

Abstract
Background and purpose:
Previous studies demonstrated that intraplaque haemorrhage increased the contents of cholesterol and oxidants in atherosclerotic plaques. The present study was aimed to test the hypothesis that enhanced expression of haem oxygenase-1 (HO-1) may stabilize vulnerable plaques.
Experimental approach:
Intravascular ultrasound (IVUS) was performed to identify three similar abdominal aortic plaques in each of 58 fat-fed New Zealand rabbits after aortic balloon injury. With the guidance of IVUS, 50 µL autologous erythrocytes (RBC) or normal saline (NS) were injected from adventitia into two of the pre-selected plaques, respectively, whereas the third plaque served as a blank control. All rabbits were randomly divided into two groups, receiving intraperitoneal injection of haemin and saline respectively.
Key results:
Compared with NS or control plaques, RBC plaques had more macrophage infiltration and lipid content, thinner plaque fibrous cap, and higher expression of inflammatory factors and incidence of plaque rupture. RBC plaques in the haemin group had about a 50% lower incidence of plaque rupture than those in the control group.
Conclusions and implications:
Haem oxygenase-1 may eliminate haem or other oxidants, exert unexpected anti-oxidative and anti-inflammatory effects and serve as a promising approach to the direct inhibition of erythrocyte-induced plaque instability.
Keywords: atherosclerosis, erythrocyte, vulnerable plaque, haem oxygenase-1, intraplaque hemorrhage
Introduction
Pathological studies have demonstrated that vulnerable plaques induced by intraplaque hemorrhage are frequently associated with increased density of microvessels (Burke et al., 1999; Kockx et al., 2003) and presence of erythrocyte membranes within the necrotic core (Kolodgie et al., 2003). The number of vasa vasorum was increased twofold and fourfold in vulnerable and ruptured plaques, respectively, as compared with stable plaques. The cholesterol content of the erythrocyte membrane was also found to contribute to the progression of atherosclerosis (Torkhovskaia et al., 1983; Miwa et al., 2003). Lipid contents derived from erythrocytes were associated with large necrotic cores of atherosclerotic plaques with intraplaque hemorrhage (Kolodgie et al., 2003; Takaya et al., 2005). These findings suggest that microvascular disruption or leakage may promote lesion progression and erythrocytes may represent a potent atherogenic stimulus leading to plaque destabilization. In the pathogenesis of erythrocyte-induced atherosclerosis, peroxidation may play an important role. Erythrocyte ‘ghosts’ may accumulate membrane lipid hydroperoxides and render low-density lipoprotein (LDL) more sensitive to free radical-mediated oxidative modification (Vila et al., 2002). Haemoglobin-derived haem acts as a catalyst for the oxidation of LDL (Jeney et al., 2002). The free haem can also lead to a dose-dependent peroxidation of unsaturated lipids in the presence of H2O2 and microsomal membrane fractions (Vincent et al., 1988). In contrast, haem oxygenase-1 (HO-1), as the first and rate-limiting enzyme of haem catabolism, has remarkable anti-oxidative and anti-inflammatory properties (Kadl et al., 2002; Asatryan et al., 2003). Furthermore, HO-1 has anti-atherogenic properties (Hayashi et al., 1999; Siow et al., 1999; Ishikawa et al., 2001a,b; Goto et al., 2002; Liu et al., 2002) that depend not only on elimination of haem and other oxidants but also on its catabolic production of biliverdin IXα, carbon monoxide and bilirubin IX (Siow et al., 1999; Jeney et al., 2002; Liu et al., 2002; Otterbein et al., 2003). In intraplaque hemorrhage, for example, haem and other oxidants derived from erythrocytes accumulate in the plaque and trigger peroxidization and inflammation, and HO-1 is induced in the plaque at the same time. However, little is known about the role of HO-1 in this situation and whether erythrocyte-induced plaque instability would be affected by enhanced expression of HO-1 remains unclear.
