

Abstract
Background and purpose:
P-glycoprotein (Pgp) efflux assays are widely used to identify Pgp substrates. The kidney cell lines Madin-Darby canine kidney (MDCK)-II and LLC-PK1, transfected with human MDR1 (ABCB1) are used to provide recombinant models of drug transport. Endogenous transporters in these cells may contribute to the activities of recombinant transporters, so that drug transport in MDR1-transfected cells is often corrected for the transport obtained in parental (wildtype) cells. However, expression of endogenous transporters may vary between transfected and wildtype cells, so that this correction may cause erroneous data. Here, we have measured the expression of endogenous efflux transporters in transfected and wildtype MDCK-II or LLC cells and the consequences for Pgp-mediated drug transport.
Experimental approach:
Using quantitative real-time RT-PCR, we determined the expression of endogenous Mdr1 mRNA and other efflux transporters in wildtype and MDR1-transfected MDCK-II and LLC cells. Transcellular transport was measured with the test substrate vinblastine.
Key results:
In MDR1-transfected MDCK cells, expression of endogenous (canine) Mdr1 and Mrp2 (Abcc2) mRNA was markedly lower than in wildtype cells, whereas MDR1-transfected LLC cells exhibited comparable Mdr1 but strikingly higher Mrp2 mRNA levels than wildtype cells. As a consequence, transport of vinblastine by human Pgp in efflux experiments was markedly underestimated when transport in MDR1-transfected MDCK cells was corrected for transport obtained in wildtype cells. This problem did not occur in LLC cells.
Conclusions and implications:
Differences in the expression of endogenous efflux transporters between transfected and wildtype MDCK cells provide a potential bias for in vitro studies on Pgp-mediated drug transport.
Keywords: multidrug resistance, transwell, ABCB1, multidrug resistance proteins, kidney cell lines
Introduction
The efflux transporter P-glycoprotein (Pgp), the product of the multidrug resistance 1 (MDR1; ABCB1) gene, affects the absorption, distribution and excretion of a variety of drugs (Schinkel and Jonker, 2003; Thuerauf and Fromm, 2006). Thus, Pgp substrate assays have become important tools, used during drug discovery to aid drug candidate selection and optimization (Polli et al., 2001; Yamazaki et al., 2001; Feng et al., 2008). Furthermore, the recent U.S. Food and Drug Administration (FDA) concept paper on drug interactions recommends that new drug candidates be evaluated as substrates, inhibitors and inducers of Pgp to assess the potential for clinical drug-drug interactions (FDA, U.S. Department of Health and Human Services, 2006). There are several in vitro methods that can evaluate whether a drug candidate is a substrate or inhibitor of Pgp. The most commonly used and definitive methods are transcellular transport assays, in which drug transport is determined between an apical and basolateral compartment separated by a polarized cell monolayer that expresses human Pgp (Yamazaki et al., 2001; Schwab et al., 2003; Löscher and Potschka, 2005; Rautio et al., 2006; Feng et al., 2008; Jin and Di, 2008). Typically, epithelial kidney cell lines such as the canine kidney-derived Madin-Darby canine kidney (MDCK)-II or the porcine-kidney derived LLC-PK1 cells, either untransfected (parental) or transfected with human MDR1, are used in such assays. Because these cells also express endogenous drug transporters, transcellular transport determined in MDR1-transfected cells is often corrected by transport obtained in parental (wildtype) cells (see Schwab et al., 2003; Food and Drug Administration (FDA), U.S. Department of Health and Human Services, 2006; Feng et al., 2008). The basis for this analysis is the assumption that expression of endogenous transporters is the same in wildtype and transfected cells. However, to our knowledge, it is not known whether this assumption is correct. In other words, if the expression of endogenous transporters varies between parental and transfected cells, this phenomenon could cause a significant bias in identifying Pgp substrates.
MDCK and LLC cells express various endogenous transporters, including Pgp and members of the multidrug resistance protein (MRP; ABCC) family of efflux transporters (Horio et al., 1989; Evers et al., 1996; Evers et al., 1998; Flanagan et al., 2002; Goh et al., 2002; Tang et al., 2002a,b; Baltes et al., 2007a; nomenclature follows Alexander et al., 2009). Flanagan et al. (2002) previously reported that the expression of endogenous (pig) Pgp seemed to increase with MRP1 (ABCC1) transfection in LLC cells, whereas no Pgp up-regulation was observed in the results from the original publication (Evers et al., 1996) using the same primary antibody (C219). The main goal of the present study was to determine whether the expression of endogenous Mdr1 differs between MDR1-transfected MDCK-II and LLC and the corresponding wildtype cells. In addition, based on the report of Flanagan et al. (2002), we also determined whether endogenous Pgp expression varies between MDCK cells transfected with different human MRPs. Transcellular transport assays with the test substrate vinblastine were used to investigate the potential consequences of this variation in the expression of endogenous transporters.
Methods
Cell lines and cell cultures
Madin-Darby canine kidney type II cells transfected with human MDR1 (ABCB1), MRP1 (ABCC1), MRP2 (ABCC2) or MRP5 (ABCC5) and respective MDCK-II wildtype cells were kindly provided by Prof Piet Borst (the Netherlands Cancer Institute, Amsterdam, Netherlands). In addition, we used LLC-PK1 cells transfected with human MDR1 and respective wildtype LLC cells, which were also kindly provided by Prof Borst. The cells were cultured as previously described (Baltes et al., 2007a,b;). Cells were used within 10 passages or less after thawing from liquid nitrogen, and at a maximum of 13 passages after receipt. Because transfected LLC cells may lose the transporter cDNA in the absence of a selection agent such as vincristine, they were regularly tested for vincristine resistance (640 nmol) before experimentation (for details see Baltes et al., 2007a).
Western blotting
Cells were suspended in 10 volumes of protein lysis buffer (25 mM Tris, 50 mM NaCl, 0.5% Na-deoxycholate, 0.5% Triton X-100, 1x Complete® Protease Inhibitor Cocktail, pH = 8.0) on ice. Total cell proteins (25–50 µg) were separated on 7.5% polyacrylamide gel and transferred onto PVDF membrane. Pgp was probed using the C219 (Signet Laboratories, Dedham, MA, USA; dilution 1:200)/anti-mouse IgG-HRP (Dako Cytomatics, Glostrup, Denmark; dilution 1:1000) antibodies. Mrp2 was probed with the M2III-6 (Alexis Biochemicals; Axxora Deutschland GmbH, Lörrach, Germany; dilution 1:250)/anti-mouse IgG-HRP (1:1000) antibodies. Actin (loading control) was probed with anti-actin (Sigma–Aldrich, Steinheim, Germany; dilution 1:5000)/anti-rabbit IgG-HRP (Dako Cytomatics, Glostrup, Denmark; dilution 1:10000). Bands were visualized by ECL Super-signal Pico assay (Pierce Biotechnology, Rockford, IL, USA) and X-ray film (Pierce Biotechnology). As an alternative loading control, we used membrane staining with Ponceau S. Blots were then analysed by densitometry using ScionImage 4.0 software (Scion Corp., Frederick, MD, USA). It is important to note that the C219 antibody does not distinguish canine, porcine or human Pgp, so that only total Pgp expression could be determined in MDR1-transfected MDCK or LLC cells. However, expression of canine, porcine and human Mdr1/MDR1 mRNA could be differentiated by quantitative real-time RT-PCR.
