysed, by immunoblotting, the distribution of PKC-ε and PKC-δ in MCF-7 treated with 0.5 µmol·L−1 Pt(O,O′-acac)(γ-acac)(DMS)] for different incubation times (0–30 min). As shown in Figure 7C, a cytosol-to-membrane translocation of PKC-δ and PKC-ε was observed. Then, we used molecular (PKC-ε–siRNA and PKC-δ–siRNA) and pharmacological [the PKC-ε translocation inhibitor peptide εV1 (Yedovitzky et al., 1997) and the PKC-δ inhibitor rottlerin] techniques, in order to specifically inhibit PKC-ε and PKC-δ, and establish its role in p38MAPK control. Preliminary experiments by Western blotting demonstrated that PKC-ε–siRNA and PKC-δ–siRNA were able to reduce PKC-ε and PKC-δ expressions, and that non-specific siRNA (siRNA-NS) had no silencing effect on PKC-ε and PKC-δ expressions (Figure 7C, inset). The PKC-ε–siRNA (10 nmol·L−1) inhibited [Pt(O,O′-acac)(γ-acac)(DMS)]-induced p38MAPK phosphorylation (Figure 7C), and antagonized the effect of [Pt(O,O′-acac)(γ-acac)(DMS)] on anoikis and migration of MCF-7 cells (Figure 7D,E). This result was confirmed by using εV1 (Figure 7D,E). The inhibition of PKC-δ had no effects on the phosphorylation state of p38MAPK in [Pt(O,O′-acac)(γ-acac)(DMS)]-treated cells (Figure 7A,C).
[Pt(O,O′-acac)(γ-acac)(DMS)] activates p38MAPK in a ROS-dependent manner
There is also evidence that oxidants activate MAPK signalling pathways (Finkel, 2003). These findings led us to investigate the mechanism underlying the [Pt(O,O′-acac)(γ-acac)(DMS)]-induced activation of p38MAPK, and to determine the role of ROS in this process. ROS production in MCF-7 cells was assessed following exposure to [Pt(O,O′-acac)(γ-acac)(DMS)]. [Pt(O,O′-acac)(γ-acac)(DMS)] increased the level of ROS in a time-dependent manner with ROS generation starting after 15 min and peaking at 60 min (Figure 8A). [Pt(O,O′-acac)(γ-acac)(DMS)]-provoked ROS production was markedly suppressed by two structurally unrelated inhibitors of NADPH oxidase, DPI and apocynin (Figure 8B). DPI and apocynin were also able to inhibit the [Pt(O,O′-acac)(γ-acac)(DMS)]-induced PKC-ε translocation (Figure 8C) and p38MAPK phosphorylation (Figure 8D), thus suggesting the involvement of ROS in these processes. In addition, the pretreatments of MCF-7 cells with DPI suppressed the inhibition of cell migration provoked by [Pt(O,O′-acac)(γ-acac)(DMS)] (Figure 5). These observations indicate that some ROS-mediated event, initiated by Pt(O,O′-acac)(γ-acac)(DMS)], leads to inhibition of the migration of mammary tumour cells.
Figure 8.
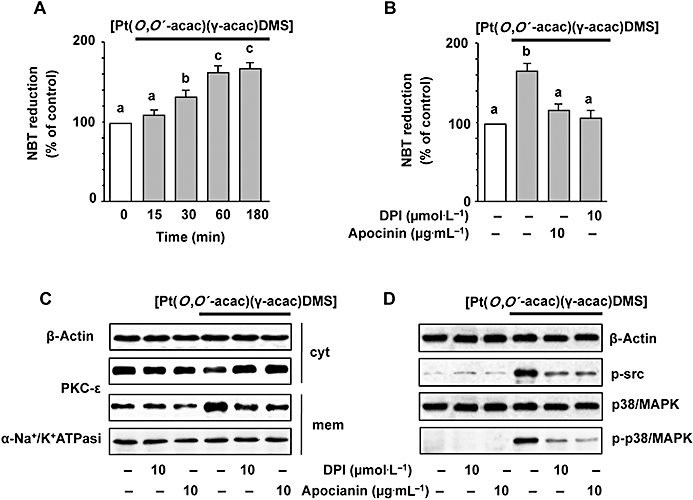
[Pt(O,O′-acac)(γ-acac)(DMS)] induces p38MAPK activation through ROS production. (A) MCF-7 cells were treated or not with 0.50 µmol·L−1[Pt(O,O′-acac)(γ-acac)(DMS)] for the indicated time intervals. Cells pre-incubated with apocynin or DPI for 30 min were exposed to [Pt(O,O′-acac)(γ-acac)(DMS)] for 30 min (C) or 1 h (D). ROS production was measured by NBT reduction. Values with shared letters are not significantly different according to Bonferroni/Dunn post hoc tests. (C) Cell fractions (cytosol and membranes for translocation studies) were analysed by Western blotting with specific anti-PKC-ε antibody. The purity of fractions was tested by immunoblotting with anti β-actin and anti-α subunit of Na+/K+ATPase monoclonal antibodies. (D) Cell lysates were analysed by Western blotting with anti-phosphorylated-Src or anti-total (unphosphorylated and phosphorylated) p38MAPK and phosphorylated-p38MAPK (p-p38MAPK) antibodies. Control loadings are shown by β-actin. The figures are representative of four independent experiments.