In the present study, we used haemin as a tool to enhance HO-1 expression in an intraplaque hemorrhage rabbit model to investigate the relation between HO-1 expression and histological and inflammatory changes in plaques, and to test the hypothesis that enhanced expression of HO-1 may increase the stability of erythrocyte-induced vulnerable plaques.
Methods
Animal model
All animal care and the experimental procedures in this investigation conformed to the Guide for the Care and Use of Laboratory Animals published by the Chinese National Institutes of Health. A total of 58 New Zealand white rabbits, 3–4 months old, were obtained from the Animal Center of Shandong Agriculture Science Academy.
The rabbit model of intraplaque hemorrhage was modified from that previously reported (Kolodgie et al., 2003). After 1 week of a high-cholesterol diet (1% cholesterol and 2% pork lard), 58 rabbits received balloon-induced endothelial injury in the abdominal aorta. The rabbits were continually fed high cholesterol diet for 9 weeks (a total of 10 weeks of atherogenic diet), and then standard rabbit chow for 8 weeks. At the end of week 18, abdominal aortas of rabbits under general anesthesia (intravenous injection of 30 mg·kg−1 sodium pentobarbital through the ear vein) were exposed, and from each rabbit, three similar plaques with similar shape and size were selected under the guidance of intravascular ultrasound (IVUS) (Galaxy, Boston Scientific Corporation, Boston, MA, USA). Blood was drawn from the ear vein of the rabbits and washed three times by normal saline to obtain pure autologous erythrocytes (≈100%). An amount of 50 µL of autologous erythrocytes (RBC) and an equal volume of normal saline (NS) were injected into two of the plaques, respectively, with the third plaque serving as a blank control. The positions of these plaques were marked by an ilio-psoas stitch, and their distances from the bifurcation of the iliac artery were recorded. Infusion of remifentanil at a constant rate of 0.3 µg·kg−1·min−1 was initiated 30 min after wakening from anesthesia in each rabbit.
All rabbits were then randomly divided into two groups. Rabbits in the haemin group (n = 29) were injected i.p. with haemin (Sigma; 25 mg·kg−1 body weight, four times per week) for 6 weeks, and those in the control group (n = 29) were treated with NS for 6 weeks, as previously reported (Ishikawa et al., 2001b). At the end of 24 weeks, after the shape and size of plaques were examined by IVUS again, the abdominal aortas were removed under general anesthesia as mentioned above. Plaques treated with erythrocytes (RBC plaques), NS (NS plaques) or nothing (blank plaques) were separated for histological staining or molecular biology studies.
Biochemical assay
At the beginning of the experiment, week 10 (the end of high cholesterol diet), week 18 (time for erythrocyte injection) and week 24 (the end of the experiment), blood was collected from rabbits fasting overnight to detect plasma levels of total cholesterol (TC), triglycerides (TG), LDL and high-density lipoprotein (HDL). TC, TG, LDL and HDL concentrations were determined enzymatically by use of cholesterol esterase and cholesterol oxidase. Erythrocyte membrane cholesterol was measured at the beginning of the experiment and at 10 and 18 weeks according to a modified procedure (Dodge et al., 1963; Macchia et al., 1991). Briefly, after being removed from plasma and platelets by centrifugation, erythrocytes were dispersed and washed three times with NS. Then double-volume double-distilled water was added and mixed with the erythrocytes. Tubes were left overnight at 4°C to complete haemolysis. Lipids were extracted as described (Macchia et al., 1991) and membrane cholesterol content was determined enzymatically.
Measurement of erythrocyte oxidative status
At the beginning of the experiment, week 10, week 18 weeks and week 24, the superoxide dismutase (SOD) activity and malondialdehyde (MDA) level of erythrocytes was measured by SOD and MDA kits following the manufacturer's instructions (Nanjing Jiancheng Bioengineering Institute, Nanjing, China).