Quantitative real-time RT-PCR (qRT-PCR)
Total RNA was extracted from the cells using the NucleoSpin RNA II kit (Macherey-Nagel, Düren, Germany) according to the manufacturer's instructions. The concentrations and purity of RNA were estimated spectrophotometrically at 260 and 280 nm, and the integrity of the RNA was checked by electrophoresis on 1% agarose gels. RNA was reverse-transcribed with oligo(dT)20 and random hexamer primers in a final volume of 10 µL using SuperScript III First-Strand cDNA synthesis kit (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's instructions. Gene expression of MDR1 (human), Mdr1 (dog), Mdr1 (pig) and dog Mrp1, Mrp2 and Mrp5 was evaluated by qPCR. Amplification was detected by SYBR® Green fluorescence on a Mx3005P instrument (Stratagene, La Jolla, CA, USA). cDNA of each sample was diluted 1:24 in RNase-free water. Thermocycling was carried out in a final volume of 20 µL containing 10 µL Precision™-MX-SY MasterMix 2x (PrimerDesign Ltd, Southampton, UK), 1 µL of each forward and reverse primer (final concentration 0.5 µM) and 8 µL diluted cDNA. The thermal cycling conditions were 10 min at 95°C to activate DNA polymerase, followed by 45 amplification cycles at 94°C for 10 s, 58°C for 10 s and 72°C for 30 s, with the last cycle finished by 3 min elongation at 72°C and dissociation curve analysis to ensure the specificity of the PCR product. As negative controls, we used ‘no template’ (NTC) and ‘no reverse transcriptase’ (–RT) probes, which produced negligible signals (usually >40 in Ct value), suggesting that primer–dimer formation and genomic DNA contamination effects were negligible. Dissociation curve analysis revealed that no side products have been formed. The GAPDH gene was used as an endogenous reference for normalizing target gene mRNAs. All primers were designed using PRIMER3 software (web-based version http://fokker.wi.mit.edu, Whitehead Institute for Biomedical Research, Cambridge, MA, USA) and checked for specificity using GenBank databases by BLAST tool (http://blast.ncbi.nlm.nih.gov, National Center for Biotechnology Information, Bethesda, MD, USA). In order to ensure that all qPCR products are generated from cDNA but not from genomic DNA, all primers were analysed by UCSC IN-SILICO PCR software (web-based version http://genome.ucsc.edu/cgi-bin/hgPcr, University of California, Santa Cruz, CA, USA), and products of PCR reaction were checked on 2% agarose gel. Primer sequences are shown in Table 1.
Table 1.
Sequence of primers used for qPCR
| Protein name | Target gene name | GenBank accession numbera | Amplicon length, bp | Forward primer, 5′→3′ | Reverse primer, 5′→3′ |
|---|---|---|---|---|---|
| Pgp (MDR1), human | Abcb1 | NM_000927.3 | 217 | CCGAACACATTGGAAGGAA | CTTTGCCATCAAGCAGCAC |
| Pgp (Mdr1), dog | Abcb1 | DQ068953.1 | 190 | TTGCTGGTTTTGATGATGGA | CTGGACCCTGAATCTTTTGG |
| Mrp1, dog | Abcc1 | NM_001002971.1 | 165 | GGCTCTGCTTCCCCTTCTAC | GGATTTTGCCCCAACTTCTT |
| Mrp2, dog | Abcc2 | NM_001003081.1 | 187 | TTGGCTTACTCCTGCCTGTT | CCAGTGTCAGAGGTTGCTTG |
| Mrp5, dog | Abcc5 | DQ985741.1 | 204 | CCTACAACAAAGGGCAGGAA | AGATGGCAAGACCCGAATAG |
| GAPDH, dog | Gapdh | AB038240.1 | 117 | ATTCCACGGCACAGTCAAG | TACTCAGCACCAGCATCACC |
| Pgp (Mdr1), pig | Abcb1 | AY825267.1 | 280 | AGAGTATCAAGGGACCACAAGG | CCAAGGATTAGAAACAAGAGTGAAA |
| Mrp1, pig | Abcc1 | AF403246.1 | 163 | CCCTGTTCGCCGTAATCT | AATACTCCTTTAGCACTCTCCACA |
| Mrp2, pig | Abcc2 | XM_001929359.1 | 182 | TGGGGTGATGGTGCTTCT | GGTTGTGGACTTGGTTTTGG |
| GAPDH, pig | Gapdh | AF017079.1 | 200 | TCAAGGCTGAGAATGGGAAG | AGCAGAAGGGGCAGAGATG |
Gene acession number at http://www.ncbi.nlm.nih.gov/Genbank/.
MDR1, multidrug resistance 1; Pgp, P-glycoprotein; qPCR, quantitative PCR.
Transport experiments
Cells were seeded on transparent polyester membrane filters (Transwell-Clear®, 24 mm diameter, 0.4 µm pore size, Corning Costar Corporation, Cambridge, MA, USA) at a density of 0.3 × 106 cells·cm−2 (LLC) or 0.4 × 106 cells·cm−2 (MDCK-II), cultured for 1–2 days to confluence and used for transport assays between days 5 and 7 after confluence (for details see Baltes et al., 2007a,b;). Transport studies were performed with the filter inserts in Transwell® multiwell culture plates that allow studying drug transport between an apical and basolateral compartment. For the present experiments, 6-well plates were used. Before starting the transport experiments, the medium was replaced with Opti-MEM® (Gibco™/Invitrogen Corporation, Eggenstein, Germany) and the transwells were pre-incubated for 1 h (with or without transport inhibitor respectively; see below). This reduced serum medium was used without any additives according to the protocol of the laboratory that provided the cell lines (Prof P. Borst) in order to minimize protein binding of the drugs. At the beginning of the experiment (t = 0), the pre-incubation medium was replaced by fresh Opti-MEM® containing the drug in both chambers (see below). The volumes in the upper and lower compartment were 2000 µL and 2700 µL respectively. For drug analysis, samples were taken at 60, 120, 240 and 360 min. The transport assays including pre-incubation were performed at 37°C in a humidified incubator (5% CO2) with shaking the transwells gently at 50 rpm. Monolayers were checked for integrity by measuring transepithelial electrical resistance (TEER) of the polarized cells before and after each transport experiment and by using [14C]-mannitol (in separate wells) as described recently (Luna-Tortós et al., 2008). The TEER measurements after each experiment indicated that none of the treatments (vinblastine, MK571, tariquidar) exerted cytotoxic effects at the concentrations used in the experiments.
Bi-directional permeability studies were initiated by adding the drug to either the apical (for apical to basolateral transport, a-B) or the basolateral (for basolateral to apical, b-A) side of the monolayer. For drug analysis, 100 µL aliquots were collected from the receiver compartment (which contained medium with the same concentration of the drug solvent as the medium in the donor chamber). The volume of the donor chamber was adjusted after each sampling to avoid ‘transport’ effects by hydrostatic pressure. Each experiment was performed in triplicate. Wildtype and transfected cells were run in parallel in each experiment. The experiments were repeated several times and representative experiments are illustrated.
In the transport experiments performed in this study, the Pgp substrate vinblastine, which is also transported by Mrp2 (Evers et al., 1998; Tang et al., 2002a), was used. [3H]-vinblastine sulphate (9.8 Ci·mmol−1; Amersham, Buckinghamshire, UK) was diluted with unlabelled vinblastine sulphate (Alexis Biochemicals) to give an activity of 0.025 µCi·mL−1 and a final concentration of 2 µM in the assay. For inhibition of Pgp, tariquidar (kindly provided by Xenova, Slough, Berkshire, UK) was used at a concentration of 0.2 µM. For inhibition of Mrps, MK571 (Alexis Biochemicals) was used at a concentration of 50 µM (except one experiment with transport of vinblastine in MDCK wildtype cells, in which MK571 was used at 20 µM). The radioactivity in samples from experiments with [3H]vinblastine was quantified using a scintillation counter.
The results of the individual transport assays are presented as the percentage of the initial drug concentration in the donor chamber over time. Apical-to-basal (Papp aB) and basal-to-apical (Papp bA) permeabilities were determined according to Artursson (1990) using the following equation:
where dQ/dt [µg·min−1] is the permeability rate of the drug, A is the surface area of the monolayer and C0 is the initial drug concentration in the donor chamber. Transport ratio (TR; also termed ‘efflux ratio’, ER) was calculated by dividing Papp bA by Papp aB. Corrected transport ratios (cTRs; also termed ‘ratio of ratios’, RR) were calculated by division of the TR obtained in MDR1-transfected cells by the TR obtained in the respective wildtype/parental cells (Schwab et al., 2003; Feng et al., 2008). A cTR of at least 1.5 (Schwab et al., 2003) or 1.7 (Feng et al., 2008) has been proposed as an indicator of active, asymmetrical transport.