[Pt(O,O′-acac)(γ-acac)(DMS)] induces phosphorylation of Src
Because it is known that ROS may induce Src phosphorylation (Seko et al., 1996; Cheng et al., 2002; Kopetz et al., 2009), we performed Western analysis using antibodies recognizing total (phosphorylated and unphosphorylated) and phosphorylated Src. As shown in Figure 9A, [Pt(O,O′-acac)(γ-acac)(DMS)] induced a dose-dependent phosphorylation of Src that was greatly decreased in the presence of DPI or apocynin (Figure 8D), thus suggesting the involvement of ROS. PP1, a specific Src inhibitor, blocked the phosphorylation of Src induced by [Pt(O,O′-acac)(γ-acac)(DMS)], without influencing the expression of total Src (Figure 9B). In addition, PP1 reversed the effect of [Pt(O,O′-acac)(γ-acac)(DMS)] on both p38MAPK phosphorylation, and MMP-2 and MMP-9 inhibition (Figure 9B). Furthermore, PP1 inhibited the effects of [Pt(O,O′-acac)(γ-acac)(DMS)] on MCF-7 cell migration (Figure 9C). To examine whether the anoikis provoked by [Pt(O,O′-acac)(γ-acac)(DMS)] was dependent upon the activity of Src, MCF-7 cells, cultured on polyHEMA-coated plates, were exposed to [Pt(O,O′-acac)(γ-acac)(DMS)] in the presence and absence of PP1, and viable cell number was assessed after 24 h treatment. Results shown in Figure 9D demonstrated that PP1 blocked the effects of [Pt(O,O′-acac)(γ-acac)(DMS)] on cell anoikis. These results highlighted the role of Src in the pathways linking the generation of ROS, due to [Pt(O,O′-acac)(γ-acac)(DMS)] exposure, to the inhibition of cell migration and anoikis induction in MCF-7 cells.
Figure 9.
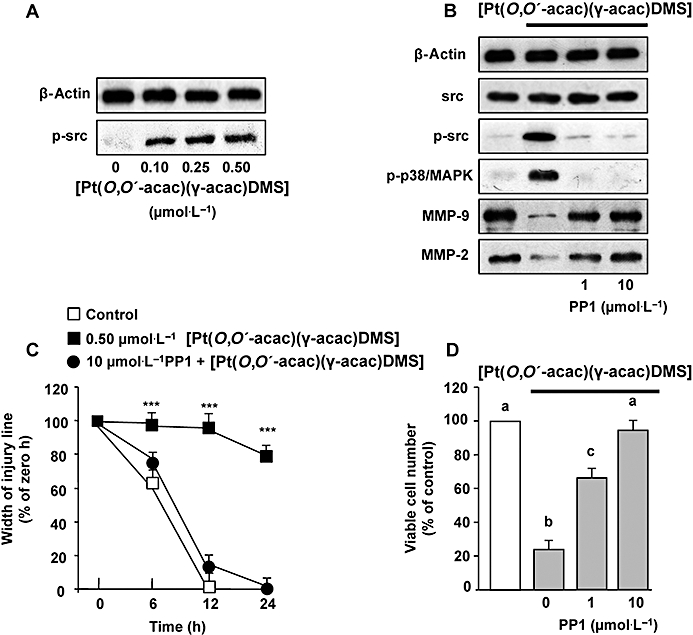
[Pt(O,O′-acac)(γ-acac)(DMS)] induces phosphorylation of Src. (A) MCF-7 cells were treated or not with 0.50 µmol·L−1[Pt(O,O′-acac)(γ-acac)(DMS)] for the indicated time intervals. (B, C and D) Cells pre-incubated with the Src inhibitor PP1 were exposed or not to [Pt(O,O′-acac)(γ-acac)(DMS)]. (B) Western blotting of total lysates was performed with specific anti-unphosphorylated (p38MAPK) and phosphorylated-p38MAPK (p-p38MAPK) or anti-unphosphorylated (Src) and phosphorylated-Src (p-Src) antibodies. Control loadings are shown by β-actin. Cell fractions (cytosol and membranes for translocation studies) were analysed by Western blotting with specific anti-PKC-ε antibody. The purity of fractions was tested by immunoblotting with anti β-actin and anti-α subunit of Na+/K+ATPase monoclonal antibodies. Representative immunoblots of three experiments are depicted. (C) Degree of wound closure was assessed by measuring the distance between wound edges at indicated time intervals in at least eight randomly chosen regions of three different experiments (means ± SD) normalized to 100% wound closure for control cells. The results are statistically different from control at *P < 0.05, **P < 0.01, ***P < 0.001. (D) Cells were plated in polyHEMA plates. The number of surviving cells was determined, after 24 h, by MTT assay and was expressed as per cent change from the initial seeding (time 0). Values with shared letters are not significantly different according to Bonferroni/Dunn post hoc tests.
