

Abstract
Background and purpose:
The formation of reactive oxygen species (ROS) is increased in heart failure (HF). However, the causal and mechanistic relationship of ROS formation with contractile dysfunction is not clear in detail. Therefore, ROS formation, myofibrillar protein oxidation and p38 MAP kinase activation were related to contractile function in failing rabbit hearts.
Experimental approach and key results:
Three weeks of rapid left ventricular (LV) pacing reduced LV shortening fraction (SF, echocardiography) from 32 ± 1% to 13 ± 1%. ROS formation, as assessed by dihydroethidine staining, increased by 36 ± 8% and was associated with increased tropomyosin oxidation, as reflected by dimer formation (dimer to monomer ratio increased 2.28 ± 0.66-fold in HF vs. sham, P < 0.05). Apoptosis (TdT-mediated dUTP nick end labelling staining) increased more than 12-fold after 3 weeks of pacing when a significant increase in the phosphorylation of p38 MAP kinase and HSP27 was detected (Western blotting). Vitamins C and E abolished the increases in ROS formation and tropomyosin oxidation along with an improvement of LVSF (19 ± 1%, P < 0.05 vs. untreated HF) and prevention of apoptosis, but without modifying p38 MAP kinase activation. Inhibition of p38 MAP kinase by SB281832 counteracted ROS formation, tropomyosin oxidation and contractile failure, without affecting apoptosis.
Conclusions and implications:
Thus, p38 MAP kinase activation appears to be upstream rather than downstream of ROS, which impacts on LV function through myofibrillar oxidation. p38 MAP kinase inhibition is a potential target to prevent or treat HF.
Keywords: mitogen-activated protein kinase, heart failure, myofilaments, reactive oxygen species, ventricular function
Introduction
In failing hearts, the formation of reactive oxygen species (ROS) is increased in both, non-cardiomyocyte cells and cardiomyocytes (Giordano, 2005). ROS impair contractile function by a number of mechanisms, including decreased nitric oxide bioavailability, decreased β-adrenoceptor expression (Qin et al., 2003) and ion channel activity (Hool, 2006), activation of protein kinases (Michel et al., 2001; Li et al., 2002) and oxidation of myofibrillar proteins; we have recently demonstrated tropomyosin oxidation in both isolated rat hearts and microembolized pig hearts and related it to contractile dysfunction (Canton et al., 2004; 2006). ROS scavenging attenuates left ventricular (LV) dysfunction following myocardial infarction (Kinugawa et al., 2000; Qin et al., 2003) or during LV pacing (Qin et al., 2003) and decreases LV hypertrophy and fibrosis (Kinugawa et al., 2000) as well as cardiomyocyte apoptosis (Qin et al., 2003).
Among other protein kinases, ROS activate p38 mitogen-activated protein kinase (p38 MAP kinase) (Michel et al., 2001; Li et al., 2002). In rabbit hearts, p38 MAP kinase is activated after myocardial infarction and its activation correlates with increased oxidative stress (Qin et al., 2005). Once activated, p38 MAP kinase contributes to LV remodelling and heart failure (HF) development (Wang, 2007), including LV hypertrophy, fibrosis and inflammation (Tenhunen et al., 2006) as well as cardiomyocyte apoptosis (Baines and Molkentin, 2005). Therefore, p38 MAP kinase provides an interesting target for HF therapy (Kerkela and Force, 2006). Indeed, pharmacological blockade of p38 MAP kinase (Wang et al., 2005) or its genetic knockout (Ren et al., 2005) improve LV function and attenuate morphological alterations following myocardial infarction. p38 MAP kinase is also activated in myocardial biopsies from HF patients (Haq et al., 2001), and the improvement of LV function by resynchronization therapy is associated with reduced p38 MAP kinase phosphorylation in human hearts (Butter et al., 2008). The mechanism underlying the improvement in LV function following blockade of p38 MAP kinase activation in failing hearts, however, remains unclear. Under physiological conditions, contractile function was reduced by pharmacological blockade of p38 MAP kinase in rat cardiomyoyctes (Liao et al., 2002) or its genetic knockout in mice hearts in vivo (Braz et al., 2003), whereas in pigs reduced contractile function was not observed (Schulz et al., 2002). In contrast, during post-ischaemic reperfusion or HF p38 MAP kinase inhibition invariably improves contractile function and myocardial viability (Clark et al., 2007).
The interaction between p38 MAP kinase activation and ROS formation is not really clear: increased ROS formation contributes to p38 MAP kinase activation (see above), but also scavenging of ROS by vitamins C and E activates p38 MAP kinase in rabbit hearts (Qin et al., 2003). The impact of the interaction of ROS with p38 MAP kinase on contractile function is particularly unclear. Therefore, the present study used a HF model of LV pacing in rabbits to address: (i) the time course of ROS formation and p38 MAP kinase activation during the progression of HF; (ii) the effect of ROS scavenging by vitamins C and E; and (iii) the effect of p38 MAP kinase inhibition on myofibrillar oxidation and MAP kinase activation and the resulting LV function and morphology. We here show that inhibition of p38 MAP kinase by SB281832 counteracts ROS formation, tropomyosin oxidation and contractile failure, without affecting apoptosis. Thus, p38 MAP kinase activation appears to be upstream rather than downstream of ROS, which impact on LV function through myofibrillar oxidation.
Methods
This study was approved by the bioethical committee of the district of Düsseldorf, Germany, and the investigation conforms with the Guide for the Care and Use of Laboratory Animals published by the US National Institute of Health (NIH publication no. 85–23, revised 1996). The drug/molecular target nomenclature conforms to BJP's Guide to Receptors and Channels (Alexander et al., 2008).
Experimental model and protocols
Instrumentation of male Chinchilla rabbits (Charles River, Kisslegg, Germany) weighing 3–4 kg body weight was performed as described in detail elsewhere (Schulz et al., 2003; Aker et al., 2004).
Time course of heart failure development
HF was induced by LV pacing (400 bpm) for 1 (HF1, n = 7), 2 (HF2, n = 7) or 3 weeks (HF3, n = 8) respectively. Seven sham-operated rabbits (Sham) served as controls. HF was evident from clinical signs, such as ascites and cachexia and echocardiographic parameters, such as a reduction of LV shortening fraction (LVSF) and an increase in LV end-diastolic diameter (LVEDD). After euthanasia of the rabbits, samples (50 mg each) were taken from the LV free wall and either frozen in liquid nitrogen and stored at −70°C or fixed in formalin and embedded in paraffin.