Statistics
The statistical significance of group differences was calculated by Student's t-test. A P < 0.05 was considered significant.
Results
Expression of endogenous (canine) Mdr1 mRNA was strikingly lower in MDR1-transfected MDCK-II cells in comparison with the parental cell line (Figure 1A). As expected, no human MDR1 mRNA was determined in MDCK wildtype cells (Figure 1B), thus confirming the specificity of the primers. The expression of human MDR1 mRNA in the transfected cells was about threefold higher than the expression of endogenous canine Mdr1 mRNA in wildtype cells (Figure 1A,B). When the Pgp protein content was determined in MDCK and MDCK-MDR1 cells by Western blot, total (endogenous canine and recombinant human) Pgp expression in the transfected cells was about three times higher than endogenous Pgp expression in the wildtype cells (Figure 1C).
Figure 1.
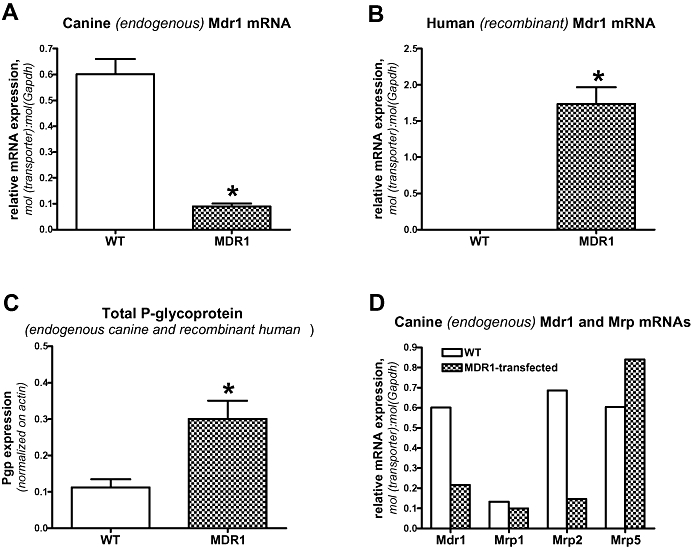
Expression of endogenous (canine) Mdr1 in parental (wildtype; WT) and MDR1-transfected MDCK-II cells. As shown in A, expression of canine Mdr1 is about seven times lower in MDR1-transfected versus wildtype cells. B demonstrates expression of human MDR1 mRNA in the transfected cells and the lack of any human MDR1 expression in wildtype cells. C illustrates the protein content of Pgp in MDCK wildtype and MDR1-transfected MDCK cells. Because it is not possible to differentiate canine and human Pgp by Western blots using the C219 antibody, total (canine and human) Pgp expression is shown. Data in (A), (B) and (C) are shown as mean ± SEM of four experiments. *Significant group differences; P < 0.001. Data in (D) illustrate a representative experiment in which qPCR was used to determine mRNAs of canine Mdr1, Mrp1, Mrp2 and Mrp5 in MDCK wildtype cells versus cells transfected with human MDR1. Note the lower expression of Mdr1 and Mrp2 mRNAs in the transfected cells. MDCK, Madin-Darby canine kidney; MDR1, multidrug resistance 1; Pgp, P-glycoprotein; qPCR, quantitative PCR.
In addition to examining whether the endogenous Mdr1 mRNA expression differs between transfected and wildtype cells, we also determined the expression of endogenous Mrps in MDCK wildtype and MDR1-transfected cells (Figure 1D). Data indicated markedly lower expression of Mrp2 mRNA in MDR1-transfected cells, but only small differences in Mrp1 and Mrp5 mRNAs between transfected and wildtype cells.
In contrast to the difference in endogenous Mdr1 mRNA expression between wildtype and transfected MDCK-II cells, such a difference was not observed in LLC cells (Figure 2). Thus, endogenous (porcine) Mdr1 mRNA expression did not differ between wildtype and MDR1-transfected LLC cells (Figure 2A). No human MDR1 mRNA was found in LLC wildtype cells, and the expression of human MDR1 mRNA in the transfected cells was about 37-fold higher than the expression of endogenous porcine Mdr1 mRNA (Figure 2B). When the Pgp protein content was determined in LLC and LLC-MDR1 cells by Western blot, total (endogenous porcine and recombinant human) Pgp expression in the transfected cells was about 27-times higher than endogenous Pgp expression in the wildtype cells (Figure 2C). In contrast to MDCK cells, expression of endogenous Pgp in LLC cells was very low (Figures 2C and 3A,B).
Figure 2.
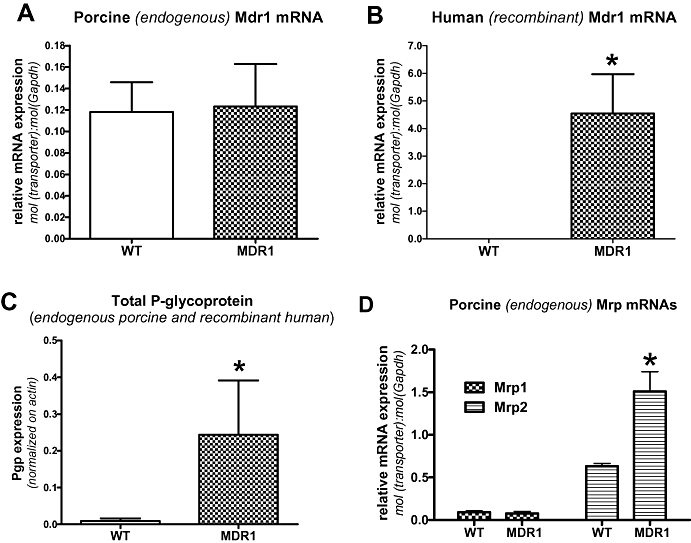
Expression of endogenous (porcine) Mdr1 in parental (wildtype; WT) and MDR1-transfected LLC cells. As shown in (A), the expression of canine Mdr1 is comparable in MDR1-transfected versus parental (WT) LLC cells. (B) demonstrates expression of human MDR1 mRNA in the transfected cells and the lack of any human MDR1 expression in wildtype cells. (C) illustrates the protein content of Pgp in LLC wildtype and MDR1-transfected MDCK cells. Because it is not possible to differentiate porcine and human Pgp by Western blots using the C219 antibody, total (porcine and human) Pgp expression is shown. Data in (A), (B) and (C) are shown as mean ± SEM of five experiments. *Significant group differences; P < 0.001. Data in (D) illustrate an experiment in which qPCR was used to determine mRNAs of porcine Mrp1 and Mrp2 in LLC wildtype cells versus cells transfected with human MDR1. Data are shown as mean ± SEM of three experiments. Note the higher expression of Mrp2 mRNAs in the transfected cells. *P = 0.0203. MDR1, multidrug resistance 1; Pgp, P-glycoprotein; qPCR, quantitative PCR.
Figure 3.
