

Abstract
Background and purpose:
Previous work has shown that NG-monomethyl-l-arginine (l-NMMA) paradoxically inhibits basal, but not ACh-stimulated activity of nitric oxide in rat aorta. The aim of this study was to determine if the endogenously produced agent, asymmetric NG, NG-dimethyl-l-arginine (ADMA), also exhibits this unusual selective blocking action.
Experimental approach:
The effect of ADMA on basal nitric oxide activity was assessed by examining its ability to enhance phenylephrine (PE)-induced tone in endothelium-containing rings. Its effect on ACh-induced relaxation was assessed both in conditions where ADMA greatly enhanced PE tone and where tone was carefully matched with control tissues at a range of different levels.
Key results:
ADMA (100 µM) potentiated PE-induced contraction, consistent with inhibition of basal nitric oxide activity. Higher concentrations (300–1000 µM) had no greater effect. Although ADMA (100 µM) also appeared to block ACh-induced relaxation when it enhanced PE tone to maximal levels, virtually no block was seen at intermediate levels of tone in the presence of ADMA. Even ADMA at 1000 µM had no effect on the maximal relaxation to ACh, although it produced a small (two- to threefold) reduction in sensitivity. ADMA and l-NMMA, like l-arginine (all at 1000 µM), protected ACh-induced relaxation against blockade by l-NAME (30 µM).
Conclusions and implications:
In the rat aorta, ADMA, like l-NMMA, blocks basal activity of nitric oxide, but has little effect on that stimulated by ACh. Further studies are required to explain these seemingly anomalous actions of ADMA and l-NMMA.
Keywords: ACh, ADMA, asymmetric dimethylarginine, endothelium, nitric oxide, NOS inhibitor, SDMA, symmetric dimethylarginine, vasodilatation
Introduction
It is well established that one of the two equivalent guanidino nitrogens of l-arginine is utilized by NOS in the synthesis of nitric oxide (Palmer et al., 1988). Since this discovery, an extensive range of guanidino (NG)-substituted analogues of l-arginine have been developed as inhibitors of NOS (Rees et al., 1989; 1990; Moore et al., 1990; Hobbs et al., 1999), and these have become valuable tools for investigating the role of the l-arginine–nitric oxide system in biological processes. Among the earliest examples developed wereNG-monomethyl-l-arginine (l-NMMA), NG-nitro-l-arginine (l-NOARG) and NG-nitro-l-arginine methyl ester (l-NAME). These remain the most commonly used investigational tools in experimental biology, probably because they are effective blockers of all three isoforms of NOS [i.e. endothelial (eNOS), neuronal (nNOS) and inducible (iNOS) forms]. In the vast majority of cases, these three agents are not metabolized by NOS, but act as classical competitive inhibitors, whose actions can be either prevented or reversed by an excess of the substrate l-arginine. Of these three agents, however, l-NMMA alone has frequently been reported to exhibit properties that are inconsistent with it acting as a simple competitive inhibitor. For example, l-NMMA acts as an alternative substrate and mechanism-based suicide inhibitor of iNOS in murine macrophages (Olken and Marletta, 1993). Furthermore, unlike l-NOARG and l-NAME, l-NMMA fails to inhibit nitrergic nerve (nNOS)-mediated relaxation in the bovine penile and ciliary arteries, and retractor penis muscle (Liu et al., 1991; Martin et al., 1993; Overend and Martin, 2007). Most surprisingly of all, however, is that l-NMMA selectively blocks basal, but not agonist (ACh or ATP)-induced activity of nitric oxide in rat aorta (Frew et al., 1993). Furthermore, this study showed that pretreatment with l-NMMA protects agonist-induced nitric oxide activity against blockade by l-NOARG in a manner similar to l-arginine, suggesting it might act as an alternative substrate for eNOS. Indeed, chemiluminescence detection has confirmed that l-NMMA, like l-arginine, but unlike l-NOARG, can fuel nitric oxide synthesis in rat aorta and pulmonary artery (Archer and Hampl, 1992).
Renewed interest in the biological actions of methylarginines was sparked by the discovery that l-NMMA, asymmetric NG, NG-dimethyl-l-arginine (ADMA) and symmetric NG, N′G-dimethyl-l-arginine (SDMA) are produced endogenously during the proteolytic digestion of methylated proteins (Vallance et al., 1992; Leiper and Vallance, 2006). These authors showed also that ADMA, like l-NMMA, inhibits NOS, but SDMA does not. Moreover, while the plasma concentration of l-NMMA is low, levels of ADMA and SDMA are around 10 times higher and accumulate further in renal failure (Vallance et al., 1992) and a wide range of pathological conditions associated with vascular dysfunction, including hypertension, pulmonary hypertension, atherosclerosis, diabetes mellitus and preeclampsia (Leiper and Vallance, 2006; Siroen et al., 2006).
In view of the growing evidence for the involvement of ADMA in cardiovascular disease, the aim of this study was to determine if this agent shared with l-NMMA the ability to block selectively basal, but not ACh-stimulated, activity of nitric oxide in rat aorta.