Discussion
Metastases are responsible for most cancer deaths, and it is therefore highly relevant to develop novel strategies to block the metastatic process. Tumour cell migration and invasion provide potential targets for therapeutic intervention, and even though these phenomena are hallmarks of metastasis, only few compounds that have some capacity to inhibit tumour cell migration have yielded, to date, interesting results in clinical trials. The biological and molecular events that regulate the invasiveness of breast tumour cells need to be further revealed to develop effective therapies that stop the breast cancer from expanding and metastasizing (Folgueras et al., 2004). Mammary tumour cell invasion and metastasis are dependent upon the ability of tumour cells to dissociate from the primary tumour, enter the bloodstream or lymphatics and reattach at distant sites. Recently, we synthesized a new platinum(II) complex containing an O,O′-acetylacetonate (acac), a σ-bonded (γ-carbon-bonded) acac, and DMS ([Pt(O,O′-acac)(γ-acac)(DMS)]) (Muscella et al., 2007). [Pt(O,O′-acac)(γ-acac)(DMS)] was able to induce apoptotic processes rapidly in human breast carcinoma MCF-7 cells which are relatively resistant to many chemotherapeutic agents, cisplatin included (Muscella et al., 2008). The cytotoxicity of [Pt(O,O′-acac)(γ-acac)(DMS)], in contrast to cisplatin, was not correlated with DNA binding, and appeared to help overcoming resistance to standard cancer therapies. Here, we have extended the earlier studies on [Pt(O,O′-acac)(γ-acac)(DMS)], and have shown its effects on MCF-7 cell migration and invasion. Generally speaking, the progression of epithelial breast cells to invasive carcinoma involves at least three independent, but highly coordinated, biological processes: (i) cell adhesion to components of the ECM; (ii) the cells' own motility, which involves the reorganization of the actin cytoskeleton; and (iii) invasion, which involves the degradation of ECM proteins by tumour-secreted proteolytic enzymes, such as MMP-2 and MMP-9 (Wolf and Friedl, 2009). We here show that [Pt(O,O′-acac)(γ-acac)(DMS)] can limit each of these processes at concentrations and treatment times that were not cytotoxic to the MCF-7 carcinoma cell line (Figure 1). Breast tumour cell migration involves numerous interactions between the tumour cell and the ECM/surface (Bartsch et al., 2003). The migration of MCF-7 cells was decreased by low concentrations of [Pt(O,O′-acac)(γ-acac)(DMS)] in a time-dependent manner (Figure 4). Such decreased migration correlated with the ability of cells to attach the ECM, as [Pt(O,O′-acac)(γ-acac)(DMS)] inhibited the in vitro adhesion of MCF-7 cells to type I collagen, one of the most common components of the basement membrane (Figure 3A). These results may suggest that the ability of tumour breast cells to attach and grow at sites distant from the primary tumour may be inhibited by [Pt(O,O′-acac)(γ-acac)(DMS)].
Anoikis, a form of apoptosis that is induced by anchorage-dependent cells detaching from the surrounding ECM, acts as a physiological barrier to metastasis (Valentijn et al., 2004). We here show that [Pt(O,O′-acac)(γ-acac)(DMS)] significantly inhibited the anchorage-independent growth of MCF-7 cells (Figure 2A,B). When cultured on soft agar, MCF-7 cells exposed to low, non-apoptogenic doses of [Pt(O,O′-acac)(γ-acac)(DMS)] underwent a rapid and substantial anoikis (Figure 2B). The interest in understanding the molecular mechanisms of anoikis has increased significantly, as it became evident that resistance to anoikis is a critical requirement for invasion and metastasis in cancers derived from epithelial cells (Evan and Vousden, 2001). It is known that Src is one of the key protein tyrosine kinases involved in cell survival and growth (Schlessinger, 2000); therefore, there are mounting data that Src signalling is critical for the metastatic process facilitating cancer cell survival and neovascularization of metastatic sites (Weis et al., 2004). Here, inhibition of Src reversed the anti-metastatic actions of [Pt(O,O′-acac)(γ-acac)(DMS)] (Figure 9), and [Pt(O,O′-acac)(γ-acac)(DMS)] provoked the generation of intracellular ROS (Figure 8A). As ROS potently activate Src (Seko et al., 1996; Cheng et al., 2002; Kopetz et al., 2009), we inhibited NADPH oxidase with the antioxidants apocynin or DPI and blocked [Pt(O,O′-acac)(γ-acac)(DMS)]-induced Src activation (Figure 8D), thus concluding that [Pt(O,O′-acac)(γ-acac)(DMS)] activated Src through a ROS-dependent mechanism.