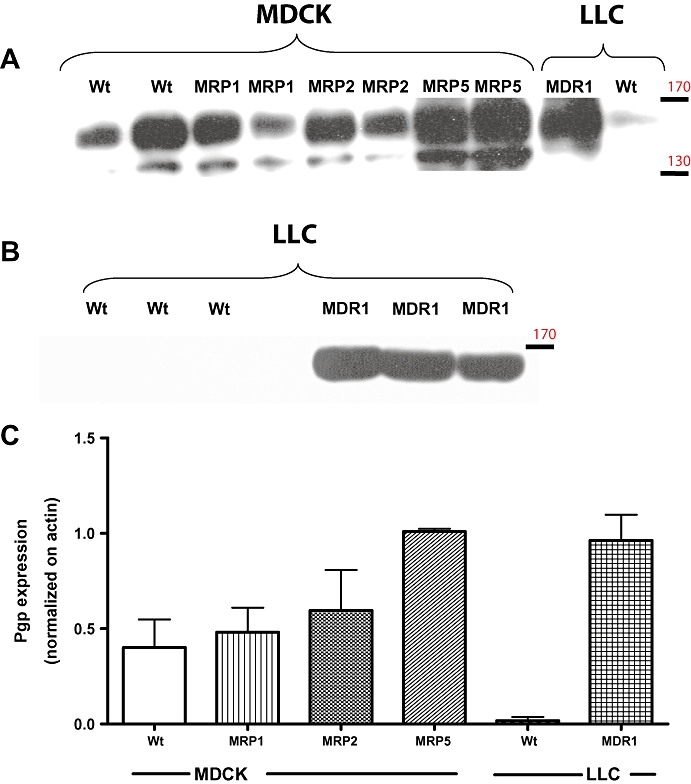
Comparison of Pgp expression in different cell lines. Data are from a representative experiment in which Pgp expression was determined by Western blot in MDCK-II wildtype and MDCK-II cells transfected with human MRP1, MRP2 or MRP5. For comparison, Pgp expression in LLC wildtype and MDR1-transfected LLC cells is shown. All data shown in (A) are from one gel, whereas data in B are from another gel with a lower amount of protein added, which led to disappearance of the Pgp band in LLC wildtype cells. For each cell line, two different samples were tested except for LLC cells, for which four different samples are shown in (A) and (B). Note that Pgp appeared as one band at around 160 kDa in Western blots from LLC cells (A), while two bands at about 155 and 140 kDa were labelled with the C219 Pgp antibody in MDCK cells. It has been suggested previously that the lower band Pgp isoform in MDCK cells is less glycosylated than the upper band (Tang et al., 2002a), which represents a mature form of Pgp. The upper band was therefore used for the calculations of Pgp expression in MDCK cells shown in C. In (C), Pgp expression was normalized on actin expression to allow comparison of Pgp expression between cell lines. Data are shown as means ± SEM. In contrast to MDCK wildtype cells, which expressed a relatively high amount of endogenous (canine) Pgp, very low Pgp expression was determined in LLC wildtype cells at the conditions of the assay. Transfection with human MDR1 led to a marked Pgp expression in LLC cells. However, a comparable Pgp expression was also observed in MRP5-transfected MDCK cells. This experiment was repeated several times on different gels (not illustrated), confirming that MRP5-transfected MDCK cells exhibit high expression of endogenous Pgp. MDCK, Madin-Darby canine kidney; MDR1, multidrug resistance 1; MRP, multidrug resistance protein; Pgp, P-glycoprotein.
Similar to the experiments in MDCK cells, we also determined expression of endogenous Mrps in LLC cells (Figure 2D). While Mrp1 mRNA expression was comparable in wildtype and MDR1-transfected cells, expression of Mrp2 mRNA was significantly higher in MDR1-transfected cells (Figure 2D).
In addition to the analyses in MDR1-transfected cells, we also determined Pgp expression in MRP-transfected MDCK cells. A representative example is shown in Figure 3, including Western blots of Pgp in wildtype and MDR1-transfected LLC cells. Pgp expression was comparable in MDCK wildtype cells and MDCK cells transfected with MRP1 and MRP2, but was about threefold higher (compared with wildtype) in MRP5-transfected MDCK cells, so that the Pgp expression of MRP5-transfected MDCK cells was similar to the Pgp expression in MDR1-transfected LLC cells. As shown in Figure 3A,B, Pgp was revealed as one single band of an apparent molecular weight of 160 kDa in LLC cells. In MDCK cells, two protein bands of approximately 155 kDa and 140 kDa reacted efficiently with the C219 antibody. Such differences in the size of Pgp betweeen different cell types have previously been reported and they are suggested to result from differences in the carbohydrate content of the protein (Jettéet al., 1997). Hence, the two bands of Pgp in MDCK cells most likely represent Pgp isoforms with a different degree of glycosylation (Tang et al., 2002b).
Prompted by the differences in Mrp2 mRNA expression between MDR1-transfected and wildtype MDCK and LLC cells (Figures 1D and 2D), we performed Western blots of Mrp2 protein in these cells (Figure 4). Corresponding to the lower Mrp2 mRNA expression in MDR1-transfected MDCK cells (Figure 1D), lower expression of Mrp2 protein was demonstrated in the transfected cells (Figure 4A,C). Similarly, the higher Mrp2 mRNA expression in MDR1-LLC cells (Figure 2D) was associated with markedly higher expression of Mrp2 protein in the transfected cells (Figure 4B,C). MRP1- and MRP5-transfected MDCK cells as well as LLC wildtype cells expressed hardly any endogenous Mrp2 at the conditions of the assay (Figure 4).
Figure 4.
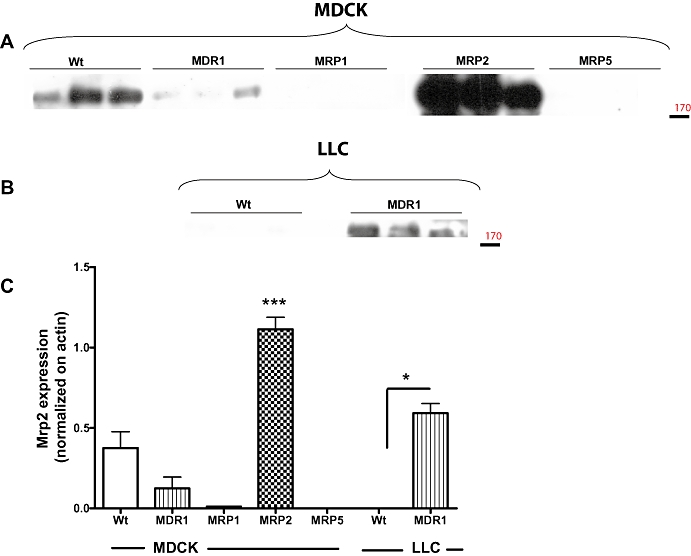
Comparison of Mrp2 expression in different cell lines. Data are from a representative experiment in which Mrp2 expression was determined by Western blot in MDCK-II wildtype and MDCK-II cells transfected with human MRP1, MRP2 or MRP5. For each cell line, three different samples are shown. As shown in (A) (MDCK-II cells) endogenous (canine) Mrp2 and recombinant human MRP2 appeared as one band at around 190 kDa. For comparison, Mrp2 expression in LLC wildtype and MDR1-transfected LLC cells is shown in (B). Note the increased expression of endogenous (pig) Mrp2 in MDR1-transfected LLC cells. In (C), Mrp2 expression was normalized on actin expression to allow comparison of Mrp2 expression between cell lines. Data are shown as mean ± SEM of three samples per cell line except MDCK wildtype, which is mean ± SEM from nine samples. In contrast to MDCK wildtype cells, which expressed a relatively high amount of endogenous (canine) Mrp2, very low Mrp2 expression was determined in LLC wildtype cells at the conditions of the assay. Transfection with human MRP2 led to a marked MRP2 expression in MDCK-II cells. *P < 0.05; ***P < 0.001; significant differences between transfected cells and respective wildtype cells. MDCK, Madin-Darby canine kidney; MDR1, multidrug resistance 1; MRP, multidrug resistance protein.