Methods
Preparation of aortic rings and tension recording
All animal care and experimental procedures complied with the UK Home Office regulations. The preparation of rat aortic rings for tension recording was essentially similar to that described earlier (Frew et al., 1993). Briefly, female Wistar rats weighing 150–200 g were killed by stunning and exsanguination. The aorta was removed, cleared of adhering fat and connective tissue and cut into 2.5 mm wide transverse rings using a device with parallel razor blades. Endothelial cells were removed from some rings by gently rubbing the intimal surface with a moist wooden stick for 30 s. Successful removal of the endothelium was confirmed by the inability of ACh (1 µM) to elicit relaxation. The aortic rings were mounted under 10 mN resting tension on stainless steel hooks in 10 mL tissue baths, and bathed at 37°C in Krebs solution containing (mM): NaCl 118, KCl 4.8, CaCl2 2.5, MgSO4 1.2, KH2PO4 1.2, NaHCO3 24 and glucose 11, and gassed with 95% O2 and 5% CO2. Tension was recorded isometrically with Grass FTO3C transducers and displayed on a PowerLab (ADInstruments, Hastings, UK).
Experimental protocols with the rat aorta
The ability of ADMA to block basal activity of nitric oxide was assessed from its enhancement of agonist-induced contraction in endothelium-containing aortic rings (Frew et al., 1993). This was performed in two ways. The first, by obtaining a low-level contraction (∼2–5 mN) to phenylephrine (PE) (30–60 nM), and then measuring the enhancement of tone produced by a range of ADMA concentrations (0.3–300 µM), each for 1 h. The ability of l-arginine (10 mM, 1 h) to either protect against or reverse the enhancement by ADMA (100 µM) was also assessed. The effects of ADMA were also examined on PE-induced tone in endothelium-denuded rings. We also determined if submaximal tone induced by 5-HT (3 µM) or by prostaglandin F2α (3 µM) in endothelium-containing rings was enhanced when subsequently treated with ADMA (100 µM). The second protocol for examining blockade of basal nitric oxide activity involved pretreating endothelium-containing rings for 1 h with ADMA (100, 300 or 1000 µM), and then constructing full concentration–response curves to PE (1 nM–10 µM), together with time-matched controls.
Agonist-stimulated activity of nitric oxide was assessed by measuring the relaxation to ACh (1 nM–10 µM) in endothelium-containing rings. When the effects of ADMA or other NOS inhibitors were to be examined, they were added for 1 h before construction of the ACh concentration–response curve. As will be seen in the Results, the level of PE tone was critical in determining the magnitude of ACh-induced relaxation both in control and ADMA-treated tissues. In some experiments with ADMA, its effects on relaxation were examined after it had enhanced PE-induced tone, whereas in others, the tone was carefully matched with those of control tissues at low, intermediate or maximal levels. In other experiments, the ability of ADMA (100 µM) to affect ACh-induced relaxation was also assessed on submaximal tone induced by 5-HT or prostaglandin F2α.
In some experiments, l-NAME (30 µM, 1 h) was used to inhibit maximal ACh (1 nM–10 µM)-induced relaxation by ∼50%. In these, the ability of l-arginine, l-NMMA, ADMA or SDMA (all at 1000 µM; pretreatment or post-treatment for 1 h) either to protect against or reverse this inhibition by l-NAME was also assessed.
Data analysis
Contractions were measured in millinewtons. Papaverine (300 µM) was added at the end of each experiment to fully relax tissues, and ACh-induced relaxations were calculated as % relaxation of PE-induced tone with respect to this baseline. Data are expressed as the mean ± SEM of n separate observations, each from a separate tissue. Graphs were drawn and statistical comparisons were made using one-way anova and Bonferroni's post-test with the aid of a computer program, Prism (GraphPad, San Diego, CA, USA). A probability (P) less than or equal to 0.05 was considered significant.
Materials
ACh chloride, ADMA, l-arginine hydrochloride, 5-HT, l-NAME, l-NMMA, papaverine hydrochloride, PE hydrochloride and prostaglandin F2α were all obtained from Sigma, Poole, UK. SDMA was obtained from Enzo Life Sciences, Exeter, UK. All drugs were dissolved and diluted in 0.9% saline.
Results
Effects of ADMA on basal nitric oxide activity
Following induction of a low level of contraction (3.1 ± 0.6 mN) of endothelium-containing rings of rat aorta using PE (30–60 nM), subsequent addition of ADMA (0.3–300 µM) led to an immediate further elevation of tone, consistent with inhibition of basal nitric oxide activity (Figures 1 and 2B). The threshold concentration was 0.3–1 µM, and the maximum was obtained at 100 µM. The enhancement of PE-induced contraction by ADMA (100 µM) was both prevented and reversed by treatment with l-arginine (10 mM). ADMA did not enhance PE-induced contraction in endothelium-denuded rings. In contrast to ADMA, SDMA (100 µM) had no effect on PE-induced contraction in endothelium-containing rings. Submaximal contraction induced by 5-HT (3 µM) or prostaglandin F2α (3 µM) was also enhanced by ADMA (100 µM).
Figure 1.