In eukaryotic cells, multiple MAPK pathways normally actively regulate diverse vital processes. Furthermore, the cell type, cell differentiation status and the phase of cell cycle determine the cell response to genotoxic agents, and, in recent years, the importance of MAPKs for the final outcome of cell treatment with cisplatin has become more obvious. As far as [Pt(O,O′-acac)(γ-acac)(DMS)] is concerned, we provided evidence that, in HeLa cells, the activation of ERK1/2 is important for the induction of apoptosis (Muscella et al., 2007). In fact, [Pt(O,O′-acac)(γ-acac)(DMS)] treatment also resulted in high and sustained activation of ERK1/2, and inhibition of ERK1/2 activation decreased the [Pt(O,O′-acac)(γ-acac)(DMS)]-mediated cell death. We here showed that all MAPKs were activated by low doses of [Pt(O,O′-acac)(γ-acac)(DMS)] (Figure 1D) with p38MAPK showing the highest response (significantly activated by 0.1 µmol·L−1[Pt(O,O′-acac)(γ-acac)(DMS)] for 15 min). Indeed, when the involvement of MAPKs in ROS/Src-mediated effects of [Pt(O,O′-acac)(γ-acac)(DMS)] was investigated using specific pharmacological inhibitors, it was found that whereas PD98059 and SP600125 did not have any effects (data not shown), SB203580 (or siRNAs to p38α) reversed the [Pt(O,O′-acac)(γ-acac)(DMS)]-provoked inhibition of cell growth in soft agar (Figure 2C) and cell migration (Figure 5). Thus, the effects of [Pt(O,O′-acac)(γ-acac)(DMS)] on tumour cell migration and anoikis (Figure 2D) are likely to be associated with p38MAPK.
The p38MAPK has been reported to elicit apparently conflicting roles in cancer cell invasion, giving either stimulatory (Huang et al., 2004; Zhang and Zhang, 2006) or inhibitory (Zhou et al., 2008) effects. In addition, p38MAPK mediates ligand-specific actions in breast cancer cells. For example, p38MAPK mediated apoptosis in response to oestrogen-, 4-hydroxytamoxifen- and activin in oestrogen receptor-positive breast cancer cells (Zhang and Shapiro, 2000; Cocolakis et al., 2001). In addition, p38MAPK mediated heregulin-induced up-regulation of vascular endothelial growth factor in several breast cancer cell lines (Xiong et al., 2001). Many studies have described the link between p38MAPK activation and apoptosis in response to chemotherapies (Valentijn et al., 2004). Thus, p38MAPK may play dual roles in cancer progression, depending on cell types and cellular environment. We here showed that p38MAPK activation inhibits cell motility and also mediates anoikis, and determined the exact position of p38MAPK in the signalling pathway chain from ROS production to anoikis and migration inhibition. The p38MAPK activation is an intermediate event mediated up-stream by PKC-ε, because PKC-ε inhibition (by siRNA–PKC-ε) blocked p38MAPK activation (Figure 7D) and reversed the [Pt(O,O′-acac)(γ-acac)(DMS)]-provoked inhibition of cell migration and anoikis (Figure 7E,F). Further upstream, the inhibition of NADPH oxidase blocked the cytosol-to-membrane translocation of PKC-ε induced by [Pt(O,O′-acac)(γ-acac)(DMS)] (Figure 8C).
With regard to upstream mediators implicated in the [Pt(O,O′-acac)(γ-acac)(DMS)]-induced ERK1/2 activation in HeLa cells, the PKCs appeared to be crucial elements in the pathway linking [Pt(O,O′-acac)(γ-acac)(DMS)] to the ERK1/2 cascade. In addition, it has been previously shown that the PKC signal transduction pathway regulates cell death by both cisplatin (Basu and Tu, 2005) and [Pt(O,O′-acac)(γ-acac)(DMS)] (Muscella et al., 2007).
Although PKC-ε has been reported to be able to mediate cell adhesion and motility (Gorin and Pan, 2009), there are also studies showing that PKC-ε does not enhance tumour cell proliferation, and does not participate in signalling pathways involved in neoplastic transformation or malignant progression (Evans et al., 2003). In particular, the anti-proliferative influence of tamoxifen on MCF-7 cells may be related to the PKC-ε cytosol-to-membrane translocation (Lavie et al., 1998). In the thyroid PC Cl3 cell line, a role for PKC-ε in apoptosis signalling pathways caused by many stimuli (Knauf et al., 1999) is well known, and we previously described its linkage with p38MAPK-mediated apoptosis (Muscella et al., 2009). However, as far as the anoikis process is concerned, to our knowledge our study is the first showing such a relationship. Finally, Src is known to interact, through the SH2 domain, with many signalling proteins including PKC-ε (Song et al., 2002).