In order to illustrate how the lower expression of endogenous Pgp and Mrp2 in MDR1-transfected MDCK cells may affect the interpretation of transport studies, we performed transcellular transport experiments with vinblastine, which is transported by Pgp and Mrp2 (Evers et al., 1998; Tang et al., 2002a). In MDCK-II wildtype cells, a marked basolateral to apical transport was determined, for which a TR of 29.3 was calculated (Figure 5A). Transport could be markedly reduced by both tariquidar (TR was reduced to 1.8) and MK571 (TR = 2.9), indicating that both Pgp and Mrps (most likely Mrp2) were involved in the basolateral-to-apical transport of vinblastine in MDCK wildtype cells (not illustrated). Despite the transfection with human MDR1 and the resulting higher expression of Pgp (see Figure 1C), transport of vinblastine in MDR1-transfected MDCK cells was similar to wildtype cells, resulting in a TR of 38.5 and a corrected TR (cTR) of only 1.31 (Figure 5B). This cTR would thus not meet the criteria proposed by Schwab et al. (2003) and Feng et al. (2008) as an indicator of active, asymmetrical transport. Transport of vinblastine in MDR1-transfected MDCK cells was almost completely blocked by the Pgp inhibitor tariquidar (Figure 5C), suggesting that transport of vinblastine in MDR1-MDCK cells was predominantly mediated by Pgp. This inference was substantiated by testing vinblastine transport in MDR1-MDCK cells in the presence of MK571, which resulted in a TR of 18.65 (not illustrated). Furthermore, combined exposure to tariquidar and MK571 (Figure 5D) was not more effective in inhibiting transport than tariquidar alone.
Figure 5.
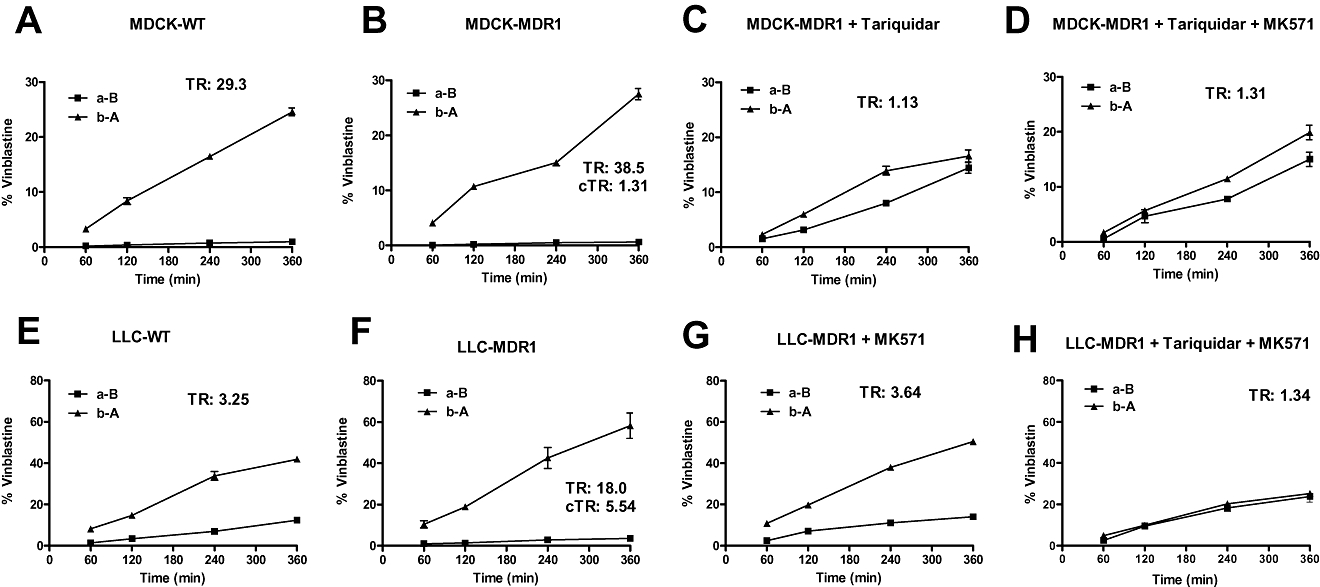
Representative transcellular transport experiments with vinblastine (2 µM) in MDCK (A–D) and LLC (E–H) cells. Experiments were performed by the conventional bi-directional (concentration gradient) assay, in which the drug is added to either the apical (for apical to basolateral transport, a-B) or the basolateral (for basolateral to apical transport, b-A) side of the monolayer. Data are given as the percentage of the initial drug concentration in the donor chamber versus time. Vinblastine transport was either determined alone or in the presence of the Mrp inhibitor MK571 (50 µM) or the Pgp inhibitor tariquidar (0.2 µM) or a combination of both inhibitors. For each experiment, transport ratios (TR) are given (for calculation, see Methods). Corrected transport ratios (cTRs) were calculated by dividing the TR obtained in MDR1-transfected cells by the TR obtained in wildtype/parental cells. A cTR of ≥1.5 is considered as an indicator of active, asymmetrical transport (Schwab et al., 2003). All experiments were performed in triplicate and values are shown as mean ± SEM. When no error bar is visible, the deviation was within the size of the symbols. MDCK, Madin-Darby canine kidney; MDR1, multidrug resistance 1; Pgp, P-glycoprotein.
Transport experiments with vinblastine in LLC cells are illustrated in Figure 5E–H. In LLC wildtype cells, an asymmetrical, basolateral to apical transport was determined with a TR of 3.25 (Figure 5E). Transport could be completely blocked by MK571 (TR was reduced to 1.2), indicating that Mrps were involved in the basolateral-to-apical transport of vinblastine in LLC wildtype cells (not illustrated). In MDR1-transfected LLC cells, transport was more marked than in wildtype cells, resulting in a TR of 18.0 and a cTR of 5.54 (Figure 5F), thus meeting the criteria proposed as an indicator of active, asymmetrical transport (Schwab et al., 2003; Feng et al., 2008). Transport of vinblastine in MDR1-transfected LLC cells was reduced to a TR of 3.64 by the MRP inhibitor MK571 (Figure 5G), and completely blocked by combining MK571 with the Pgp inhibitor tariquidar (Figure 5H), indicating that both Pgp and Mrps, most likely Mrp2, were involved in the basolateral-to-apical transport of vinblastine in MDR1-LLC cells.
Discussion
Madin-Darby canine kidney cells express endogenous Pgp and Mrp2 at their apical membrane, while Mrp1 and Mrp5 are expressed basolaterally (Horio et al., 1989; Goh et al., 2002; Tang et al., 2002a,b; Baltes et al., 2007a; Borst et al., 2007). Our studies show that, in the batches of wildtype and MDR1-transfected MDCK-II cells used, the expression of endogenous (canine) Pgp and Mrp2 mRNAs was markedly lower in the transfected cells. The data on Mrp2 protein determination showed that the difference between wildtype and transfected cells in mRNA expression was associated with a corresponding difference at the protein level. These differences in endogenous Pgp and Mrp2 expression will affect transport studies with drugs that are either transported by Pgp or Mrp2 or both, thus resulting in a potential bias when transcellular transport determined in MDR1-transfected MDCK-II cells is corrected by transport obtained in wildtype cells. Several reports have proposed calculating cTRs by dividing the TR obtained in MDR1-transfected cells by the TR obtained in the respective wildtype/parental cells (see Schwab et al., 2003; Feng et al., 2008). cTRs of at least 1.5 (Schwab et al., 2003) or >1.7 (Feng et al., 2008) have been proposed as an indicator of active, asymmetrical transport. This correction, however, is only valid if MDR1-transfected cells exhibit about the same expression of endogenous transporters as the wildtype cells. Our results, using the Pgp/Mrp2 substrate vinblastine in MDCK-MDR1 and wildtype cells and applying this correction, would produce a cTR value <1.5, suggesting that this drug is not transported by human Pgp. However, vinblastine is well-known to be a substrate for human Pgp (Silverman, 1999; Szakács et al., 2006), so that this cTR calculation would lead to a ‘false-negative’ result. The explanation is that vinblastine exhibits a pronounced basolateral-to-apical transport in MDCK wildtype cells, which is mediated by endogenous Pgp and Mrp2, resulting in a high TR. This transport can be inhibited by both tariquidar and MK571, substantiating that both Pgp and Mrp2 are involved. In contrast, in MDR1-transfected cells, endogenous Mdr1 and Mrp2 mRNAs are strikingly lower, so that vinblastine is now almost exclusively transported by human Pgp. The lack of contribution of endogenous Mrp2 to apical transport of vinblastine in MDR1-transfected MDCK cells is substantiated by the almost complete inhibition of transport with the Pgp inhibitor tariquidar, which does not inhibit Mrps (Fox and Bates, 2007).