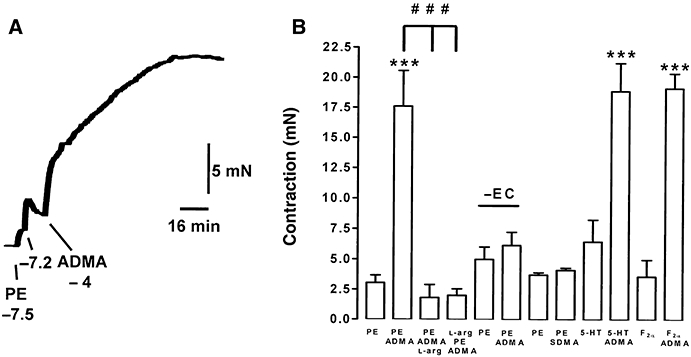
ADMA (100 µM) enhances tone induced by PE, 5-HT or prostaglandin F2α (F2α) in endothelium-containing, but not endothelium-denuded (–EC) rings of rat aorta. The enhancement of PE-induced tone is both prevented and reversed by treatment with l-arginine (10 mM). In contrast to ADMA, SDMA (100 µM) does not enhance tone in endothelium-containing rings. In the trace (A), concentrations are given in log molar units. In the histogram (B), the order of listing of drugs (PE, ADMA, l-arg; l-arg, PE, ADMA, etc.) reflects their order of addition to tissues; data are the mean ± SEM of six observations. ***P < 0.001 indicates a significant enhancement by ADMA; ###P < 0.001 indicates a significant difference in the presence of l-arginine.
Figure 2.
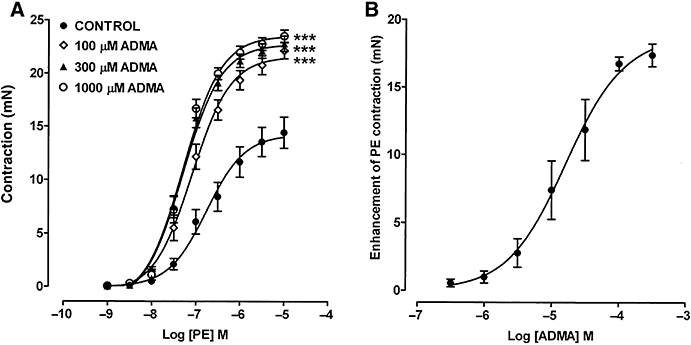
(A) Concentration–response curves showing the contractile effects of PE on endothelium-containing rings of rat aorta and the enhancement of this tone following pretreatment for 1 h with ADMA (100, 300 and 1000 µM). (B) Concentration–response curve showing the ability of ADMA to enhance low-level contraction to PE (30–60 nM); the response to each concentration of ADMA was measured after 1 h. Each point is the mean ± SEM of 6–15 observations. ***P < 0.001 indicates a significant difference from control.
A full concentration–response curve to PE (1 nM–10 µM) in endothelium-containing rings gave an Emax of 14.4 ± 1.5 mN and a pEC50 of 6.75 ± 0.07 (Figure 2). Pretreatment with ADMA (100 µM) for 1 h enhanced both the Emax (22.2 ± 0.8 mN) and sensitivity (pEC50 7.88 ± 0.04) to PE. Increasing the concentration of ADMA to 300 or 1000 µM produced little additional enhancement.
Effects of ADMA on ACh-induced relaxation
Following induction of intermediate PE-induced tone (11.9 ± 2.1 mN) on endothelium-containing rings, ACh (1 nM–10 µM) produced concentration-dependent relaxation (Emax 77.5 ± 7.8%; pEC50 7.30 ± 0.03; Figure 3A,B). When ADMA (100 µM, 1 h) was added on top of this intermediate PE-induced contraction, raising tone to a higher level (18.8 ± 1.5 mN), ACh-induced relaxation was depressed (Emax 22.1 ± 3.1%; pEC50 6.91 ± 0.04). However, when these same tissues were washed, with restoration of intermediate PE-induced tone (7.6 ± 0.9 mN) in the presence of ADMA, the blockade of ACh-induced relaxation was almost completely abolished: maximal relaxation was similar to controls, but there was a 1.7-fold reduction in sensitivity (Emax 77.6 ± 4.0%; pEC50 7.06 ± 0.04). Concentrations of ADMA below 100 µM at submaximal PE-induced tone had no effect on ACh-induced relaxation. ADMA (100 µM) at intermediate levels of tone induced by 5-HT or prostaglandin F2α also failed to affect maximal ACh-induced relaxation, but produced modest reductions in sensitivity of 2.6- and 2.3-fold (data not shown).
Figure 3.
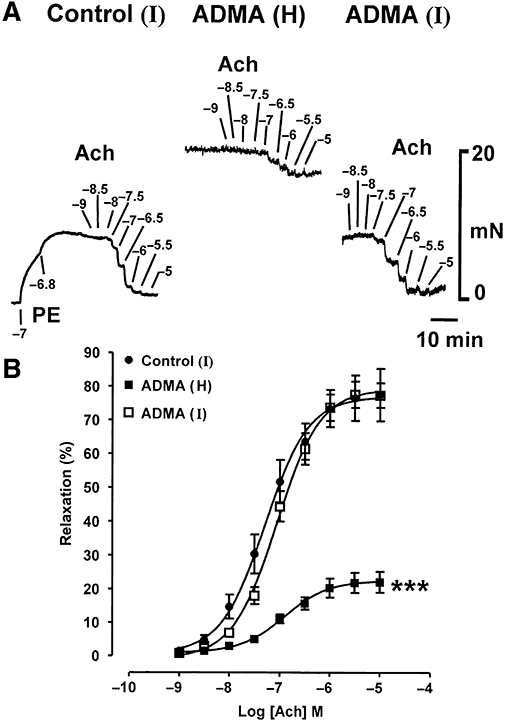
Experimental traces (A) and concentration–response curves (B) showing that when ADMA (100 µM, 1 h) enhances PE (0.1–0.3 µM)-induced tone to a high (H) level, ACh-induced relaxation appears powerfully inhibited. However, when these same tissues are washed, with intermediate PE (10–30 nM)-induced tone (I) re-established in the presence of ADMA, the blockade of ACh-induced relaxation is almost entirely abolished. Each point is the mean ± SEM of six observations. ***P < 0.001 indicates a significant difference from control.