Vitamins C and E treatment
In four additional groups, rabbits received either placebo or vitamins C (100 mg·kg−1·day−1 p.o.) and E (200 mg·day−1 p.o.) for 3 weeks without (Sham, n = 7 or Sham-Vit, n = 7) or with LV pacing (HF3, n = 8 or HF3-Vit, n = 6).
SB281832 treatment
In four additional groups, rabbits received either placebo or the p38 MAP kinase inhibitor SB281832 at a dose of 50 mg·kg−1·day−1 p.o. for 3 weeks without (Sham, n = 7 or Sham-SB, n = 7) or with LV pacing (HF3, n = 8 or HF3-SB, n = 6). To detect potential blood pressure changes following p38 MAP kinase blockade, measurements of systolic and diastolic blood pressures were performed using a sphygmomanometer cuff (Memoprint, Babenhausen, Germany) in five rabbits of each group, while conscious. There was no significant effect of p38 MAP kinase blockade with SB281832 on blood pressure in sham (placebo: 106 ± 1/69 ± 1 mmHg vs. SB: 106 ± 1/71 ± 1 mmHg) or HF (placebo: 99 ± 3/65 ± 1 mmHg vs. SB: 92 ± 6/66 ± 1 mmHg) rabbits.
Echocardiography
Heart rate and LV function were measured (Supervision 7000, Toshiba, Neuss, Germany) as described in detail elsewhere (Schulz et al., 2003; Aker et al., 2004). LVSF was calculated from LVEDD and end-systolic diameter: (LVEDD − end-systolic diameter)/LVEDD × 100.
Histology
Apoptosis was determined using the TdT-mediated dUTP nick end labelling (TUNEL) technique (In Situ Cell Death Detection Kit, La Roche Diagnostics, Mannheim, Germany) as described recently (Schulz et al., 2003; Aker et al., 2004). TUNEL-positive cardiomyocyte nuclei were counted using fluorescence microscopy (Leica DMLB, Bensheim, Germany). Three fields of 0.075 mm2 each were analysed; TUNEL-positive cardiomyocyte nuclei were counted and calculated per mm2.
Cardiomyocyte cross-sectional area was measured in haematoxylin and eosin-stained tissue sections (three fields of 0.075 mm2 each) as described in detail before (Schulz et al., 2003; Aker et al., 2004).
Western blot
The tissue samples were processed as described previously (Schulz et al., 2003; Aker et al., 2004).
p38 MAP kinase and JNK MAP kinase phosphorylation
The blots were incubated for 2 h either with an antiserum recognizing total p38 MAP kinase or with an antiserum specific for the dually phosphorylated form of p38 MAP kinase (New England Biolabs, Beverly, MA, USA). In addition, the phosphorylated forms of JNK1 and 2 (46/54) MAP kinase (Thr183/Tyr185) or total JNK1 and 2 MAP kinase (Thr183/Tyr185) (Cell Signaling, Invitrogen, Life Technologies Corporation, Carlsbad, CA, USA) were assessed. The resulting autoradiographs were analysed by quantitative two-dimensional densitometry, using commercially available software (Herolab, Wiesloch, Germany). To quantify the extent of MAP kinase phosphorylation, the phosphorylated MAP kinase band intensity was expressed relative to the intensity of the total MAP kinase band. Data were expressed relative to the mean value of sham rabbits.
p38 MAP kinase activation
After blocking, the membranes were incubated overnight at 4°C with mouse monoclonal antibodies against HSP27 phosphorylated at Ser83 (1:100, kindly provided by Professor M. Marber). After incubation with the respective secondary antibodies, immunoreactivity was detected using the SuperSignal West Femto Maximum Sensitivity Substrate (Pierce, Rockford, IL, USA). After detection of phosphorylated HSP27, membranes were reprobed with monoclonal mouse anti-chicken HSP25/27 antibodies (1:1750, HyTest, Turku, Finland). Signal intensities were quantified using the Scion Image software (Frederick, MD, USA).
Reactive oxygen species
Samples were snap frozen in liquid nitrogen, embedded in O.C.T. (Cryomatrix, Shandon, Pittsburgh, PA, USA) and cut into 10 µm sections at −20°C. Sections were mounted on glass slides. Dihydroethidine (DHE, D-11347, Molecular Probes Inc., Eugene, OR, USA) at a concentration of 2 × 10−6 M was incubated at 37°C for 30 min in a humidified chamber. Negative controls were obtained by blocking the reaction with 100 mM N-Acetyl-L-Cysteine (NAC, A9165 Sigma-Aldrich, Taufkirchen, Germany) for 30 min prior to DHE staining. Images were taken with a laser scanning microscope (LSM Pascal 5, Zeiss, Oberkochem, Germany) including a krypton/argon laser; fluorescence was detected by a 585 nm long-pass filter. Three fields of 0.075 mm2 each were analysed.
Tropomyosin oxidation
The tissue samples were processed as described previously (Canton et al., 2004; 2006;). Purified human cardiac tropomyosin (Abcam, Cambridge, UK) oxidized with 2 mM 5,5′-dithiobis(2-nitrobenzoate) in a buffer containing 1 M NaCl, 1 mM EDTA and 0.05 M sodium phosphate served as positive control.
Serum noradrenaline (NA) concentration and myocardial β-adrenoceptor density
Peripheral arterial blood was drawn through a catheter into ice-cold tubes before euthanasia. The β-adrenoceptor density was assessed in frozen tissue. The blood and tissue samples were processed as described previously (Leineweber et al., 2006).
Statistics
Data are expressed as mean values ± SEM. Comparison of data before and during LV pacing was performed by one-way anova. Comparison of haemodynamic data before and during the 3 weeks of pacing between the different treatment groups was performed by two-way anova. Cardiomyocyte cross-sectional area, the number of TUNEL-positive cardiomyocytes, β-adrenoceptor density, MAP kinase phosphorylation and activation and tropomyosin oxidation were compared between untreated and treated sham and HF rabbits by two-way anova. When a significant overall effect was detected by anova, individual mean values were compared using Bonferroni's method. All statistical calculations were performed using the Prism programme. A P-value < 0.05 was considered significant.
Results
Time course
There was a progressive increase in LVEDD and a reduction of LVSF with increasing duration of LV pacing (Table 1). Heart rate and serum NA concentration increased, while β-adrenoceptor density decreased already after 1 week of LV pacing. Cardiomyocyte cross-sectional area remained unchanged at 1 week, but increased after 2 and 3 weeks of LV pacing, and the number of TUNEL-positive cardiomyocytes was increased more than 12-fold after 3 weeks of LV pacing. The ROS concentration was significantly increased after 3 weeks of LV pacing (Figure 1), and such increase in ROS was associated with increased tropomyosin oxidation, as indicated by an increased tropomyosin dimer to monomer ratio (Figure 2). p38 MAP kinase and HSP27 phosphorylation were increased only after 3 weeks of LV pacing (Figure 3; Table 1). JNK1 phosphorylation remained unchanged throughout 3 weeks of LV pacing, while JNK2 phosphorylation was increased after 3 weeks of pacing (7.03 ± 2.24-fold compared with sham).