IVUS examination
IVUS study was performed before and after plaque injection by use of a 3.2 F catheter containing a single rotating element transducer of 40 MHz connected to an IVUS system (Galaxy, Boston Scientific Corp, Boston, MA, USA). The image frame frequency was 32 f/s and the axial and lateral resolution was 150 µm and 300 µm respectively. After the catheter reached the aortic arch, it was withdrawn by use of a motorized pullback device at a constant speed of 0.5 mm·s−1. Each IVUS study was carried out according to a standard procedure (Mintz et al., 2001).
At week 18, three similar plaques in the same aorta were selected for injection with erythrocytes, NS or nothing under guidance of IVUS. The process of plaque injection was shown in Figure 1A–C. IVUS was performed at week 18 and week 24 to measure the external elastic membrane area (EEMA) and the lumen area (LA) from 10 equidistant cross-sectional views along the abdominal aorta, and the values were averaged. The percentage of plaque burden (PB%) was calculated as PB% = (EEMA − LA)/EEMA.
Figure 1.
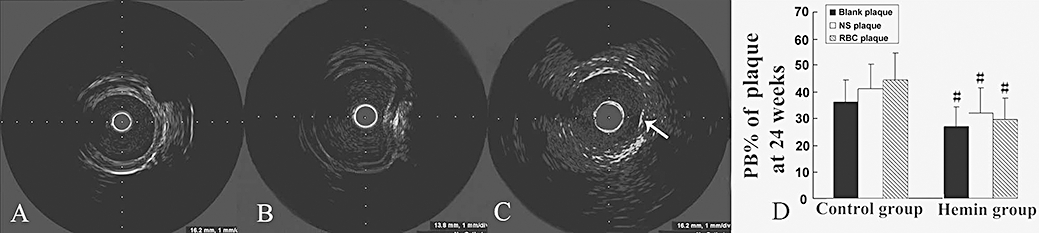
IVUS images of plaque injection and plaque burden (PB%) in two groups of rabbits. (A) before injection; (B) during injection; (C) after injection, a small haematoma appeared in the plaque (arrow); (D) PB% at week 24 in the control and haemin groups. #P < 0.05 versus the same treatment in the control group (control group n = 28, haemin group n = 26). IVUS, intravascular ultrasound; PB, plaque burden.
Histopathological and immunohistochemical staining
Abdominal aortic segments were fixed in 4% formaldehyde, embedded in paraffin, and 5 µm sections were stained with haematoxylin and eosin, Masson's trichrome, Movat pentachrome, Perls (to determine iron), picrosirius red or for immunohistochemical analysis. Cryosections (6 µm) were stained with oil red O to determine lipid content.
Immunohistochemical staining was performed as described previously (Torzewski et al., 1998). Briefly, endogenous peroxidase activity was inhibited by incubation with 3% H2O2. After sections were blocked with 5% (v/v) goat serum in phosphate buffered saline (PBS), sections were incubated overnight at 4°C with the primary antibodies. After a PBS wash, the sections were incubated with secondary antibody at 37°C for 30 min. Visualization of a positive reaction was by means of a peroxidase substrate solution containing 0.02% (wt/vol) H2O2 and 0.1% (wt/vol) 3,3′-diaminobenzidine tetrahydrochloride (ZSBIO, Beijing, China) in PBS to give the reaction product a brown colour, and then sections were counterstained with hematoxylin. A negative control, with the primary antibody replaced by mouse IgG antibody, was always included. The primary antibodies used were monoclonal antibody against rabbit alveolar macrophages (RAM11) (Laboratory Vision Neomakers, Fremont, CA, USA) diluted 1:400 to identify macrophages; isolectin B4 from Bandeiraea simplicifolia conjugated with biotin (GSL-B4) (Vector, Burlingame, CA, USA) diluted at 10 µg·mL−1 to identify erythrocyte membranes; mouse anti-rabbit HO-1 monoclonal antibody (OSA-111) (StressGen, Victoria, BC, Canada) diluted 