In contrast to the differences in endogenous Mdr1 in wildtype and transfected MDCK-II cells, the expression of endogenous Mdr1 mRNA was comparable in wildtype and MDR1-transfected LLC cells. Furthermore, results of transport experiments with vinblastine in LLC cells differed from those obtained in MDCK cells in that a less marked basolateral-to-apical transport of vinblastine was determined in LLC wildtype cells. The transport observed in LLC wildtype cells is most likely to be mediated by both endogenous Mrp2 and Pgp, although expression of these transporters in LLC cells is low (Chen et al., 1999; Goh et al., 2002; Baltes et al., 2007a; present study). Asymmetrical transport of vinblastine in MDR1-transfected LLC cells was much higher than transport observed in wildtype cells, resulting in a cTR of >5. Thus, MDR1-LLC cells correctly identified vinblastine as a substrate of human Pgp. However, unexpectedly, MDR1-transfected LLC cells exhibited a much higher expression of endogenous Mrp2 than wildtype cells, so that both recombinant human Pgp and endogenous porcine Mrp2 were involved in the basolateral-to-apical transport of vinblastine in MDR1-transfected LLC cells, which was substantiated by the partial inhibition of transport by the Mrp inhibitor MK571.
The mechanisms involved in the marked difference in endogenous Mdr1 expression in transfected versus wildtype MDCK cells, but a lack of such a difference in LLC cells, are yet to be elucidated. One plausible explanation for this finding may be the difference in endogenous Pgp expression between MDCK and LLC wildtype cells. Expression of endogenous (canine) Mdr1 mRNA and Pgp protein in MDCK wildtype cells is high, whereas expression of endogenous (porcine) Mdr1 mRNA and Pgp protein in LLC cells is low or undetectable. Transfection with human MDR1 could cause MDCK cells to reach a Pgp protein threshold, which may potentially cause negative feedback and trigger down-regulation of endogenous Mdr1 in these cells and as a consequence, endogenous Pgp protein is replaced by human Pgp. However, due to low endogenous Pgp expression in LLC cells, a Pgp protein threshold may not be reached and such a scenario may not take place in these cells.
Down-regulation of endogenous mRNA and protein expression caused by transfection with an exogenous construct is a commonly observed event, indicating a feedback-regulatory response from the endogenous gene to satisfy cellular demand (see Lloyd et al., 1992). Interestingly, in addition to lower Mdr1 mRNA levels, endogenous Mrp2 mRNA was also down-regulated in MDR1-transfected MDCK cells, whereas expression of Mrp1 and Mrp5 was comparable with parental cells. Down-regulation of both Mdr1 and Mrp2 may suggest a functional synergism between these transporters in kidney cells, as was recently suggested by Oswald et al. (2009).
An alternative explanation for lower Mdr1 mRNA expression in MDR1-MDCK-II versus wildtype cells could be that the differences in expression of endogenous transporter between wildtype and transfected cells is a chance event, e.g. possibly due to naturally occurring batch-to-batch variations in transporter expression of parental and transfected cells. MDCK-II cells do not show stable expression of various endogenous drug transporters, which leads to substantial variation in canine Mdr1 mRNA and Pgp protein expression between different batches of the parental population. The MDR1-transfected MDCK-II cells used in the present study were derived from a single clone, and only one MDCK-II parental population was used for transfection. Of course, this does not formally exclude the possibility that low expression of endogenous canine Mdr1 in the MDCK-II-MDR1 cell line is the result of clonal variation. Furthermore, cell passaging may affect transporter expression, but all cell line experiments reported here were performed at comparable passage numbers. However, irrespective of the mechanism, the consequences of differences in transporter expression between MDR1-transfected and wildtype MDCK-II cells remain the same. This phenomenon affects the results of transport studies as illustrated by the present findings. Thus, to avoid erroneous inferences, expression of endogenous transporters, particularly Pgp and Mrp2, has to be analysed in both wildtype and transfected MDCK or LLC cells before performing transport studies A more detailed analysis of the mechanisms causing differences in endogenous transporter expression between wildtype and transfected cell lines as shown here is beyond the scope of our study. However, such experiments could include (i) transfection of cells with tagged constructs (e.g. green fluorescent protein-tag) that would allow differentiating exogenous and endogenous Pgp at the protein level; (ii) analysis of several clones transfected with MDR1 cDNA; (iii) comparison of different cell populations derived from the same clone; and (iv) selective knock-down of either endogenous or recombinant Pgp by species-specific siRNAs.
Based on the present data with MDCK and LLC cells, one may argue that MDR1-transfected LLC cells are better suited for studies on drug transport mediated by human Pgp, because these cells only express low levels of endogenous Pgp (Chen et al., 1999; Goh et al., 2002; Baltes et al., 2007a; present study) and endogenous Mdr1 expression is comparable in MDR1-transfected and wildtype LLC cells. However, we found that MDR1-transfected LLC cells, at least the batch used in the present study, express strikingly more endogenous Mrp2 than wildtype cells. Furthermore, LLC cells in general express more endogenous breast cancer-related protein (BCRP; ABCG2) at their apical membrane than MDCK cells (Takada et al., 2005; Giri et al., 2009), which might contribute to basolateral-to-apical drug transport in MDR1-transfected LLC cells. Pgp and BCRP mediated drug transport cannot be differentiated by Pgp inhibitors such as tariquidar or elacridar, because these ‘third generation’ Pgp inhibitors also inhibit BCRP (Kühnle et al., 2009). However, BCRP-selective inhibitors, such as Ko143, are available (Allen et al., 2002) and can be used to avoid interference from BCRP in transport studies with LLC cells. In MDCK-MDR1 cells, no BCRP-mediated drug transport was found (Wang et al., 2008), which is in line with the low BCRP expression in these cells. Thus, in view of the low expression of endogenous Mdr1 and Mrp2 in MDR1-transfected MDCK cells found in the present study, MDR1-MDCK cells may have advantages versus MDR1-LLC cells in identifying substrates of human Pgp. In both cell lines, Mrp inhibitors such as MK571 can be used to block endogenous Mrps when using these cells for identifying Pgp substrates (Baltes et al., 2007a,b; Luna-Tortós et al., 2008; present study). However, it should be noted that MK571 has been reported to cause partial inhibition of the basolateral-to-apical efflux of the specific Pgp substrate digoxin at concentrations of 30–50 µM (Luna-Tortós et al., 2008; Wang et al., 2008), indicating that MK571 is less specific than previously thought. When used at a concentration of 50 µM in transwell transport experiments, MK571 reduced digoxin transport in MDR1-transfected LLC cells by 43% (Luna-Tortós et al., 2008) compared with complete blockade of vinblastine transport in MRP2-transfected MDCK cells (in the presence of tariquidar), indicating a ∼2.3-fold higher inhibitory potency of MK571 to block MRP2 than Pgp (Baltes et al., 2007b; Luna-Tortós et al., 2008). Similarly, using uptake assays for determining inhibition of MRP2 and Pgp, a 2.6-fold higher inhibitory potency of MK571 to block MRP2 than Pgp has recently been reported by Matsson et al. (2009).
Rather than comparing drug transport in wildtype and MDR1-transfected cell lines, Polli et al. (2001) proposed that involvement of a Pgp-mediated efflux mechanism is indicated if the basolateral-to-apical/apical-to-basolateral TR in the transfected cell line is >2 (see also Food and Drug Administration (FDA), U.S. Department of Health and Human Services, 2006). For compounds with a TR of 1.5–2.0, Polli et al. (2001) proposed to perform a follow-up experiment with a specific Pgp inhibitor to confirm that the compound is a Pgp substrate. By using this strategy, vinblastine would have been identified as Pgp substrate in both MDR1-MDCK and MDR1-LLC cell lines (see Figure 5). For this strategy, tariquidar rather than elacridar should be used, because elacridar blocks both Pgp and BCRP at about the same concentration (IC50s are 193 and 250 nM), whereas tariquidar is about 4-times more potent to block Pgp (IC50 = 223 nM) than BCRP (IC50 = 916 nM) in efflux assays (Kühnle et al., 2009).