One important class of ECM-degrading enzymes, the MMPs, is implicated in tumour cell metastasis (Wolf and Friedl, 2009), and among the MMPs, gelatinases MMP-2 and MMP-9 are correlated with an aggressive, invasive or metastatic tumour phenotype (Somiari et al., 2006). Increased levels of MMPs have also been reported in breast tumour cells, as well as in the surrounding non-cancerous breast tissue (Wang et al., 2000). Here, we found that [Pt(O,O′-acac)(γ-acac)(DMS)] provoked a clear concentration- and time-dependent suppression of MMP-2 and MMP-9 protein levels (Figure 6A,B). These results correlated to zymography of conditioned media of [Pt(O,O′-acac)(γ-acac)(DMS)]-treated cells (Figure 6A), suggesting that [Pt(O,O′-acac)(γ-acac)(DMS)] may inhibit processes that lead to mammary tumour metastasis. Cell invasion is characteristic of cancer cells with actin remodelling being one of the crucial elements of a deregulated actin system in malignant transformation and progression. Initial signals controlling actin cytoskeleton rearrangements may include modification of FAK, a non-receptor tyrosine kinase that is localized in focal adhesions (Rodriguez-Fernandez et al., 1999). Less attention has been given to the role of intrinsic FAK activity in promoting tumour progression, which may be due to the fact that there is no evidence to date that FAK is mutated in cancer. However, elevated FAK expression and/or activity are associated with malignancy in a variety of cancer cells (Mitra and Schlaepfer, 2006). Notably, RNAi-mediated knock-down of FAK in aggressive breast carcinoma cells did not affect proliferation or apoptosis in culture, but FAK-lacking carcinoma cells did not exhibit invasive activity in vitro or spontaneous mammary-to-lung metastasis in vivo (Mitra and Schlaepfer, 2006). In transformed cells, FAK–Src activity has been also shown to promote invasion by influencing actin cytoskeleton dynamics that in turn modify cell–substratum adhesion (Jones et al., 2000; Mitra and Schlaepfer, 2006).
In an effort to further understand the mechanism that regulates activation of FAK in human breast cancer cells, we observed changes on the phosphorylation of Tyr397 upon detachment and the actin cytoskeleton remodelling, suggesting that phosphorylation of Tyr397 was the major mechanism for the detachment-induced increase of FAK activity in MCF-7 cells (Mitra and Schlaepfer, 2006). Treatment with [Pt(O,O′-acac)(γ-acac)(DMS)] inhibited FAK phosphorylation on Tyr397 (Figure 3C), and such inhibition was completely reversed by the NADPH oxidase inhibitor, DPI (Figure 3C). DPI also restored the inhibition of the cell adhesion to collagen after [Pt(O,O′-acac)(γ-acac)(DMS)] administration (Figure 3D).
The data presented herein are the first to indicate that [Pt(O,O′-acac)(γ-acac)(DMS)] prevents events leading to metastasis via alterations in cell migration; loss of anchorage independency; stromal interactions; and altered MMP production, secretion and/or activity. The key points of this model are summarized in Figure 10. More importantly, [Pt(O,O′-acac)(γ-acac)(DMS)] was effective after both short- and long-term treatment by exerting toxic effects at high doses and suppression of breast cancer metastasis at sublethal doses. Altogether, these findings suggest that [Pt(O,O′-acac)(γ-acac)(DMS)] is a promising therapeutic agent for preventing growth and metastasis of breast cancer.
Figure 10.
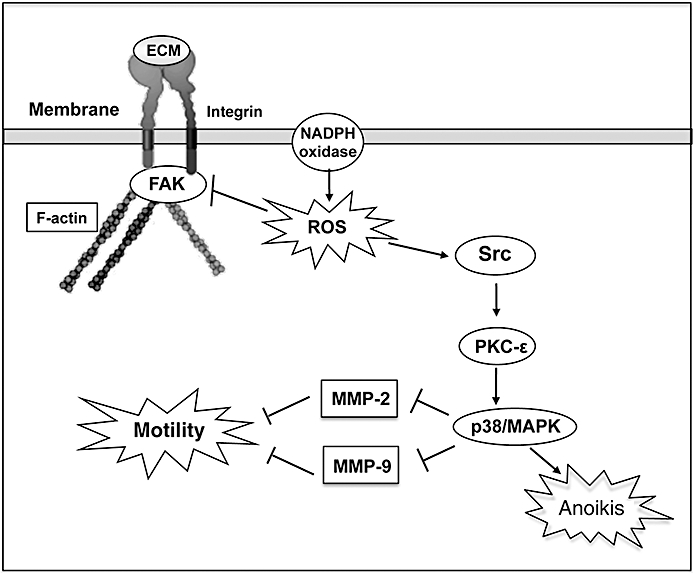
A proposed model for MCF-7 cell responses to [Pt(O,O′-acac)(γ-acac)(DMS)]. Oxidant stress provokes activation of Src and inhibition of FAK phosphorylation. Src activation leads to PKC-ε and p38MAPK activation which increases [Pt(O,O′-acac)(γ-acac)(DMS)]-mediated anoikis. Activated p38MAPK also inhibited MMP-2 and MMP-9 secretion, and cell migration. FAK dephosphorylation provoked actin cytoskeleton rearrangements leading to inhibition of detached MCF-7 cell invasion.
Acknowledgments
The research was supported by University of Salento (Fondi di Ateneo per la Ricerca di Base, 2009).
Glossary
Abbreviations:
- DAPI
4,6-diamino-2-phenylindole
- DMEM
Dulbecco's modified Eagle's medium
- DMS
dimethylsulphide
- ECL
enhanced chemiluminescence
- ERK
extracellular signal-regulated kinase
- MAPK
mitogen-activated protein kinase
- MEK
MAPK/ERK kinase
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenol tetrazolium bromide
- NBT
nitroblue tetrazolium
- PARP
poly(ADP-ribose) polymerase
- PBS
phosphate-buffered saline
- PKC-ε
protein kinase C-epsilon
- PVDF
polyvinylidene difluoride membrane
- ROS
reactive oxygen species
- SDS
sodium dodecyl sulphate
- siRNA
small interfering RNA
Conflict of interest
The authors state no conflict of interest.