1:100 to detect HO-1; mouse anti-nuclear transcription factor κB (NF-κB) P65 subunit monoclonal antibody (MAB3026) (Chemicon, Temecula, CA, USA) diluted 1:100 to detect NF-κB, which increases the transcription of cytokines and acute phase proteins; mouse anti-rabbit α-smooth muscle actin (BM0002) (Boster, Wuhan, China) diluted 1:100 to detect α-actins; mouse anti-collagen I monoclonal antibody (ab6308) (Abcam, Cambridge, MA, USA) diluted 1:400 to detect collagen I; mouse anti-collagen III monoclonal antibody (CP19L) (Merck & Co., Inc., Whitehouse, NJ, USA) diluted 1:2000 to detect collagen III; mouse anti-matrix metalloproteinase 9 (MMP-9) monoclonal antibody (sc-21733) (Santa Cruz Biotechnology, Santa Cruz, CA, USA) diluted 1:100 to detect MMP-9, the prediluted mouse monoclonal antibody (Abcam ab74604) to detect CD163 and mouse IgG antibody (ab17890) (Abcam) diluted 1:100 for negative control. Biotinylated secondary antibodies (zb-2020) (Zymed, San Francisco, CA, USA), and avidin-biotin-peroxidase complex (Vectastain ABC KIT, Vector, Burlingame, CA, USA) were also used to detect HO-1 as described previously (Torzewski et al., 1998). Apoptosis was assessed by a rabbit polyclonal antibody against caspase 3 (ab5694, Abcam) and a terminal deoxynucleotidyl transferase end-labelling kit (TUNEL, TA4625, R&D, Minneapolis, MN, USA). The apoptosis rate was expressed as the proportion of apoptotic cells to total number of cells in a given area.
Positive staining area was measured and expressed as a mean percentage of the plaque area in at least 10 high-power fields (×400 magnification) by use of Image-Pro Plus 5.1 software. The mean fibrous cap thickness, measured in 10 equidistant sites per section, and the relative lipid content of plaque was measured as the percentage of immunoreactive area to the total plaque area. Plaque rupture was defined as a buried fibrous cap or thrombi overlying fissured plaques (Williams et al., 2002). The vulnerability index, a well-accepted pathological index of the probability of plaque rupture, was calculated by dividing the relative positive staining area of (macrophages +lipids) by the relative positive staining area of (SMCs + collagen) in a given plaque (Shiomi et al., 2001).
Gelatin zymography
Gelatin zymography was performed as described previously (Lockwood et al., 2008). Briefly, protein extracts were subjected to sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) on gelatin-containing acrylamide gels under non-reducing conditions, and then analysed by electrophoresis at 50 V 4°C for 5 h in Tris-glycine SDS running buffer. To enable the enzymes to re-nature, the gel was incubated for 45 min in 2.5% Triton X-100 at room temperature and then placed in fresh zymogram development buffer with gentle shaking (50 r.p.m.) at 37°C for at least 24 h. The gel was stained with 0.5% Coomassie Billiant Blue solution of methanol/acetic acid/water for 1 h at room temperature and then destained with methanol/acetic acid/water (30:10:60 v/v) at room temperature. The presence of clear bands in the gels at the appropriate molecular weights reflects gelatinolytic activity of MMP 9.