In addition to measuring Mdr1 mRNA and Pgp expression in MDR1-transfected MDCK and LLC cells, we determined Pgp expression in MDCK cells transfected with either MRP1, MRP2 or MRP5. This strategy was prompted by the report of Flanagan et al. (2002), indicating that expression of endogenous Pgp was increased in LLC cells transfected with human MRP1. In our experiments, endogenous Pgp expression was similar to the parental cells in MRP1- and MRP2-transfected cells, whereas we found a markedly higher expression of endogenous Pgp in MRP5-transfected cells. In addition to potential effects of transfection or inter-batch variation, an alternative explanation for the findings of Flanagan et al. (2002) and the present study would be clonal variation in expression of endogenous transporters, which may occur in MDCK and other immortalized kidney cell lines, further complicating transport analysis (Borst and Elferink, 2002). These data thus demonstrate that it is very important to determine the expression of endogenous transporters not only in MDR1-transfected but also in MRP-transfected cells, because differences in the expression of transporters such as Pgp can significantly influence the results and interpretation of transport data in transcellular transport assays.
In conclusion, calculation of cTR by comparison of the TR determined in MDR1-MDCK versus wildtypeMDCK cells is a commonly used method to enable the assessment of whether a compound is a specific human Pgp substrate (Feng et al., 2008). However, as demonstrated by the present data, this method may lead to false negative data because the expression of endogenous Pgp (and Mrp2) may differ markedly between wildtype and transfected cells. Furthermore, interspecies differences in binding site affinities on human and canine Pgp that are responsible for recognition and transport or possibly differences in the relative functional activities of human and canine Pgp towards test substrates introduce additional complexity when comparing drug transport in MDR1-MDCK versus MDCK wildtype cells (Taub et al., 2005). Thus, while among various in vitro assays, the transwell assay was found to be the most reliable for predicting the Pgp efflux liability in vivo and is considered the method of choice for evaluating drug candidates (Yamazaki et al., 2001; Food and Drug Administration (FDA), U.S. Department of Health and Human Services, 2006; Feng et al., 2008), cTR calculation should be avoided in this assay, at least when using MDCK cells. An additional caveat of the monolayer efflux assay with MDCK or LLC cells is to fail with highly permeable Pgp substrates (Papp aB > 300 nm·s−1; Polli et al., 2001), but this limitation can be resolved by using a modified (concentration equilibrium) transport assay (CETA) that minimizes the bias of passive diffusion (Luna-Tortós et al., 2008; Luna-Tortós et al., 2009).
Acknowledgments
We thank Prof Piet Borst for providing cell lines and Dr Alfred Schinkel, Dr Ursula Mönning and Dr Björn Bauer for helpful discussion. The skilful technical assistance of Mrs Britta Sterzik is gratefully acknowledged. The study was supported by a grant from the Deutsche Forschungsgemeinschaft (Bonn, Germany). Carlos Luna-Tortós receives a PhD scholarship from the DAAD (German Academic Exchange Service; Bonn, Germany).
Glossary
Abbreviations
- BCRP
breast cancer-related protein
- cTR
corrected transport ratio
- FDA
Food and Drug Administration
- MDCK
Madin-Darby canine kidney
- MDR1
multidrug resistance 1
- MRP
multidrug resistance protein
- Pgp
P-glycoprotein
- qRT-PCR
quantitative real-time RT-PCR
- TEER
transepithelial electrical resistance
- TR
transport ratio
Conflict of interest
The authors state no conflict of interest.
References
- Alexander SPH, Mathie A, Peters JA. Guide to receptors and channels (GRAC), 4th edition. Br J Pharmacol. 2009;158(Suppl 1):S1–S254. doi: 10.1111/j.1476-5381.2009.00499.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen JD, van Loevezijn A, Lakhai JM, van der Valk M, van Tellingen O, Reid G, et al. Potent and specific inhibition of the breast cancer resistance protein multidrug transporter in vitro and in mouse intestine by a novel analogue of fumitremorgin C. Mol Cancer Ther. 2002;1:417–425. [PubMed] [Google Scholar]
- Artursson P. Epithelial transport of drugs in cell culture. I: a model for studying the passive diffusion of drugs over intestinal absorptive (Caco-2) cells. J Pharm Sci. 1990;79:476–482. doi: 10.1002/jps.2600790604. [DOI] [PubMed] [Google Scholar]
- Baltes S, Gastens AM, Fedrowitz M, Potschka H, Kaever V, Löscher W. Differences in the transport of the antiepileptic drugs phenytoin, levetiracetam and carbamazepine by human and mouse P-glycoprotein. Neuropharmacology. 2007a;52:333–346. doi: 10.1016/j.neuropharm.2006.07.038. [DOI] [PubMed] [Google Scholar]
- Baltes S, Fedrowitz M, Luna-Tortós C, Potschka H, Löscher W. Valproic acid is not a substrate for P-glycoprotein or multidrug resistance proteins 1 and 2 in a number of in vitro and in vivo transport assays. J Pharmacol Exp Ther. 2007b;320:331–343. doi: 10.1124/jpet.106.102491. [DOI] [PubMed] [Google Scholar]
- Borst P, Elferink RO. Mammalian ABC transporters in health and disease. Annu Rev Biochem. 2002;71:537–592. doi: 10.1146/annurev.biochem.71.102301.093055. [DOI] [PubMed] [Google Scholar]
- Borst P, de Wolf C, van de Wetering K. Multidrug resistance-associated proteins 3, 4, and 5. Pflugers Arch. 2007;453:661–673. doi: 10.1007/s00424-006-0054-9. [DOI] [PubMed] [Google Scholar]
- Chen ZS, Kawabe T, Ono M, Aoki S, Sumizawa T, Furukawa T, et al. Effect of multidrug resistance-reversing agents on transporting activity of human canalicular multispecific organic anion transporter. Mol Pharmacol. 1999;56:1219–1228. doi: 10.1124/mol.56.6.1219. [DOI] [PubMed] [Google Scholar]
- Evers R, Zaman GJ, van Deemter L, Jansen H, Calafat J, Oomen LC, et al. Basolateral localization and export activity of the human multidrug resistance-associated protein in polarized pig kidney cells. J Clin Invest. 1996;97:1211–1218. doi: 10.1172/JCI118535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evers R, Kool M, van Deemter L, Janssen H, Calafat J, Oomen LC, et al. Drug export activity of the human canalicular multispecific organic anion transporter in polarized kidney MDCK cells expressing cMOAT (MRP2) cDNA. J Clin Invest. 1998;101:1310–1319. doi: 10.1172/JCI119886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng B, Mills JB, Davidson RE, Mireles RJ, Janiszewski JS, Troutman MD, et al. In vitro P-glycoprotein assays to predict the in vivo interactions of P-glycoprotein with drugs in the central nervous system. Drug Metab Dispos. 2008;36:268–275. doi: 10.1124/dmd.107.017434. [DOI] [PubMed] [Google Scholar]