Table 1.
Haemodynamics, β-adrenoceptor density and noradrenaline concentration, morphology and MAP kinase activation during the progression of HF
| Sham | HF1 | HF2 | HF3 | |
|---|---|---|---|---|
| Heart rate (bpm) | 224 ± 5 | 263 ± 10A | 277 ± 10A | 258 ± 15 |
| LVEDD (mm) | 17.0 ± 0.3 | 18.7 ± 0.4A | 19.3 ± 0.3A | 19.6 ± 0.5AB |
| Fractional shortening (%) | 30.0 ± 1.7 | 19.1 ± 0.7A | 15.9 ± 0.7A | 10.3 ± 1.3AB |
| Noradrenaline (ng·mL−1) | 1.33 ± 0.17 | 2.87 ± 0.69A | 4.03 ± 0.81A | 3.83 ± 0.64A |
| ß-Adrenoceptor density (fmol·mg−1) | 67 ± 5 | 45 ± 2A | 47 ± 2A | 41 ± 3A |
| Cardiomyocyte cross-sectional area (µm2) | 242 ± 11 | 266 ± 4 | 299 ± 8AB | 334 ± 7ABC |
| Number of TUNEL-positive cardiomyocytes [/(1000*mm2)] | 4 ± 2 | 23 ± 5A | 16 ± 4 | 50 ± 8ABC |
| Phospho p38-MAPK/total p38-MAPK (relative to sham) | 1.00 ± 0.24 | 0.54 ± 0.09 | 0.65 ± 0.24 | 2.34 ± 0.54ABC |
| Phospho HSP27/total HSP27 (relative to sham) | 1.00 ± 0.42 | 1.75 ± 0.24 | 1.01 ± 0.14 | 2.58 ± 0.60AC |
Mean ± SEM.
p < 0.05 versus Sham.
p < 0.05 versus HF1.
p < 0.05 versus HF2.
HF1, heart failure rabbits, 1 week (n = 7); HF2, heart failure rabbits, 2 weeks (n = 7); HF3, heart failure rabbits, 3 weeks (n = 8); LVEDD, left ventricular end-diastolic diameter; Sham, sham-operated rabbits (n = 7); TUNEL, TdT-mediated dUTP nick end labelling.
Figure 1.
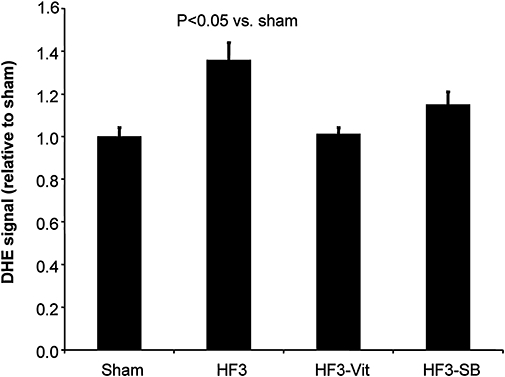
The myocardial ROS concentration (DHE signal) increased following 3 weeks of LV pacing (n = 8) compared with sham rabbits (n = 7). Such increase in the myocardial ROS concentration was prevented by both, pretreatment with vitamins C and E (n = 6) and p38 MAP kinase blockade with SB281832 (n = 6). Data are expressed as mean ± SEM. DHE, dihydroethidine; HF3, heart failure rabbits after 3 weeks of pacing; LV, left ventricular; ROS, reactive oxygen species.
Figure 2.
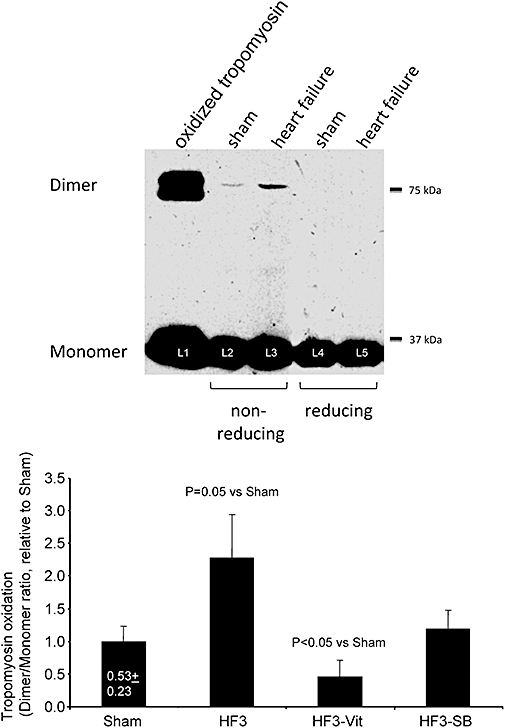
Upper panel: representative Western blots of tropomyosin oxidation. Lane 1 (L1) illustrates the highest degree of oxidation obtained by adding 1 mM H2O2 for 15 min to isolated tropomyosin. Lanes 3 and 5 illustrate typical examples of Western blots from failing rabbit hearts after 3 weeks of LV pacing as compared with control hearts from sham operated rabbits (lanes 2, L2 and 4, L4). Electrophoreses of lanes 2 and 3 were carried out under non-reducing conditions as opposed to conventional reducing conditions used for lanes 4 and 5. The comparison between reducing and non-reducing electrophoresis permits the attribution of tropomyosin dimers in lanes 2 and 3 (L2, L3) to disulfide cross-bridge formation. Lower panel: tropomyosin oxidation increased following 3 weeks of LV pacing (n = 8) compared with sham rabbits (n = 7). Such increases in tropomyosin oxidation were prevented by both, pretreatment with vitamins C and E (n = 6) and p38 MAP kinase blockade with SB281832 (n = 6). Data are expressed as mean ± SEM. HF3, heart failure rabbits after 3 weeks of pacing; LV, left ventricular.
Figure 3.