Quantitative real-time RT-PCR
RNA from RBC plaques, NS plaques or control plaques was isolated by use of Trizol reagent (Invitrogen). Total RNA was quantified by spectrophotometry and reversely transcribed with use of the M-MLV Reverse Transcriptase System (Promega, Madison, WI) with oligo(dT) (16) primers. The mRNA sequences of the investigated genes were obtained from GenBank. Quantitative real-time RT-PCR involved use of LightCycler (Roche Applied Science, Indianapolis, IN, USA) following the manufacturer's instructions. The SYBR Green I kit (TaKaRa Biotechnology, Dalian, China) was used for amplification and detection of the expression of HO-1 mRNA, and TaqMan probes were used for detecting the mRNA expression of monocyte chemoattractant protein-1 (MCP-1), vascular cell adhesion molecule-1 (VCAM-1), MMP-9 and tissue inhibitor of metalloproteinase 1 (TIMP-1) (Table 1). Quantitative values were obtained from the threshold cycle value (Ct), the point at which a significant increase of fluorescence was first detected. The transcript number of glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was quantified as an internal control. Experiments were performed in triplicate for each data point and the data were analysed with the 2−ΔΔ CT method (Livak and Schmittgen, 2001). The results of RT-PCR were confirmed by gel electrophoresis. The primers and probes used are listed in Table 1.
Table 1.
Primer and probe for RT-PCR
| Gene | Primer and probe sequence | GenBank accession No. | TA(°C) | |
|---|---|---|---|---|
| GAPDH | S | GAA CGG GAA ACT CAC TGG CAT | AB231852 | 60 |
| A | CCT TCT TGA TGT CGT CAT ACT TAGC | |||
| Probe | CTC CAG GCG GCA GGT CAG GTC CAC | |||
| MCP-1 | S | TCA TAG CAG TCG CCT TCA GC | M57440 | 58 |
| A | GTG TGT TCT TGG GTT GTG GAA TAA | |||
| Probe | CTT GCC CAG CCA GAT GCC GTG AAT | |||
| VCAM-1 | S | AGA ACC CAG ATA GAT AGC CCT CT | AY212510 | 60 |
| A | ACT CAC AGG ACT CAA TGT TAG CA | |||
| Probe | TCC CTT CAC TTG TCA CCT GCC CATT | |||
| TIMP-1 | S | TAG GCT CTG ACA AGG GCT TCC | AY829731 | 56 |
| A | GTA GGC TTC GGC TTC CAA CAG | |||
| Probe | CGC CAT CTC GCC TGC CTG CCAC | |||
| MMP-9 | S | TGT GTC TTC CCC TTC GTC TTC C | D26514 | 66 |
| A | GCC CCA CTT CTT GTC GTC GCT GT | |||
| Probe | GAA GTG GTG GCA CAC CAG AGG CG | |||
| HO-1 | S | AGG TGA CTG CCG AGG GT | AY421756 | 50 |
| A | GGC GTG TAG GGG ATG GT | |||
GAPDH, glyceraldehyde 3-phosphate dehydrogenase; HO-1, haem oxygenase-1; MCP-1, monocyte chemoattractant protein-1; MMP, matrix metalloproteinase; TIMP-1, tissue inhibitor of metalloproteinase 1; VCAM-1, vascular cell adhesion molecule-1.
Statistical analysis
Data were analysed with use of SPSS 11.5 for Windows. All values were expressed as mean ± SD. Plasma lipids, erythrocyte cholesterol, erythrocyte MDA and SOD activity, and gelatinolytic activity of MMP-9 were evaluated by one-way analysis of variance (anova). The incidence of plaque rupture between the two groups was compared by the χ2 test. Measurements of different kinds of plaques in the same groups were analysed by anova for randomized block design, with measurements between groups analysed by unpaired t-test. PB% at week 18 and week 24 was compared by paired t-test. A P-value < 0.05 was considered significant.
Results
Plasma lipid and erythrocyte membrane cholesterol level, erythrocyte MDA level and SOD activity
The mean plasma level of TC, TG, LDL and HDL increased significantly after 10 weeks of atherogenic diet but returned to the baseline level after 8 weeks of normal diet (Figure S1). However, after a high cholesterol diet for 10 weeks, the erythrocyte membrane cholesterol level and MDA level were markedly increased and maintained at a high level up to week 24, even though the plasma lipid level had decreased with the changed diet. In contrast, the erythrocyte SOD activity showed opposing changes.