References
- Bartsch JE, Staren ED, Appert HE. Matrix metalloproteinase expression in breast cancer. J Surg Res. 2003;110:383–392. doi: 10.1016/s0022-4804(03)00007-6. [DOI] [PubMed] [Google Scholar]
- Basu A, Tu H. Activation of ERK during DNA damage-induced apoptosis involves protein kinase C-d. Biochem Biophys Res Commun. 2005;334:11068–11073. doi: 10.1016/j.bbrc.2005.06.199. [DOI] [PubMed] [Google Scholar]
- Blaschke F, Stawowy P, Goetze S, Hintz O, Gräfe M, Kintscher U, et al. Hypoxia activates β1-integrin via ERK 1/2 and p38 MAP kinase in human vascular smooth muscle cells. Biochem Biophys Res Commun. 2002;296:890–896. doi: 10.1016/s0006-291x(02)02033-8. [DOI] [PubMed] [Google Scholar]
- Bracke ME, Boterberg T, Bruyneel EA, Mareel MM. Collagen vasion assay. In: Brooks S, Schumacher U, editors. Metastasis Research Protocols. Totowa: Humana Press; 2001. pp. 81–89. [Google Scholar]
- Carragher NO, Frame MC. Focal adhesion and actin dynamics: a place where kinases and proteases meet to promote invasion. Trends Cell Biol. 2004;14:241–249. doi: 10.1016/j.tcb.2004.03.011. [DOI] [PubMed] [Google Scholar]
- Chen L, Mayer JA, Krisko TI, Speers CW, Wang T, Hilsenbeck SG, et al. Inhibition of the p38 kinase suppresses the proliferation of human negative breast cancer cells. Cancer Res. 2009;69:8853–8861. doi: 10.1158/0008-5472.CAN-09-1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng JJ, Chao YJ, Wang DL. Cyclic strain activates redox-sensitive proline-rich tyrosine kinase 2 (PYK2) in endothelial cells. J Biol Chem. 2002;277:48152–48157. doi: 10.1074/jbc.M110937200. [DOI] [PubMed] [Google Scholar]
- Cocolakis E, Lemay S, Ali S, Lebrun JJ. The p38 MAPK pathway is required for cell growth inhibition of human breast cancer cells in response to activin. J Biol Chem. 2001;276:18430–18436. doi: 10.1074/jbc.M010768200. [DOI] [PubMed] [Google Scholar]
- De Pascali SA, Papadia P, Ciccarese A, Pacifico C, Fanizzi FP. First examples of β-diketonate platinum(II) complexes with sulfoxide ligands. Eur J Inorg Chem. 2005;5:788–796. [Google Scholar]
- Evan GI, Vousden KH. Proliferation, cell cycle and apoptosis in cancer. Nature. 2001;411:342–348. doi: 10.1038/35077213. [DOI] [PubMed] [Google Scholar]
- Evans JD, Cornford PA, Dodson A, Neoptolemos JP, Foster CS. Expression patterns of protein kinase C isoenzymes are characteristically modulated in chronic pancreatitis and pancreatic cancer. Am J Clin Pathol. 2003;119:392–402. doi: 10.1309/bkpc9dx98r781b87. [DOI] [PubMed] [Google Scholar]
- Ferlay J, McCarron P, Parkin DM. The changing global patterns of female breast cancer incidence and mortality. Breast Cancer Res. 2004;6:229–239. doi: 10.1186/bcr932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkel T. Oxidant signals and oxidative stress. Curr Opin Cell Biol. 2003;15:247–254. doi: 10.1016/s0955-0674(03)00002-4. [DOI] [PubMed] [Google Scholar]
- Folgueras AR, Pendás AM, Sánchez LM, López-Otín C. Matrix metalloproteinases in cancer: from new functions to improved inhibition strategies. Int J Dev Biol. 2004;48:411–424. doi: 10.1387/ijdb.041811af. [DOI] [PubMed] [Google Scholar]
- Folkman J, Moscona A. Role of cell shape in growth control. Nature. 1978;273:345–349. doi: 10.1038/273345a0. [DOI] [PubMed] [Google Scholar]
- Gorin MA, Pan Q. Protein kinase C-ε: an oncogene and emerging tumor biomarker. Mol Cancer. 2009;8:1–9. doi: 10.1186/1476-4598-8-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homma Y, Henning-Chubb CB, Huberman E. Translocation of protein kinase C in human leukemia cells susceptible or resistant to differentiation induced by phorbol 12-myristate 13-acetate. Proc Natl Acad Sci USA. 1986;83:7316–7319. doi: 10.1073/pnas.83.19.7316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C, Jacobson K, Schaller MD. MAP kinases and cell migration. J Cell Sci. 2004;117:4619–4628. doi: 10.1242/jcs.01481. [DOI] [PubMed] [Google Scholar]
- Jones RJ, Brunton VG, Frame MC. Adhesion-linked kinases in cancer; emphasis on Src, focal adhesion kinase and PI 3-kinase. Eur J Cancer. 2000;36:1595–1606. doi: 10.1016/s0959-8049(00)00153-2. [DOI] [PubMed] [Google Scholar]