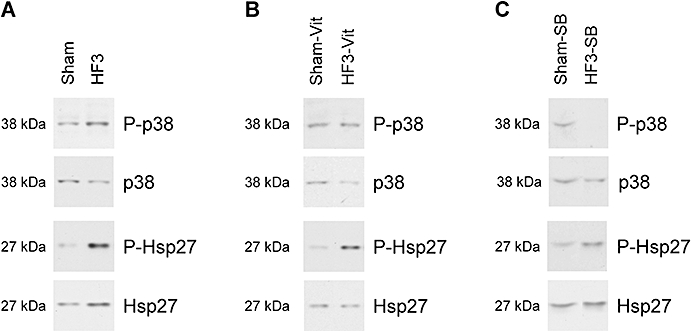
(A) Representative Western blots of phosphorylated p38 MAP kinase, total p38 MAP kinase, phosphorylated HSP27 and total HSP27 in Sham and HF3 rabbits. (B) Representative Western blots of phosphorylated p38 MAP kinase, total p38 MAP kinase, phosphorylated HSP27 and total HSP27 in Sham and HF3 rabbits, treated with vitamins C and E. (C) Representative Western blots of phosphorylated p38 MAP kinase, total p38 MAP Kinase, phosphorylated HSP27 and total HSP27 in Sham and HF3 rabbits, blocked with SB281832. HF3, heart failure rabbits after 3 weeks of pacing.
ROS scavenging
Administration of antioxidant vitamins did not significantly alter any analysed parameter in sham rabbit hearts, except that p38 MAP kinase phosphorylation was increased, however, without a significant concomitant increase in HSP27 phosphorylation (Table 2).
Table 2.
Haemodynamics, morphology, β-adrenoceptor density and noradrenaline concentration without and with ROS scavenging by vitamins C and E in sham and HF rabbits
| Sham | Sham-Vit | HF3 | HF3-Vit | |
|---|---|---|---|---|
| Heart rate (bpm) | 223 ± 6 | 220 ± 4 | 259 ± 11A | 241 ± 5 |
| LVEDD (mm) | 16.6 ± 0.2 | 16.7 ± 0.2 | 19.8 ± 0.5A | 18.8 ± 0.2A |
| Fractional shortening (%) | 31.9 ± 1.0 | 31.1 ± 0.7 | 12.9 ± 0.7A | 18.7 ± 0.9AB |
| Noradrenaline (ng·mL−1) | 2.19 ± 0.37 | 1.96 ± 0.26 | 5.82 ± 0.76A | 5.37 ± 1.12A |
| ß-Adrenoceptor density (fmol·mg−1) | 62 ± 5 | 67 ± 4 | 37 ± 3A | 53 ± 7AB |
| Cardiomyocyte cross-sectional area (µm2) | 227 ± 6 | 211 ± 4 | 306 ± 6A | 259 ± 9AB |
| Number of TUNEL-positive cardiomyocytes [/(1000*mm2)] | 4 ± 3 | 19 ± 7 | 64 ± 4A | 18 ± 4A |
| Phospho p38-MAPK/total p38-MAPK (relative to sham) | 1.00 ± 0.30 | 4.79 ± 0.80A | 2.47 ± 0.71A | 3.25 ± 0.55A |
| Phospho HSP27/total HSP27 (relative to sham) | 1.00 ± 0.25 | 1.52 ± 0.55 | 2.70 ± 0.66A | 2.90 ± 0.28A |
Mean ± SEM.
p < 0.05 versus Sham.
p < 0.05 versus HF3.
HF3, heart failure rabbits, 3 weeks (n = 8); HF3-Vit, heart failure rabbits (3 weeks) receiving vitamins C and E (n = 6); LVEDD, left ventricular end-diastolic diameter; ROS, reactive oxygen species; Sham, sham-operated rabbits (n = 7); Sham-Vit (n = 7), sham-operated rabbits receiving vitamins C and E; TUNEL, TdT-mediated dUTP nick end labelling.
Administration of antioxidant vitamins throughout the 3 weeks of LV pacing completely abolished the increases in ROS concentration and tropomyosin oxidation (Figures 1 and 2), and it increased LVSF. The serum NA concentration remained elevated despite an improvement in LVSF, while β-adrenoceptor density returned back to baseline. Cardiomyocyte cross-sectional area and number of TUNEL-positive cardiomyocytes were reduced following 3 weeks of LV pacing with antioxidant vitamins. In contrast, p38 MAP kinase and HSP27 phosphorylation remained elevated after 3 weeks of LV pacing with antioxidant vitamins (Table 2). Similarly, enhancement of JNK2 phosphorylation was unaffected by vitamin treatment (9.80 ± 1.88-fold increase compared with sham).
p38 MAP kinase inhibition
Blockade of p38 MAP kinase with SB281832 did not alter any analysed parameter in sham rabbit hearts (Table 3). SB281832 abolished the increased p38 MAP kinase, HSP27 and JNK2 phosphorylation following 3 weeks of LV pacing as well as the HF-induced increase in ROS concentration and tropomyosin oxidation (Figures 1 and 2); LVSF was better preserved following 3 weeks of LV pacing. As with vitamin treatment, the β-adrenoceptor density returned back to baseline in the presence of an unchanged serum NA concentration. Cardiomyocyte hypertrophy was attenuated with SB281832 while the increased number of TUNEL-positive cardiomyocytes remained unaffected following 3 weeks of LV pacing.
Table 3.
Haemodynamics, morphology, β-adrenoceptor density and noradrenaline concentration without and with blockade of p38 MAP kinase in sham and HF rabbits
| Sham | Sham-SB | HF3 | HF3-SB | |
|---|---|---|---|---|
| Heart rate (bpm) | 224 ± 5 | 228 ± 7 | 258 ± 15A | 264 ± 18A |
| LVEDD (mm) | 17.0 ± 0.3 | 16.8 ± 0.2 | 19.6 ± 0.5A | 18.9 ± 0.3A |
| Fractional shortening (%) | 30.0 ± 1.7 | 30.3 ± 0.6 | 10.3 ± 1.3A | 17.0 ± 1.3AB |
| Noradrenaline (ng·mL−1) | 1.33 ± 0.17 | 1.47 ± 0.27 | 3.83 ± 0.64A | 3.95 ± 1.11A |
| ß-Adrenoceptor density (fmol·mg−1) | 67 ± 5 | 69 ± 6 | 41 ± 3A | 63 ± 10AB |
| Cardiomyoycte cross-sectional area (µm2) | 242 ± 11 | 253 ± 6 | 334 ± 7A | 295 ± 4AB |
| Number of TUNEL-positive cardiomyocytes [/(1000*mm2)] | 4 ± 2 | 14 ± 5 | 50 ± 8A | 54 ± 14A |
| Phospho p38-MAPK/total p38-MAPK (relative to sham) | 1.00 ± 0.24 | 1.01 ± 0.21 | 2.34 ± 0.54A | 0.46 ± 0.24 |
| Phospho HSP27/total HSP27 (relative to sham) | 1.00 ± 0.42 | 0.49 ± 0.15 | 2.58 ± 0.60A | 1.13 ± 0.52 |
Mean ± SEM.
p < 0.05 versus Sham.
p < 0.05 versus HF3.