These findings prompted us to examine the effect the level of tone had on the magnitude of ACh-induced relaxation in control and ADMA (100 µM)-treated rings. In control rings, ACh (1 nM–10 µM)-induced relaxation was similar at two different levels of submaximal PE-induced tone (3.7 ± 0.4 and 10.3 ± 0.5 mN; Figure 4A). Relaxation was, however, significantly depressed at a maximal level of tone (14.1 ± 0.5 mN). The ability of ADMA (100 µM) to enhance PE-induced contraction-permitted examination of a wider range of tone levels in the presence of this agent. At levels of PE-induced tone of 4.1 ± 0.6 or 10.2 ± 0.5 mN in the presence of ADMA, maximal ACh-induced relaxation was not significantly different from controls at similar levels of tone (Figure 4A,B). However, at 15.7 ± 0.5 mN tone, ACh-induced relaxation was depressed, but to the same extent as control tissues at the same level of tone. At 20.3 ± 0.7 mN and 24.7 ± 0.6 mN tone, which cannot be achieved in control tissues, ACh-induced relaxation was even more depressed.
Figure 4.
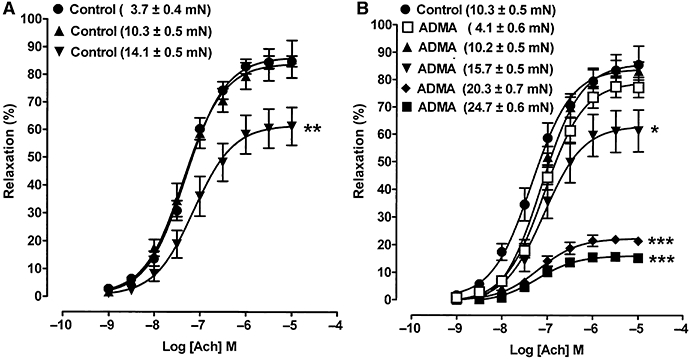
Concentration–response curves showing ACh-induced relaxation in aortic rings taken to different levels of PE tone (indicated in mN) in (A) control tissues and (B) in tissues treated with ADMA (100 µM, 1 h). Both control and ADMA-treated tissues exhibit depression of relaxation, but only at high levels of tone. Each point is the mean ± SEM of six to nine observations. *P < 0.05, **P < 0.01 and ***P < 0.001 indicate significant differences from the first control.
With a higher concentration of ADMA (1000 µM, 1 h) at intermediate PE-induced tone, the Emax to ACh was unaffected, but there was a twofold decrease in sensitivity when compared to controls (Figure 5; Table 1). Testing these tissues again at 3 and 4.5 h showed a slight progressive decrease in sensitivity to ACh in controls, but in ADMA-treated tissue the Emax remained unchanged when compared to time-matched controls, with a roughly similar decrease in sensitivity (threefold) to that seen at 1 h.
Figure 5.
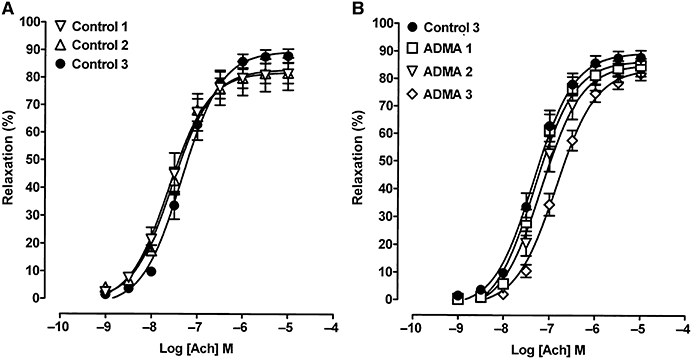
Concentration–response curves showing ACh-induced relaxation at intermediate PE tone in control tissues (control 1) and tissues treated with ADMA (1000 µM) for 1 h (ADMA 1). These same tissues were tested again at 3 h (control 2; ADMA 2) and at 4.5 h (control 3; ADMA 3), but there was little evidence of progressive blockade with time using ADMA. Each point is the mean ± SEM of five to six observations.
Table 1.
Effects of NOS inhibitors on ACh-induced relaxation of rat aorta, together with time-matched controls
| Treatment | Concentration | Emax (%) | pEC50 |
|---|---|---|---|
| Control | – | 81.3 ± 5.9 | 7.61 ± 0.02 |
| ADMA | 1000 µM (1 h) | 83.8 ± 2.8 | 7.31 ± 0.07** |
| Control | – | 87.9 ± 2.5 | 7.33 ± 0.04## |
| ADMA | 1000 µM (4.5 h) | 81.9 ± 2.3 | 6.85 ± 0.04*** |
| Control | – | 93.3 ± 2.8 | 7.51 ± 0.03 |
| SDMA | 1000 µM (1 h) | 91.2 ± 4.4 | 7.52 ± 0.04 |
| Control | – | 86.0 ± 5.8 | 7.54 ± 0.05 |
| l-NMMA | 1000 µM (1 h) | 89.0 ± 4.3 | 7.12 ± 0.04*** |
All experiments were conducted at submaximal levels of PE-induced tone. Data are expressed at mean ± SEM of n = 6–9 observations.