- Knauf JA, Elisei R, Mochly-Rosen D, Liron T, Chen XN, Gonsky R, et al. Involvement of protein kinase C-epsilon (PKCepsilon) in thyroid cell death. A truncated chimeric PKCepsilon cloned from a thyroid cancer cell line protects thyroid cells from apoptosis. J Biol Chem. 1999;274:23414–23425. doi: 10.1074/jbc.274.33.23414. [DOI] [PubMed] [Google Scholar]
- Kopetz S, Lesslie DP, Dallas NA, Park SI, Johnson M, Parikh NU, et al. Synergistic activity of the SRC family kinase inhibitor dasatinib and oxaliplatin in colon carcinoma cells is mediated by oxidative stress. Cancer Res. 2009;69:3842–3849. doi: 10.1158/0008-5472.CAN-08-2246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavie Y, Zhang Z, Cao H, Han T, Jones RC, Liu Y, et al. Tamoxifen induces selective membrane association of protein kinase c epsilon in MCF-7 human breast cancer cells. Int J Cancer. 1998;77:928–932. doi: 10.1002/(sici)1097-0215(19980911)77:6<928::aid-ijc22>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- Li QS, Lee GY, Ong CN, Lim CT. AFM indentation study of breast cancer cells. Biochem Biophys Res Commun. 2008;374:609–613. doi: 10.1016/j.bbrc.2008.07.078. [DOI] [PubMed] [Google Scholar]
- McPherson I. Soft agar techniques. In: Kruse PF, Patterson MK, editors. Tissue Culture Methods and Applications. New York: Academic Press; 1973. pp. 276–280. [Google Scholar]
- Martiny-Baron G, Kazanietz MG, Mischak H, Blumberg PM, Kochs G, Hug H, et al. Selective inhibition of protein kinase C isozymes by the indolocarbazole Gö 6976. J Biol Chem. 1993;268:9194–9197. [PubMed] [Google Scholar]
- Mendes O, Kim HT, Lungu G, Stoica G. MMP-2 role in breast cancer brain metastasis development and its regulation by TIMP 2 and ERK 1/2. Clin Exp Metastasis. 2007;24:341–351. doi: 10.1007/s10585-007-9071-0. [DOI] [PubMed] [Google Scholar]
- Mitra SK, Schlaepfer DD. Integrin-regulated FAK-Src signaling in normal and cancer cells. Curr Opin Cell Biol. 2006;18:516–523. doi: 10.1016/j.ceb.2006.08.011. [DOI] [PubMed] [Google Scholar]
- Muscella A, Greco S, Elia MG, Storelli C, Marsigliante S. Angiotensin II stimulation of Na+/K+ATPase activity and cell growth by calcium-independent pathway in MCF-7 breast cancer cells. J Endocrinol. 2002;173:315–323. doi: 10.1677/joe.0.1730315. [DOI] [PubMed] [Google Scholar]
- Muscella A, Storelli C, Marsigliante S. Atypical PKC-zeta and PKC-iota mediate opposing effects on MCF-7 Na+/K+ATPase activity. J Cell Physiol. 2005;205:278–285. doi: 10.1002/jcp.20396. [DOI] [PubMed] [Google Scholar]
- Muscella A, Calabriso N, De Pascali SA, Urso L, Ciccarese A, Fanizzi FP, et al. New platinum(II) complexes containing both an O,O′-chelated acetylacetonate ligand and a sulfur ligand in the platinum coordination sphere induce apoptosis in HeLa cervical carcinoma cells. Biochem Pharmacol. 2007;74:28–40. doi: 10.1016/j.bcp.2007.03.027. [DOI] [PubMed] [Google Scholar]
- Muscella A, Calabriso N, Fanizzi FP, De Pascali SA, Urso L, Ciccarese A, et al. [Pt(O,O′-acac)(γ-acac)(DMS)], a new Pt compound exerting fast cytotoxicity in MCF-7 breast cancer cells via the mitochondrial apoptotic pathway. Br J Pharmacol. 2008;153:34–49. doi: 10.1038/sj.bjp.0707576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muscella A, Urso L, Calabriso N, Vetrugno C, Fanizzi FP, Storelli C, et al. Functions of epidermal growth factor receptor in cisplatin response of thyroid cells. Biochem Pharmacol. 2009;77:979–992. doi: 10.1016/j.bcp.2008.12.001. [DOI] [PubMed] [Google Scholar]
- Oliveira HR, Verlengia R, Carvalho CR, Britto LR, Curi R, Carpinelli AR. Pancreatic beta-cells express phagocyte-like NAD(P)H oxidase. Diabetes. 2003;52:1457–1563. doi: 10.2337/diabetes.52.6.1457. [DOI] [PubMed] [Google Scholar]
- Ridley AJ. Rho GTPases and cell migration. J Cell Sci. 2001;114:2713–2722. doi: 10.1242/jcs.114.15.2713. [DOI] [PubMed] [Google Scholar]