HF3, heart failure rabbits, 3 weeks (n = 8); HF3-SB, heart failure rabbits (3 weeks) receiving SB (n = 6); LVEDD, left ventricular end-diastolic diameter; Sham, sham-operated rabbits (n = 7); Sham-SB (n = 7), sham-operated rabbits receiving SB; TUNEL, TdT-mediated dUTP nick end labelling.
The myocardial ROS concentration inversely correlated to LVSF, such that any attenuation in the ROS concentration was associated with an improvement in LVSF (Figure 4).
Figure 4.
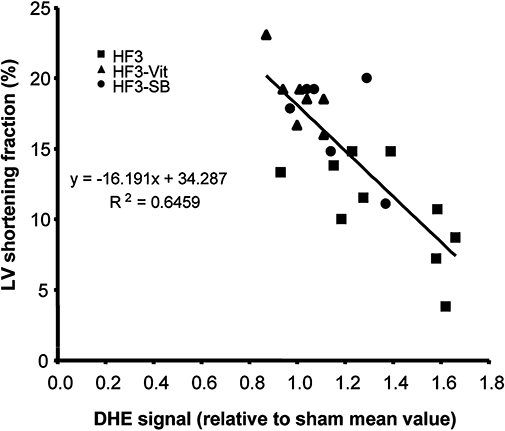
Relationship between ROS concentration and LV shortening fraction at the end of the 3 week study protocol. There is a close inverse relationship between ROS concentration and LV shortening fraction, and the beneficial effects of vitamins C and E or SB281832, respectively, fall along the relationship. DHE, dihydroethidine; HF3, heart failure rabbits after 3 weeks of pacing; LV, left ventricular; ROS, reactive oxygen species.
Discussion
The present study demonstrates that: (i) antioxidants abolish the increases in both myocardial ROS formation and tropomyosin oxidation otherwise seen in failing hearts, along with improvement of contractile function and prevention of apoptosis. Antioxidants, however, do not modify the pacing-induced activation of p38 MAP kinase; (ii) inhibition of p38 MAP kinase counteracts ROS accumulation, tropomyosin oxidation and contractile failure, while it does not affect apoptosis, indicating that p38 MAP kinase activation is upstream rather than downstream of oxidative stress; and (iii) oxidation of myofibrillar proteins is a causal link between ROS formation and contractile dysfunction.
Interaction between ROS and p38 MAP kinase
Reactive oxygen species have been proposed to trigger the activation of downstream redox-sensitive kinases, such as p38 MAP kinase (Michel et al., 2001; Li et al., 2002). In the present study, however, scavenging of ROS did not affect p38 MAP kinase phosphorylation and activity, suggesting that other signals – apart from ROS – contribute to p38 MAP kinase activation in failing hearts.
Reactive oxygen species scavenging with vitamins C and E even increased p38 MAP kinase phosphorylation in sham rabbit hearts, confirming previous data (Qin et al., 2003) and suggesting a negative feedback between ROS formation and p38 MAP kinase phosphorylation. In neuronal cells, activation of MAP kinase occurs indeed via ROS-induced inhibition of protein phosphatases 2A and 5 (Chen et al., 2008).
While ROS scavenging did not alter p38 MAP kinase phosphorylation and activity, p38 MAP kinase blockade decreased ROS formation, suggesting that ROS are downstream rather than upstream of p38 MAP kinase. Indeed, attenuated ROS formation following p38 MAP kinase blockade has previously been demonstrated in neutrophils (Sakamoto et al., 2006), kidney homogenates of hypertensive rats (Tojo et al., 2005) and vascular rings of rats with HF (Widder et al., 2004). The mechanism by which p38 MAP kinase activation increases ROS formation relates possibly to an increased expression and activity of NADPH oxidase (Bao et al., 2007).
Cardiomyocyte hypertrophy and apoptosis
p38 MAP kinase activation is involved in cardiomyocyte hypertrophy and apoptosis (Michel et al., 2001; Baines and Molkentin, 2005; Tenhunen et al., 2006). In the present study p38 MAP kinase activation increased the formation of ROS, which could then promote cell death through many pathways, notably by MPTP opening and mitochondrial dysfunction (Di Lisa and Bernardi, 2009). Both antioxidants and p38 MAP kinase inhibition prevented HF. However, antioxidants did not attenuate the HF-induced increase in p38 MAP kinase activation, and conversely, the inhibition of p38 MAP kinase kinase did not prevent apoptosis, which, in turn, was abrogated by antioxidants. The latter finding could be related to the concomitant blockade of JNK phosphorylation by SB281832, as JNK inhibition increases the extent of cardiomyocyte apoptosis in failing hamster hearts (Taniike et al., 2008). Therefore, the pro-apoptotic effect of JNK blockade is likely to counteract the anti-apoptotic effect afforded by p38 MAP kinase inhibition and the subsequent ROS formation.
Myofibrillar oxidation and failure
Although a wide array of mechanisms is set in motion in decompensated hypertrophy, our results emphasize the importance of oxidative stress and the consequent alteration of myofilaments. ROS can impair contractile function at various levels, notably by alterations of energy metabolism, calcium homeostasis and β-adrenoceptor expression (Qin et al., 2003; Bilginoglu et al., 2009). Down-regulation of β-adrenoceptor density contributes to contractile dysfunction in failing hearts, and up-regulation of β-adrenoceptors – for example by knocking out G protein-coupled receptor kinase 2 (GRK2) (Raake et al., 2008) – attenuates HF development. Importantly, ROS activates GRK2 in glioma cells, thereby favouring down-regulation of β-adrenoceptors (Cobelens et al., 2007). Accordingly, the present findings show that both antioxidant vitamins and p38 MAP kinase blockade attenuated ROS formation and β-adrenoceptor down-regulation.
There is little information on the direct impact of oxidative stress on the contractile machinery (Ide et al., 2000; Giordano, 2005). Genetic deletion of enzymes involved in ROS removal induces severe structural and functional alterations (Li et al., 1995), whereas antioxidant treatments counteract adverse remodelling in HF (Lombardi et al., 2009). We have previously demonstrated the role of ROS in myofibrillar protein oxidation and contractile dysfunction during myocardial ischaemia and reperfusion (Canton et al., 2004). Furthermore, the coronary microembolization-induced tropomyosin oxidation (Canton et al., 2006) is associated with impaired calcium responsiveness of the contractile machinery (Skyschally et al., 2008). The present study extends the relationship between tropomyosin oxidation and contractile impairment to a model of HF totally devoid of ischaemia.