P < 0.01
P < 0.001 indicate significant differences from respective controls.
P < 0.01 indicates a difference from the control at 1 h.
Effects of ADMA on the blockade of ACh-induced relaxation by l-NAME
In the presence of intermediate PE-induced tone, l-NAME (30 µM, 1 h) blocked maximal ACh (1 nM–10 µM)-induced relaxation by ∼50% (Figure 6). This blockade was both largely prevented and reversed by treatment with l-arginine (1000 µM; Figure 6A). l-NMMA (1000 µM) induced a 2.6-fold reduction in sensitivity to ACh without affecting the Emax, and, like l-arginine, both prevented and reversed the blockade induced by l-NAME (Figure 6B; Table 1). ADMA (1000 µM) too produced significant protection, but did not reverse the blockade induced by l-NAME (Figure 6C). SDMA (1000 µM) had no effect by itself on ACh-induced relaxation, and did not prevent or reverse the blockade induced by l-NAME (Figure 6D; Table 1).
Figure 6.
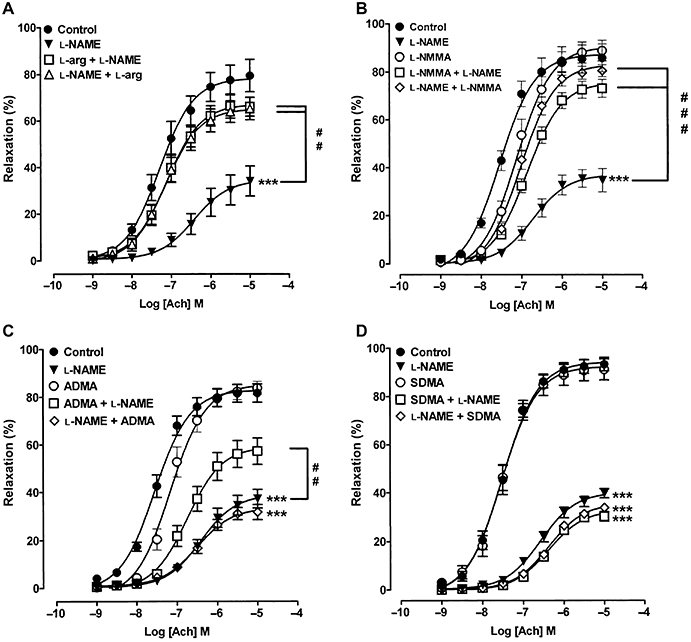
Concentration–response curves showing ACh-induced relaxation at intermediate PE tone and blockade of this relaxation with l-NAME (30 µM, 1 h). The ability of (A) l-arginine, (B) l-NMMA, (C) ADMA and (D) SDMA (all at 1000 µM) to either protect against or reverse blockade by l-NAME is also shown. Note that the order of listing of drugs reflects their order of addition to tissues. Each point is the mean ± SEM of six to nine observations. ***P < 0.001 indicates significant blockade by l-NAME; ##P < 0.001 and ###P < 0.001 indicate significant protection against or reversal of blockade induced by l-NAME.
Discussion
The main new finding in this study is that while ADMA blocks basal activity of nitric oxide in rat aorta, it has little effect on that stimulated by ACh. These seemingly anomalous findings parallel the actions of l-NMMA in rat aorta (Frew et al., 1993), but are in stark contrast to those of l-NOARG and l-NAME which, as expected of typical NOS inhibitors, uniformly abolish both basal and agonist (ACh or ATP)-stimulated activity of nitric oxide.
Basal nitric oxide activity exerts a tonic endothelium-dependent vasodilator influence in many isolated blood vessels, and is typically assessed by observing the rise in submaximal vasoconstrictor-induced tone produced by agents that inhibit either the action or synthesis of nitric oxide (Martin et al., 1986; Rees et al., 1989; Moore et al., 1990). These authors, working on rat and rabbit aorta, and rat mesentery, concluded that this basal vasodilator influence opposing vasoconstriction resulted from the spontaneous (i.e. unstimulated) release of nitric oxide from the endothelium. In these tissues, there was no evidence that the constricting adrenoceptor agonists used had stimulated release of nitric oxide through activation of endothelial α2-adrenoceptors as occurs in canine and porcine coronary and pulmonary arteries (Cocks and Angus, 1983; Miller and Vanhoutte, 1985). Others have, instead, suggested that vascular smooth muscle contraction may indirectly stimulate endothelial cells to release nitric oxide via a signal transmitted either through myoendothelial gap junctions (Dora et al., 2000; Jackson et al., 2008) or by mechanical stress (Fleming et al., 1999). However, in endothelium-containing untreated rings of rat aorta, cyclic GMP levels are two- to threefold higher than in endothelium-denuded rings (Rapoport and Murad, 1983), thus demonstrating that the tonic influence of basal nitric oxide is present in the complete absence of smooth muscle activation. Nevertheless, regardless of the mechanisms governing the basal activity of nitric oxide, there is general agreement that NOS inhibitors enhance vasoconstrictor tone by abolishing the inhibitory influence of nitric oxide. In keeping with a previous report (Vallance et al., 1992), we found that the endogenously produced NOS inhibitor, ADMA, also enhances vasoconstrictor tone induced by a number of agents (PE, 5-HT or prostaglandin F2α) in rat aorta. Both studies report a threshold effect for ADMA at 0.3–1 µM, consistent with concentrations found in the plasma in a number of disease states (Vallance et al., 1992; Siroen et al., 2006), and a maximal effect at ∼100 µM. They also show that the inactive isomer, SDMA, lacks the ability of ADMA to enhance vasoconstrictor tone. Moreover, the ability of the endogenous substrate, l-arginine, to both protect against and reverse blockade of basal nitric oxide activity by ADMA shows it occurs by simple competitive antagonism.