- Rittling SR, Denhardt DT. Osteopontin function in pathology: lessons from osteopontin-deficient mice. Exp Nephrol. 1999;7:103–113. doi: 10.1159/000020591. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Fernandez JL, Gomez M, Luque A, Hogg N, Sanchez-Madrid F, Cabanas C. The interaction of activated integrin lymphocyte function-associated antigen 1 with ligand intercellular adhesion molecule 1 induces activation and redistribution of focal adhesion kinase and proline-rich tyrosine kinase 2 in T lymphocytes. Mol Biol Cell. 1999;10:1891–1907. doi: 10.1091/mbc.10.6.1891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlessinger J. New roles for Src kinases in control of cell survival and angiogenesis. Cell. 2000;100:293–296. doi: 10.1016/s0092-8674(00)80664-9. [DOI] [PubMed] [Google Scholar]
- Seko Y, Tobe K, Takahashi N, Kaburagi Y, Kadoyaki T, Yazaki Y. Hypoxia and hypoxia reoxygenation activate Src family tyrosine kinase and p21ras in cultured cardiac myocytes. Biochem Biophys Res Commun. 1996;226:530–535. doi: 10.1006/bbrc.1996.1389. [DOI] [PubMed] [Google Scholar]
- Somiari SB, Somiari RI, Heckman CM, Olsen CH, Jordan RM, Russell SJ, et al. Circulating MMP2 and MMP9 in breast cancer – potential role in classification of patients into low risk, high risk, benign disease and breast cancer categories. Int J Cancer. 2006;119:1403–1411. doi: 10.1002/ijc.21989. [DOI] [PubMed] [Google Scholar]
- Song C, Vondriska TM, Wang GW, Klein JB, Cao X, Zhang J, et al. Molecular conformation dictates signaling module formation: example of PKCepsilon and Src tyrosine kinase. Am J Physiol Heart Circ Physiol. 2002;282:1166–11671. doi: 10.1152/ajpheart.00830.2001. [DOI] [PubMed] [Google Scholar]
- Valentijn AJ, Zouq N, Gilmore AP. Anoikis. Biochem Soc Trans. 2004;32:421–425. doi: 10.1042/BST0320421. [DOI] [PubMed] [Google Scholar]
- Wang JL, Sun Y, Wu S. Gamma-irradiation induces matrix metalloproteinase II expression in a p53-dependent manner. Mol Carcinog. 2000;27:252–258. doi: 10.1002/(sici)1098-2744(200004)27:4<252::aid-mc2>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- Weis S, Cui J, Barnes L, Cheresh D. Endothelial barrier disruption by VEGF-mediated Src activity potentiates tumor cell extravasation and metastasis. J Cell Biol. 2004;167:223–229. doi: 10.1083/jcb.200408130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf K, Friedl P. Mapping proteolytic cancer cell–extracellular matrix interfaces. Clin Exp Metastasis. 2009;26:289–298. doi: 10.1007/s10585-008-9190-2. [DOI] [PubMed] [Google Scholar]
- Xiong S, Grijalva R, Zhang L, Nguyen NT, Pisters PW, Pollock RE, et al. Up-regulation of vascular endothelial growth factor in breast cancer cells by the heregulin-beta1-activated p38 signaling pathway enhances endothelial cell migration. Cancer Res. 2001;61:1727–1732. [PubMed] [Google Scholar]
- Yedovitzky M, Mochly-Rosen D, Johnson JA, Gray MO, Ron D, Abramovitch E, et al. Translocation inhibitors define specificity of protein kinase C isoenzymes in pancreatic beta-cells. J Biol Chem. 1997;272:1417–1420. doi: 10.1074/jbc.272.3.1417. [DOI] [PubMed] [Google Scholar]
- Zhang CC, Shapiro DJ. Activation of the p38 mitogen-activated protein kinase pathway by estrogen or by 4-hydroxytamoxifen is coupled to estrogen receptor-induced apoptosis. J Biol Chem. 2000;275:479–486. doi: 10.1074/jbc.275.1.479. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Zhang B. D4-GDI, a rho GTPase regulator, promotes breast cancer cell invasiveness. Cancer Res. 2006;66:5592–5598. doi: 10.1158/0008-5472.CAN-05-4004. [DOI] [PubMed] [Google Scholar]
- Zhou J, Chen Y, Lang JY, Lu JJ, Ding J. Salvicine inactivates beta 1 integrin and inhibits adhesion of MDA-MB-435 cells to fibronectin via reactive oxygen species signaling. J Mol Cancer Res. 2008;6:194–204. doi: 10.1158/1541-7786.MCR-07-0197. [DOI] [PubMed] [Google Scholar]
- Zhu Z, Sanchez-Sweatman O, Huang X, Wiltrout R, Khokha R, Zhao Q, et al. Anoikis and metastatic potential of cloudman S91 melanoma cells. Cancer Res. 2001;61:1707–1716. [PubMed] [Google Scholar]