Activated p38 MAP kinase – potentially via increased NADPH oxidase expression and activity – contributes to increased ROS formation in failing hearts, while increased ROS formation has little impact on p38 MAP kinase activation. Increased ROS formation induces β-adrenoceptor down-regulation, possibly via activation of GRK2, and most importantly, oxidizes myofilaments, thereby reducing contractile function. These effects are not mutually exclusive, but might well be independent from each other and act in a cooperative fashion to impair contractile function and contribute to the progression of HF.
Limitations
Because LV dysfunction is closely related to the extent of ROS formation (Qin et al., 2003) and the time course of ROS formation is unknown, we pretreated rabbits with vitamins C and E in order to maximize any therapeutic effect. For the same reason and to study the interaction between p38 MAP kinase activation and ROS formation, also the p38 MAP kinase inhibition was applied prior to the initiation of LV pacing.
The focus on oxidative stress and p38 MAP kinase might represent a limitation of the present study. However, the present findings do not exclude the involvement of other mechanisms in the transition from hypertrophy to failure. Indeed, it is clear from the present study that the initial decline in contractile function early during LV pacing is not related to p38 MAP kinase activation, as significant changes in p38 MAP kinase and HSP27 phosphorylation were observed only during the transition from the second to the third week of LV pacing. Therefore, it is not surprising that blockade of p38 MAP kinase with SB281832 did not completely restore LV function but maintained it at the level achieved after 2 weeks of LV pacing. Thus, p38 MAP kinase activation contributes to the progression rather than the initiation of HF. The mechanisms by which p38 MAP kinase becomes activated during the progression of HF are numerous and might include LV dilatation, neurohumoral activation, protein kinase activation or ion alterations (Aker et al., 2004).
The present results suggest that the extent of protein oxidation in relation to contractile dysfunction is probed reliably by tropomyosin, as we have shown in microembolized hearts (Canton et al., 2006). The fact that the amount of tropomyosin undergoing DCB formation appears rather modest might suggest that this process is not the only one responsible for contractile dysfunction. Indeed, it is likely that the alteration of contractile function results from the sum of several oxidative processes affecting various proteins. Please note our attention was focused on DCB formation, as this covalent modification is suitable for quantification. It is likely that other oxidative changes affect the native band, yet they can hardly be quantified. Regarding the relatively modest amount of tropomyosin oxidized, it has to be pointed out that severe contractile abnormalities have been reported for cardiomyocytes harbouring a minor fraction of altered myofibrillar proteins. Indeed, this was the case with the original report of inborn error of tropomyosin (Bottinelli et al., 1998). As far as the relevance of tropomyosin oxidation is concerned, it is worth pointing out that the only cysteine residue of cardiac tropomyosin (Cys190) (Lewis and Smillie, 1980) is located at the interface with troponin T (White et al., 1987). Large conformational modifications that generally result from protein oxidation especially when DCB are generated are likely to alter protein interactions that are crucial for contractile activity. This concept is supported by experimental models and clinical findings relating tropomyosin alterations to contractile abnormalities (Michele and Metzger, 2000). Furthermore, other covalent changes, such (de)phosphorylation, nitration/nitrosylation and proteolysis, might also occur and impair contractile function (van der Velden, 2006; Belin et al., 2007; Vahebi et al., 2007). These changes, far from being mutually exclusive with ROS-induced alterations, can be favoured by oxidation or, in turn, render proteins more susceptible to ROS. We did not find proteolysis of tropomyosin and desmin (data not shown), yet we cannot exclude that other myofibrillar proteins are proteolysed. Evidence of tropomyosin nitrosylation was obtained (unpublished results), yet its extent was not affected by antioxidant vitamins and p38 MAP kinase inhibition. The phosphorylation status of myofibrillar proteins is important in several models of HF (Belin et al., 2007; Vahebi et al., 2007). Actually, p38 MAP kinase activation is associated with tropomyosin dephosphorylation and reduced sarcomeric function (Vahebi et al., 2007). Although we did not assess myofibrillar protein phosphorylation, the activation of p38 MAP kinase favours ROS formation, so that any functional changes resulting from tropomyosin dephosphorylation could have been secondary to oxidation or vice versa.
In summary, we show that inhibition of p38 MAP kinase by SB281832 counteracts ROS formation, tropomyosin oxidation and contractile failure, without affecting apoptosis. Thus, p38 MAP kinase activation appears to be upstream rather than downstream of ROS, which impacts on LV function through myofibrillar oxidation. However, we did not exclude other modifications of the contractile machinery nor other targets of ROS. Further studies will have to look at the potential use of p38 MAP kinase inhibition in the prevention and treatment of HF.
Acknowledgments
The study was supported by a grant form the German Research Foundation (BR 526/12-1). The SB compound was a kind gift from Robert N. Willette, GlaxoSmithKline Pharmaceuticals. This paper reports data of the MD thesis of PH.
Conflicts of interest
None.