- Flanagan SD, Cummins CL, Susanto M, Liu X, Takahashi LH, Benet LZ. Comparison of furosemide and vinblastine secretion from cell lines overexpressing multidrug resistance protein (P-glycoprotein) and multidrug resistance-associated proteins (MRP1 and MRP2) Pharmacology. 2002;64:126–134. doi: 10.1159/000056161. [DOI] [PubMed] [Google Scholar]
- Food and Drug Administration (FDA), U.S. Department of Health and Human Services. Guidance for industry. Drug interaction studies – study design, data analysis, and implications for dosing and labelling. 2006. Available at http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm072101.pdf (accessed 24 April 2010)
- Fox E, Bates SE. Tariquidar (XR9576): a P-glycoprotein drug efflux pump inhibitor. Expert Rev Anticancer Ther. 2007;7:447–459. doi: 10.1586/14737140.7.4.447. [DOI] [PubMed] [Google Scholar]
- Giri N, Agarwal S, Shaik N, Pan G, Chen Y, Elmquist WF. Substrate-dependent breast cancer resistance protein (Bcrp1/Abcg2)-mediated interactions: consideration of multiple binding sites in in vitro assay design. Drug Metab Dispos. 2009;37:560–570. doi: 10.1124/dmd.108.022046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goh LB, Spears KJ, Yao D, Ayrton A, Morgan P, Roland WC, et al. Endogenous drug transporters in in vitro and in vivo models for the prediction of drug disposition in man. Biochem Pharmacol. 2002;64:1569–1578. doi: 10.1016/s0006-2952(02)01355-2. [DOI] [PubMed] [Google Scholar]
- Horio M, Chin KV, Currier SJ, Goldenberg S, Williams C, Pastan I, et al. Transepithelial transport of drugs by the multidrug transporter in cultured Madin-Darby canine kidney cell epithelia. J Biol Chem. 1989;264:14880–14884. [PubMed] [Google Scholar]
- Jetté L, Potier M, Beliveau R. P-glycoprotein is a dimer in the kidney and brain capillary membranes: effect of cyclosporin A and SDZ-PSC 833. Biochemistry. 1997;36:13929–13937. doi: 10.1021/bi970737+. [DOI] [PubMed] [Google Scholar]
- Jin H, Di L. Permeability –in vitro assays for assessing drug transporter activity. Curr Drug Metab. 2008;9:911–920. doi: 10.2174/138920008786485056. [DOI] [PubMed] [Google Scholar]
- Kühnle M, Egger M, Muller C, Mahringer A, Bernhardt G, Fricker G, et al. Potent and selective inhibitors of breast cancer resistance protein (ABCG2) derived from the p-glycoprotein (ABCB1) modulator tariquidar. J Med Chem. 2009;52:1190–1197. doi: 10.1021/jm8013822. [DOI] [PubMed] [Google Scholar]
- Lloyd C, Schevzov G, Gunning P. Transfection of nonmuscle beta- and gamma-actin genes into myoblasts elicits different feedback regulatory responses from endogenous actin genes. J Cell Biol. 1992;117:787–797. doi: 10.1083/jcb.117.4.787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Löscher W, Potschka H. Role of drug efflux transporters in the brain for drug disposition and treatment of brain diseases. Prog Neurobiol. 2005;76:22–76. doi: 10.1016/j.pneurobio.2005.04.006. [DOI] [PubMed] [Google Scholar]
- Luna-Tortós C, Fedrowitz M, Löscher W. Several major antiepileptic drugs are substrates for human P-glycoprotein. Neuropharmacology. 2008;55:1364–1375. doi: 10.1016/j.neuropharm.2008.08.032. [DOI] [PubMed] [Google Scholar]
- Luna-Tortós C, Rambeck B, Jurgens UH, Loscher W. The antiepileptic drug topiramate is a substrate for human P-glycoprotein but not multidrug resistance proteins. Pharm Res. 2009;26:2464–2470. doi: 10.1007/s11095-009-9961-8. [DOI] [PubMed] [Google Scholar]
- Matsson P, Pedersen JM, Norinder U, Bergstrom CA, Artursson P. Identification of novel specific and general inhibitors of the three major human ATP-binding cassette transporters P-gp, BCRP and MRP2 among registered drugs. Pharm Res. 2009;26:1816–1831. doi: 10.1007/s11095-009-9896-0. [DOI] [PubMed] [Google Scholar]
- Oswald S, May K, Rosin J, Lutjohann D, Siegmund W. Synergistic influence of Abcb1 and Abcc2 on disposition and sterol lowering effects of ezetimibe in rats. J Pharm Sci. 2009;99:422–429. doi: 10.1002/jps.21821. [DOI] [PubMed] [Google Scholar]
- Polli JW, Wring SA, Humphreys JE, Huang L, Morgan JB, Webster LO, et al. Rational use of in vitro P-glycoprotein assays in drug discovery. J Pharmacol Exp Ther. 2001;299:620–628. [PubMed] [Google Scholar]
- Rautio J, Humphreys JE, Webster LO, Balakrishnan A, Keogh JP, Kunta JR, et al. In vitro p-glycoprotein inhibition assays for assessment of clinical drug interaction potential of new drug candidates: a recommendation for probe substrates. Drug Metab Dispos. 2006;34:786–792. doi: 10.1124/dmd.105.008615. [DOI] [PubMed] [Google Scholar]
- Schinkel AH, Jonker JW. Mammalian drug efflux transporters of the ATP binding cassette (ABC) family: an overview. Adv Drug Deliv Rev. 2003;55:3–29. doi: 10.1016/s0169-409x(02)00169-2. [DOI] [PubMed] [Google Scholar]
- Schwab D, Fischer H, Tabatabaei A, Poli S, Huwyler J. Comparison of in vitro P-glycoprotein screening assays: recommendations for their use in drug discovery. J Med Chem. 2003;46:1716–1725. doi: 10.1021/jm021012t. [DOI] [PubMed] [Google Scholar]
- Silverman JA. Multidrug-resistance transporters. Pharm Biotechnol. 1999;12:353–386. doi: 10.1007/0-306-46812-3_13. [DOI] [PubMed] [Google Scholar]
- Szakács G, Paterson JK, Ludwig JA, Booth-Genthe C, Gottesman MM. Targeting multidrug resistance in cancer. Nat Rev Drug Discov. 2006;5:219–234. doi: 10.1038/nrd1984. [DOI] [PubMed] [Google Scholar]
- Takada T, Suzuki H, Sugiyama Y. Characterization of polarized expression of point- or deletion-mutated human BCRP/ABCG2 in LLC-PK1 cells. Pharm Res. 2005;22:458–464. doi: 10.1007/s11095-004-1884-9. [DOI] [PubMed] [Google Scholar]
- Tang F, Horie K, Borchardt RT. Are MDCK cells transfected with the human MRP2 gene a good model of the human intestinal mucosa? Pharm Res. 2002a;19:773–779. doi: 10.1023/a:1016192413308. [DOI] [PubMed] [Google Scholar]
- Tang F, Horie K, Borchardt RT. Are MDCK cells transfected with the human MDR1 gene a good model of the human intestinal mucosa? Pharm Res. 2002b;19:765–772. doi: 10.1023/a:1016140429238. [DOI] [PubMed] [Google Scholar]
- Taub ME, Podila L, Ely D, Almeida I. Functional assessment of multiple P-glycoprotein (P-gp) probe substrates: influence of cell line and modulator concentration on P-gp activity. Drug Metab Dispos. 2005;33:1679–1687. doi: 10.1124/dmd.105.005421. [DOI] [PubMed] [Google Scholar]
- Thuerauf N, Fromm MF. The role of the transporter P-glycoprotein for disposition and effects of centrally acting drugs and for the pathogenesis of CNS diseases. Eur Arch Psychiatry Clin Neurosci. 2006;256:281–286. doi: 10.1007/s00406-006-0662-6. [DOI] [PubMed] [Google Scholar]
- Wang Q, Strab R, Kardos P, Ferguson C, Li J, Owen A, et al. Application and limitation of inhibitors in drug-transporter interactions studies. Int J Pharm. 2008;356:12–18. doi: 10.1016/j.ijpharm.2007.12.024. [DOI] [PubMed] [Google Scholar]
- Yamazaki M, Neway WE, Ohe T, Chen I, Rowe JF, Hochman JH, et al. In vitro substrate identification studies for p-glycoprotein-mediated transport: species difference and predictability of in vivo results. J Pharmacol Exp Ther. 2001;296:723–735. [PubMed] [Google Scholar]