Our most striking new finding is that ADMA, at a concentration of 100 µM which abolishes basal nitric oxide activity, has virtually no effect on ACh-induced relaxation. Although ADMA appeared to produce blockade under conditions where it had potentiated PE-induced tone to near maximal levels, this was almost certainly due to physiological antagonism rather than blockade of NOS, because matching this increased tone in control tissues with additional PE produced a similar degree of blockade. When PE-induced tone in the presence of ADMA was held at intermediate levels, virtually no blockade of ACh-induced relaxation was seen; there was a modest 1.7-fold reduction in sensitivity without any effect on the maximal relaxation. ADMA was similarly ineffective against ACh-induced relaxation in tissues contracted submaximally with 5-HT or prostaglandin F2α. Even increasing both the concentration of ADMA to 1000 µM and the time of exposure to 4.5 h at intermediate levels of PE tone failed to produce any greater blockade of ACh-induced relaxation. These findings are clearly in conflict with previous studies on rat aorta which report blockade of ACh-induced relaxation by ADMA (Vallance et al., 1992; Jin and D'Alecy, 1996; Feng et al., 1998). Because these authors made no mention of matching the tone in control and ADMA-treated tissues, it is likely that the blockade of ACh-induced relaxation they reported resulted from physiological antagonism, due to over-contraction, rather than to blockade of NOS.
Our finding that ADMA blocks basal, but not ACh-induced, nitric oxide activity in rat aorta mirrors what we reported previously for l-NMMA (Frew et al., 1993). Furthermore, just like the endogenous substrate, l-arginine, both ADMA and l-NMMA, protect ACh-induced relaxation against blockade by the NOS inhibitor, l-NAME; l-arginine and l-NMMA also reversed established blockade by l-NAME, but ADMA failed to do this after an hour of treatment, perhaps due to lower potency than the other agents. Whether or not longer treatments with ADMA would have reversed the blockade by l-NAME was not investigated. Thus, although ADMA and l-NMMA both act like conventional competitive inhibitors of basal nitric oxide synthesis, they paradoxically appear to behave more like substrates for ACh-induced and ATP-induced (Frew et al., 1993) synthesis of nitric oxide. Indeed, chemiluminescence detection has revealed that l-NMMA, just like l-arginine, fuels the synthesis of nitric oxide in rat aorta and pulmonary artery, whereas l-NOARG inhibits it (Archer and Hampl, 1992). It is unlikely that demethylation reactions to form l-arginine explain the abilities of ADMA and l-NMMA to behave as alternative substrates for agonist-stimulated eNOS, because the blockade they produce of basal NO activity is sustained, suggestive of ongoing stability of these agents.
At present, we can only speculate about the mechanisms that underlie the finding that ADMA and l-NMMA block basal, but have little effect on agonist-stimulated, activity of nitric oxide. It is possible that different isoforms of eNOS, with different structural requirements for substrates and inhibitors, might be responsible for basal and agonist-stimulated production of nitric oxide, but there is no evidence in the literature to support this. Alternatively, it is known that activation of eNOS by ACh or ATP via Ca2+–calmodulin binding induces a conformational change that permits dissociation of the enzyme from its inhibitory anchor protein, caveolin-1 (Dudzinski and Michel, 2007). It is therefore possible that a concomitant conformational change in the eNOS substrate binding site occurs such that it now recognizes ADMA and l-NMMA as substrates rather than inhibitors. In this context, there is good evidence that flow-mediated dilatation, which occurs through the Ca2+-independent activation of eNOS via phosphorylation by the phosphatidylinositol 3-kinase/Akt pathway (Fulton et al., 1999; Gallis et al., 1999), is inhibited by ADMA in humans (Boger et al., 1998; Vladimirova-Kitova et al., 2008). A direct comparison of the differential sensitivity to blockade by ADMA and l-NMMA of stimuli operating via Ca2+–calmodulin-stimulated and phosphorylation-stimulated activation of eNOS in a single tissue will be required to explore this more thoroughly.