References
- Aker S, Snabaitis AK, Konietzka I, van de Sand A, Böngler K, Avkiran M, et al. Inhibition of the Na+/H+ exchanger attenuates the deterioration of ventricular function during pacing-induced heart failure in rabbits. Cardiovasc Res. 2004;63:273–282. doi: 10.1016/j.cardiores.2004.04.014. [DOI] [PubMed] [Google Scholar]
- Alexander SP, Mathie A, Peters JA. Guide to receptors and channels (GRAC), 3rd edition. Br J Pharmacol. 2008;153(Suppl 2):S1–209. doi: 10.1038/sj.bjp.0707746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baines CP, Molkentin JD. STRESS signaling pathways that modulate cardiac myocyte apoptosis. J Mol Cell Cardiol. 2005;38:47–62. doi: 10.1016/j.yjmcc.2004.11.004. [DOI] [PubMed] [Google Scholar]
- Bao W, Behm DJ, Nerurkar SS, Ao Z, Bentley R, Mirabile RC, et al. Effects of p38 MAPK Inhibitor on angiotensin II-dependent hypertension, organ damage, and superoxide anion production. J Cardiovasc Pharmacol. 2007;49:362–368. doi: 10.1097/FJC.0b013e318046f34a. [DOI] [PubMed] [Google Scholar]
- Belin RJ, Sumandea MP, Allen EJ, Schoenfelt K, Wang H, Solaro RJ, et al. Augmented protein kinase C-alpha-induced myofilament protein phosphorylation contributes to myofilament dysfunction in experimental congestive heart failure. Circ Res. 2007;101:195–204. doi: 10.1161/CIRCRESAHA.107.148288. [DOI] [PubMed] [Google Scholar]
- Bilginoglu A, Seymen A, Tuncay E, Zeydanli E, Aydemir-Koksoy A, Turan B. Antioxidants but not doxycycline treatments restore depressed beta-adrenergic responses of the heart in diabetic rats. Cardiovasc Toxicol. 2009;9:21–29. doi: 10.1007/s12012-009-9032-8. [DOI] [PubMed] [Google Scholar]
- Bottinelli R, Coviello DA, Redwood CS, Pellegrino MA, Maron BJ, Spirito P, et al. A mutant tropomyosin that causes hypertrophic cardiomyopathy is expressed in vivo and associated with an increased calcium sensitivity. Circ Res. 1998;82:106–115. doi: 10.1161/01.res.82.1.106. [DOI] [PubMed] [Google Scholar]
- Braz JC, Bueno OF, Liang Q, Wilkins BJ, Dai Y-S, Parsons S, et al. Targeted inhibition of p38 MAPK promotes hypertrophic cardiomyopathy through upregulation of calcineurin-NFAT signaling. J Clin Invest. 2003;111:1475–1504. doi: 10.1172/JCI17295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butter C, Rastogi S, Minden HH, Meyhofer J, Burkhoff D, Sabbah HN. Cardiac contractility modulation electrical signals improve myocardial gene expression in patients with heart failure. J Am Coll Cardiol. 2008;51:1784–1789. doi: 10.1016/j.jacc.2008.01.036. [DOI] [PubMed] [Google Scholar]
- Canton M, Neverova I, Menabó R, Van Eyk J, Di Lisa F. Evidence of myofibrillar protein oxidation induced by postischemic reperfusion in isolated rat hearts. Am J Physiol Heart Circ Physiol. 2004;286:H870–H877. doi: 10.1152/ajpheart.00714.2003. [DOI] [PubMed] [Google Scholar]
- Canton M, Skyschally A, Menabo R, Boengler K, Gres P, Schulz R, et al. Oxidative modification of tropomyosin and myocardial dysfunction following coronary microembolization. Eur Heart J. 2006;27:875–881. doi: 10.1093/eurheartj/ehi751. [DOI] [PubMed] [Google Scholar]
- Chen L, Liu L, Huang S. Cadmium activates the mitogen-activated protein kinase (MAPK) pathway via induction of reactive oxygen species and inhibition of protein phosphatases 2A and 5. Free Radic Biol Med. 2008;45:1035–1044. doi: 10.1016/j.freeradbiomed.2008.07.011. [DOI] [PubMed] [Google Scholar]
- Clark JE, Sarafraz N, Marber MS. Potential of p38-MAPK inhibitors in the treatment of ischaemic heart disease. Pharmacol Ther. 2007;116:192–206. doi: 10.1016/j.pharmthera.2007.06.013. [DOI] [PubMed] [Google Scholar]
- Cobelens PM, Kavelaars A, Heijnen CJ, Ribas C, Mayor F, Jr, Penela P. Hydrogen peroxide impairs GRK2 translation via a calpain-dependent and cdk1-mediated pathway. Cell Signal. 2007;19:269–277. doi: 10.1016/j.cellsig.2006.06.009. [DOI] [PubMed] [Google Scholar]
- Di Lisa F, Bernardi P. A CaPful of mechanisms regulating the mitochondrial permeability transition. J Mol Cell Cardiol. 2009;46:775–780. doi: 10.1016/j.yjmcc.2009.03.006. [DOI] [PubMed] [Google Scholar]
- Giordano FJ. Oxygen, oxidative stress, hypoxia, and heart failure. J Clin Invest. 2005;115:500–508. doi: 10.1172/JCI200524408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haq S, Choukroun G, Lim H, Tymitz KM, del Monte F, Gwathmey J, et al. Differential activation of signal transduction pathways in human hearts with hypertrophy versus advanced heart failure. Circulation. 2001;103:670–677. doi: 10.1161/01.cir.103.5.670. [DOI] [PubMed] [Google Scholar]
- Hool LC. Reactive oxygen species in cardiac signalling: from mitochondria to plasma membrane ion channels. Clin Exp Pharmacol Physiol. 2006;33:146–151. doi: 10.1111/j.1440-1681.2006.04341.x. [DOI] [PubMed] [Google Scholar]
- Ide T, Tsutsui H, Kinugawa S, Suematsu N, Hayashidani S, Ichikawa K, et al. Direct evidence for increased hydroxyl radicals originating from superoxide in the failing myocardium. Circ Res. 2000;86:152–157. doi: 10.1161/01.res.86.2.152. [DOI] [PubMed] [Google Scholar]
- Kerkela R, Force T. p38 Mitogen-activated protein kinase. A future target for heart failure therapy. J Am Coll Cardiol. 2006;48:556–558. doi: 10.1016/j.jacc.2006.05.005. [DOI] [PubMed] [Google Scholar]
- Kinugawa S, Tsutsui H, Hayashidani S, Ide T, Suematsu N, Satoh S, et al. Treatment with dimethylthiourea prevents left ventricular remodeling and failure after experimental myocardial infarction in mice: role of oxidative stress. Circ Res. 2000;87:392–398. doi: 10.1161/01.res.87.5.392. [DOI] [PubMed] [Google Scholar]
- Leineweber K, Aker S, Beilfuss A, Rekasi H, Konietzka I, Martin C, et al. Inhibition of Na+/H+-exchanger with sabiporide attenuates the downregulation and uncoupling of the myocardial beta-adrenoceptor system in failing rabbit hearts. Br J Pharmacol. 2006;148:137–146. doi: 10.1038/sj.bjp.0706714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis WG, Smillie LB. The amino acid sequence of rabbit cardiac tropomyosin. J Biol Chem. 1980;255:6854–6859. [PubMed] [Google Scholar]
- Li J-M, Nick P, Grieve DJ, Chen M, Shah AM. Activation of NADPH oxidase during progression of cardiac hypertrophy to failure. Hypertension. 2002;40:477–484. doi: 10.1161/01.hyp.0000032031.30374.32. [DOI] [PubMed] [Google Scholar]
- Li Y, Huang TT, Carlson EJ, Melov S, Ursell PC, Olson JL, et al. Dilated cardiomyopathy and neonatal lethality in mutant mice lacking manganese superoxide dismutase. Nat Genet. 1995;11:376–381. doi: 10.1038/ng1295-376. [DOI] [PubMed] [Google Scholar]
- Liao P, Wang S-Q, Wang S, Zheng M, Zhang S-J, Cheng H, et al. p38 Mitogen-activated protein kinase mediates a negative inotropic effect in cardiac myocytes. Circ Res. 2002;90:190–196. doi: 10.1161/hh0202.104220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombardi R, Rodriguez G, Chen SN, Ripplinger CM, Li W, Chen J, et al. Resolution of established cardiac hypertrophy and fibrosis and prevention of systolic dysfunction in a transgenic rabbit model of human cardiomyopathy through thiol-sensitive mechanisms. Circulation. 2009;119:1398–1407. doi: 10.1161/CIRCULATIONAHA.108.790501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel MC, Li Y, Heusch G. Mitogen-activated protein kinases in the heart. Naunyn Schmiedebergs Arch Pharmacol. 2001;363:245–266. doi: 10.1007/s002100000363. [DOI] [PubMed] [Google Scholar]
- Michele DE, Metzger JM. Physiological consequences of tropomyosin mutations associated with cardiac and skeletal myopathies. J Mol Med. 2000;78:543–553. doi: 10.1007/s001090000161. [DOI] [PubMed] [Google Scholar]
- Qin F, Shite J, Liang C. Antioxidants attenuate myocyte apoptosis and improve cardiac function in heart failure: association with changes in the MAPK pathways. Am J Physiol Heart Circ Physiol. 2003;285:H822–H832. doi: 10.1152/ajpheart.00015.2003. [DOI] [PubMed] [Google Scholar]
- Qin F, Liang MC, Liang CS. Progressive left ventricular remodeling, myocyte apoptosis, and protein signaling cascades after myocardial infarction in rabbits. Biochim Biophys Acta. 2005;1740:499–513. doi: 10.1016/j.bbadis.2004.11.007. [DOI] [PubMed] [Google Scholar]
- Raake PW, Vinge LE, Gao E, Boucher M, Rengo G, Chen X, et al. G protein-coupled receptor kinase 2 ablation in cardiac myocytes before or after myocardial infarction prevents heart failure. Circ Res. 2008;103:413–422. doi: 10.1161/CIRCRESAHA.107.168336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren J, Zhang S, Kovacs A, Wang Y, Muslin AJ. Role of p38alpha MAPK in cardiac apoptosis and remodeling after myocardial infarction. J Mol Cell Cardiol. 2005;38:617–623. doi: 10.1016/j.yjmcc.2005.01.012. [DOI] [PubMed] [Google Scholar]
- Sakamoto K, Kuribayashi F, Nakamura M, Takeshige K. Involvement of p38 MAP kinase in not only activation of the phagocyte NADPH oxidase induced by formyl-methionyl-leucyl-phenylalanine but also determination of the extent of the activity. J Biochem. 2006;140:739–745. doi: 10.1093/jb/mvj204. [DOI] [PubMed] [Google Scholar]
- Schulz R, Belosjorow S, Gres P, Jansen J, Michel MC, Heusch G. p38 MAP kinase is a mediator of ischemic preconditioning in pigs. Cardiovasc Res. 2002;55:690–700. doi: 10.1016/s0008-6363(02)00319-x. [DOI] [PubMed] [Google Scholar]
- Schulz R, Aker S, Belosjorow S, Konietzka I, Rauen U, Heusch G. Stress kinase phosphorylation is increased in pacing-induced heart failure in rabbits. Am J Physiol Heart Circ Physiol. 2003;285:H2084–H2090. doi: 10.1152/ajpheart.01038.2002. [DOI] [PubMed] [Google Scholar]
- Skyschally A, Gres P, van CP, van de SA, Boengler K, Schulz R, et al. Reduced calcium responsiveness characterizes contractile dysfunction following coronary microembolization. Basic Res Cardiol. 2008;103:552–559. doi: 10.1007/s00395-008-0732-1. [DOI] [PubMed] [Google Scholar]
- Taniike M, Yamaguchi O, Tsujimoto I, Hikoso S, Takeda T, Nakai A, et al. Apoptosis signal-regulating kinase 1/p38 signaling pathway negatively regulates physiological hypertrophy. Circulation. 2008;117:545–552. doi: 10.1161/CIRCULATIONAHA.107.710434. [DOI] [PubMed] [Google Scholar]
- Tenhunen O, Rysä J, Ilves M, Soini Y, Ruskoaho H, Leskinen H. Identification of cell cycle regulatory and inflammatory genes as predominant targets of p38 mitogen-activated protein kinase in the heart. Circ Res. 2006;99:485–493. doi: 10.1161/01.RES.0000238387.85144.92. [DOI] [PubMed] [Google Scholar]
- Tojo A, Onozato ML, Kobayashi N, Goto A, Matsuoka H, Fujita T. Antioxidative effect of p38 mitogen-activated protein kinase inhibitor in the kidney of hypertensive rat. J Hypertens. 2005;23:165–174. doi: 10.1097/00004872-200501000-00027. [DOI] [PubMed] [Google Scholar]
- Vahebi S, Ota A, Li M, Warren CM, de Tombe PP, Wang Y, et al. p38-MAPK induced dephosphorylation of alpha-tropomyosin is associated with depression of myocardial sarcomeric tension and ATPase activity. Circ Res. 2007;100:408–415. doi: 10.1161/01.RES.0000258116.60404.ad. [DOI] [PubMed] [Google Scholar]
- van der Velden J. Functional significance of myofilament protein oxidation. Eur Heart J. 2006;27:764–765. doi: 10.1093/eurheartj/ehi742. [DOI] [PubMed] [Google Scholar]
- Wang M, Tsai BM, Turrentine MW, Mahomed Y, Brown JW, Meldrum DR. p38 mitogen activated protein kinase mediates both death signaling and functional depression in the heart. Ann Thorac Surg. 2005;80:2235–2241. doi: 10.1016/j.athoracsur.2005.05.070. [DOI] [PubMed] [Google Scholar]
- Wang Y. Mitogen-activated protein kinases in heart development and diseases. Circulation. 2007;116:1413–1423. doi: 10.1161/CIRCULATIONAHA.106.679589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White SP, Cohen C, Philips GNJ. Structure of co-crystals of tropomyosin and troponin. Nature. 1987;325:826–828. doi: 10.1038/325826a0. [DOI] [PubMed] [Google Scholar]
- Widder J, Behr T, Fraccarollo D, Hu K, Galuppo P, Tas P, et al. Vascular endothelial dysfunction and superoxide anion production in heart failure are p38 MAP kinase-dependent. Cardiovasc Res. 2004;63:161–167. doi: 10.1016/j.cardiores.2004.03.008. [DOI] [PubMed] [Google Scholar]