Although ADMA and l-NMMA block basal, but not agonist-induced, activity of nitric oxide in rat aorta, both elevate blood pressure when infused into anaesthetized rats (Rees et al., 1990; Jin and D'Alecy, 1996; De Gennaro Colonna et al., 2007). It would therefore be interesting to explore in vivo the sensitivity to blockade by ADMA and l-NMMA of the components of nitric oxide activity (basal vs. flow stimulated vs. agonist stimulated) that contribute to the regulation of blood pressure. Indeed, because ADMA and l-NMMA both elevate vascular tone in human volunteers (Vallance et al., 1989; 1992;), and the former accumulates in a number of disease states (Vallance et al., 1992; Leiper and Vallance, 2006; Siroen et al., 2006), it will be valuable to re-assess the effects of ADMA and l-NMMA on isolated blood vessels from a range of species, including humans, to determine if the differential sensitivity of basal and agonist-stimulated activity of nitric oxide seen in the rat occurs elsewhere. We would caution that such studies should be conducted at submaximal tone with matched controls to ensure the outcomes are not compromised by physiological antagonism, resulting from over-contraction of inhibitor-treated tissues.
In conclusion, our findings demonstrate that ADMA, like l-NMMA, preferentially blocks basal activity of nitric oxide in rat aorta, but has little effect on that stimulated by ACh. Previous reports suggesting blockade of ACh-induced relaxation of this tissue by ADMA are likely to have been due to physiological antagonism rather than blockade of NOS. Further studies will be required to determine the mechanism by which ADMA and l-NMMA produce this paradoxically selective action.
Acknowledgments
Dr Mohammed J Al-Zobaidy is in receipt of a PhD Scholarship funded by the Iraqi Ministry of Higher Education and Scientific Research.
Glossary
Abbreviations
- ADMA
asymmetric NG, NG-dimethyl-l-arginine
- l-NAME
NG-nitro-l-arginine methyl ester
- l-NMMA
NG-monomethyl-l-arginine
- SDMA
symmetric NG, N′G-dimethyl-l-arginine
Conflict of interest
None.
References
- Archer SL, Hampl V. NG-monomethyl-l-arginine causes nitric oxide synthesis in isolated arterial rings: trouble in paradise. Biochem Biophys Res Commun. 1992;188:590–596. doi: 10.1016/0006-291x(92)91097-a. [DOI] [PubMed] [Google Scholar]
- Boger RH, Bode-Boger SM, Szuba A, Tsao PS, Chan JR, Tangphao O, et al. Asymmetric dimethylarginine (ADMA): a novel risk factor for endothelial dysfunction – its role in hypercholesterolemia. Circulation. 1998;98:1842–1847. doi: 10.1161/01.cir.98.18.1842. [DOI] [PubMed] [Google Scholar]
- Cocks TM, Angus JA. Endothelium-dependent relaxation of coronary arteries by noradrenaline and serotonin. Nature. 1983;305:627–630. doi: 10.1038/305627a0. [DOI] [PubMed] [Google Scholar]
- De Gennaro Colonna V, Bonomo S, Ferrario P, Bianchi M, Berti M, Guazzi M, et al. Asymmetric dimethylarginine (ADMA) induces vascular endothelium impairment and aggravates post-ischemic ventricular dysfunction in rats. Eur J Pharmacol. 2007;557:178–185. doi: 10.1016/j.ejphar.2006.11.034. [DOI] [PubMed] [Google Scholar]
- Dora KA, Hinton JM, Walker SD, Garland CJ. An indirect influence of phenylephrine on the release of endothelium-derived vasodilators in rat small mesenteric artery. Br J Pharmacol. 2000;129:381–387. doi: 10.1038/sj.bjp.0703052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudzinski DM, Michel T. Life history of eNOS: partners and pathways. Cardiovasc Res. 2007;75:247–260. doi: 10.1016/j.cardiores.2007.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng QP, Lu XG, Fortin AJ, Pettersson A, Hedner T, Kline RL, et al. Elevation of an endogenous inhibitor of nitric oxide synthesis in experimental congestive heart failure. Cardiovasc Res. 1998;37:667–675. doi: 10.1016/s0008-6363(97)00242-3. [DOI] [PubMed] [Google Scholar]
- Fleming I, Bauersachs J, Schafer A, Scholz D, Aldershvile J, Busse R. Isometric contraction induces the Ca2+-independent activation of the endothelial nitric oxide synthase. Proc Natl Acad Sci USA. 1999;96:1123–1128. doi: 10.1073/pnas.96.3.1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frew JD, Paisley K, Martin W. Selective inhibition of basal but not agonist-stimulated activity of nitric oxide in rat aorta by NG-monomethyl-l-arginine. Br J Pharmacol. 1993;110:1003–1008. doi: 10.1111/j.1476-5381.1993.tb13913.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulton D, Gratton JP, McCabe TJ, Fontana J, Fujio Y, Walsh K, et al. Regulation of endothelium-derived nitric oxide production by the protein kinase Akt. Nature. 1999;399:597–601. doi: 10.1038/21218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallis B, Corthals GL, Goodlett DR, Ueba H, Kim F, Presnell SR, et al. Identification of flow-dependent endothelial nitric-oxide synthase phosphorylation sites by mass spectrometry and regulation of phosphorylation and nitric oxide production by the phosphatidylinositol 3-kinase inhibitor LY294002. J Biol Chem. 1999;274:30101–30108. doi: 10.1074/jbc.274.42.30101. [DOI] [PubMed] [Google Scholar]
- Hobbs AJ, Higgs A, Moncada S. Inhibition of nitric oxide synthase as a potential therapeutic target. Annu Rev Pharmacol Toxicol. 1999;39:191–220. doi: 10.1146/annurev.pharmtox.39.1.191. [DOI] [PubMed] [Google Scholar]
- Jackson WF, Boerman EM, Lange EJ, Lundback SS, Cohen KD. Smooth muscle α1d-adrenoceptors mediate phenylephrine-induced vasoconstriction and increases in endothelial cell Ca2+ in hamster cremaster arterioles. Br J Pharmacol. 2008;155:514–524. doi: 10.1038/bjp.2008.276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin JS, D'Alecy LG. Central and peripheral effects of asymmetric dimethylarginine, an endogenous nitric oxide synthetase inhibitor. J Cardiovasc Pharmacol. 1996;28:439–446. doi: 10.1097/00005344-199609000-00014. [DOI] [PubMed] [Google Scholar]
- Leiper JM, Vallance P. The synthesis and metabolism of asymmetric dimethylarginine (ADMA) Eur J Clin Pharmacol. 2006;62:33–38. [Google Scholar]
- Liu X, Gillespie JS, Gibson IF, Martin W. Effects of NG-substituted analogues of l-arginine on NANC relaxation of the rat anococcygeus and bovine retractor penis muscles and the bovine penile artery. Br J Pharmacol. 1991;104:53–58. doi: 10.1111/j.1476-5381.1991.tb12384.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin W, Villani GM, Jothianandan D, Furchgott RF. Depression of contractile responses in rat aorta by spontaneously released endothelium-derived relaxing factor. J Pharmacol Exp Ther. 1986;237:529–538. [PubMed] [Google Scholar]
- Martin W, Gillespie JS, Gibson IF. Actions and interactions of NG-substituted analogues of l-arginine on NANC neurotransmission in the bovine retractor penis and rat anococcygeus muscles. Br J Pharmacol. 1993;108:242–247. doi: 10.1111/j.1476-5381.1993.tb13469.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller VM, Vanhoutte PM. Endothelial α2-adrenoceptors in canine pulmonary and systemic blood vessels. Eur J Pharmacol. 1985;118:123–129. doi: 10.1016/0014-2999(85)90670-3. [DOI] [PubMed] [Google Scholar]
- Moore PK, al-Swayeh OA, Chong NWS, Evans RA, Gibson A. l-NG-nitro-arginine (l-NOARG), a novel, l-arginine-reversible inhibitor of endothelium-dependent relaxation in vitro. Br J Pharmacol. 1990;99:408–412. doi: 10.1111/j.1476-5381.1990.tb14717.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olken NM, Marletta MA. NG-monomethyl-l-arginine functions as an alternative substrate and mechanism-based inhibitor of nitric oxide synthase. Biochemistry. 1993;32:9677–9685. doi: 10.1021/bi00088a020. [DOI] [PubMed] [Google Scholar]
- Overend J, Martin W. Differential effects of nitric oxide synthase inhibitors on endothelium-dependent and nitrergic nerve-mediated vasodilatation in the bovine ciliary artery. Br J Pharmacol. 2007;150:488–493. doi: 10.1038/sj.bjp.0707113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer RMJ, Ashton DS, Moncada S. Vascular endothelial cells synthesise nitric oxide from l-arginine. Nature. 1988;333:664–666. doi: 10.1038/333664a0. [DOI] [PubMed] [Google Scholar]
- Rapoport RM, Murad F. Agonist-induced endothelium-dependent relaxation may be mediated through cyclic GMP. Circ Res. 1983;52:352–357. doi: 10.1161/01.res.52.3.352. [DOI] [PubMed] [Google Scholar]
- Rees DD, Palmer RMJ, Hodson HF, Moncada S. A specific inhibitor of nitric oxide formation form l-arginine attenuates endothelium-dependent relaxations. Br J Pharmacol. 1989;96:418–424. doi: 10.1111/j.1476-5381.1989.tb11833.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rees DD, Palmer RMJ, Schulz R, Hodson HF, Moncada S. Characterisation of three inhibitors of endothelial nitric oxide synthase in vitro and in vivo. Br J Pharmacol. 1990;101:746–752. doi: 10.1111/j.1476-5381.1990.tb14151.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siroen MPC, Teerlink T, Nijveldt RJ, Prins HA, Richir MC, Leeuwen PAM. The clinical significance of asymmetric dimethylarginine. Annu Rev Nutr. 2006;26:203–228. doi: 10.1146/annurev.nutr.26.061505.111320. [DOI] [PubMed] [Google Scholar]
- Vallance P, Collier J, Moncada S. Effects of endothelium-derived nitric oxide on peripheral arteriolar tone in man. Lancet. 1989;2:997–1000. doi: 10.1016/s0140-6736(89)91013-1. [DOI] [PubMed] [Google Scholar]
- Vallance P, Leone A, Calver A, Collier J, Moncada S. Accumulation of an endogenous inhibitor of nitric oxide synthesis in chronic renal failure. Lancet. 1992;339:572–575. doi: 10.1016/0140-6736(92)90865-z. [DOI] [PubMed] [Google Scholar]
- Vladimirova-Kitova L, Deneva T, Angelova E, Nikolov F, Marinov B, Mateva N. Relationship of asymmetric dimethylarginine with flow-mediated dilatation in subjects with newly detected severe hypercholesterolemia. Clin Physiol Funct Imaging. 2008;28:417–425. doi: 10.1111/j.1475-097X.2008.00825.x. [DOI] [PubMed] [Google Scholar]