Plaque measurements by IVUS
At week 24, the plaque burden (PB%) tended to be higher in RBC plaques than in NS or blank plaques in either control or haemin group, although the difference was not significant. However, the PB% in the RBC, NS or blank plaques of the haemin group were all significantly lower than that in the corresponding type of plaques of the control group (Figure 1D).
Histopathological and immunohistochemical staining
Picrosirius red staining showed a thinner fibrous cap in RBC plaques than in NS plaques or blank plaques in both control and haemin groups (all P < 0.05), with no difference between NS and blank plaques. However, the haemin group did not differ from the control group in fibrous cap thickness. The level of total collagen expression was markedly increased in RBC plaques of the haemin group in comparison with those of the control group (Figures 2A,B, 3E). Anti-collagen I and anti-collagen III immunostaining revealed that collagen type III expression was lower whereas collagen type I expression was higher in RBC plaques of the haemin group than those of the control group (Figure 4), suggesting that RBC plaques of the control group contained more newly synthesized collagen whereas those of the haemin group comprised more mature collagen.
Figure 2.
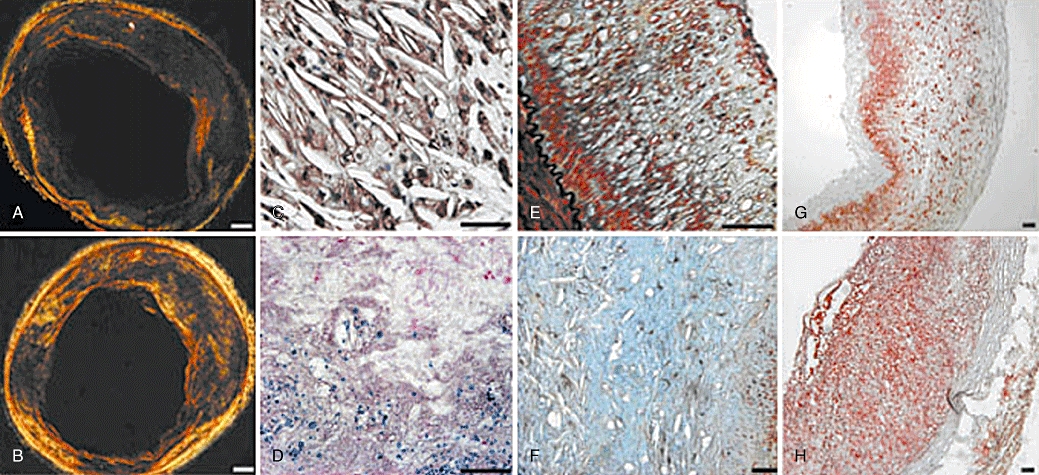
Picrosirus red, GSL-B4, Perls, Movat and Oil red staining of plaques in two groups of rabbits. (A) Picrosirius red staining of RBC plaques in the control group showing rich type III collagen in dark green, and sparse type I or II collagen in red or yellow (bar = 500 µm); (B) Picrosirius red staining of RBC plaques in the haemin group displaying less type III collagen in dark green and more type I or II collagen in red or yellow (bar = 500 µm); (C) GSL-B4 staining in RBC plaques exhibiting numerous cholesterol clefts surrounded by erythrocytes (brown) (bar = 50 µm); (D) Iron deposits (blue) on Perls staining in RBC plaques (bar = 50 µm); (E–F) Movat pentachrome staining of NS plaque (E) (bar = 200 µm) and RBC plaque (F) (bar = 50 µm) demonstrating a large pool of cholesterol clefts surrounded by proteoglycans in blue or green in RBC plaque; (G–H) Oil red staining revealing lower lipid content in NS plaque (G) and higher lipid content in RBC plaque (H) (bar = 200 µm). NS, normal saline; RBC, erythrocyte.
Figure 3.
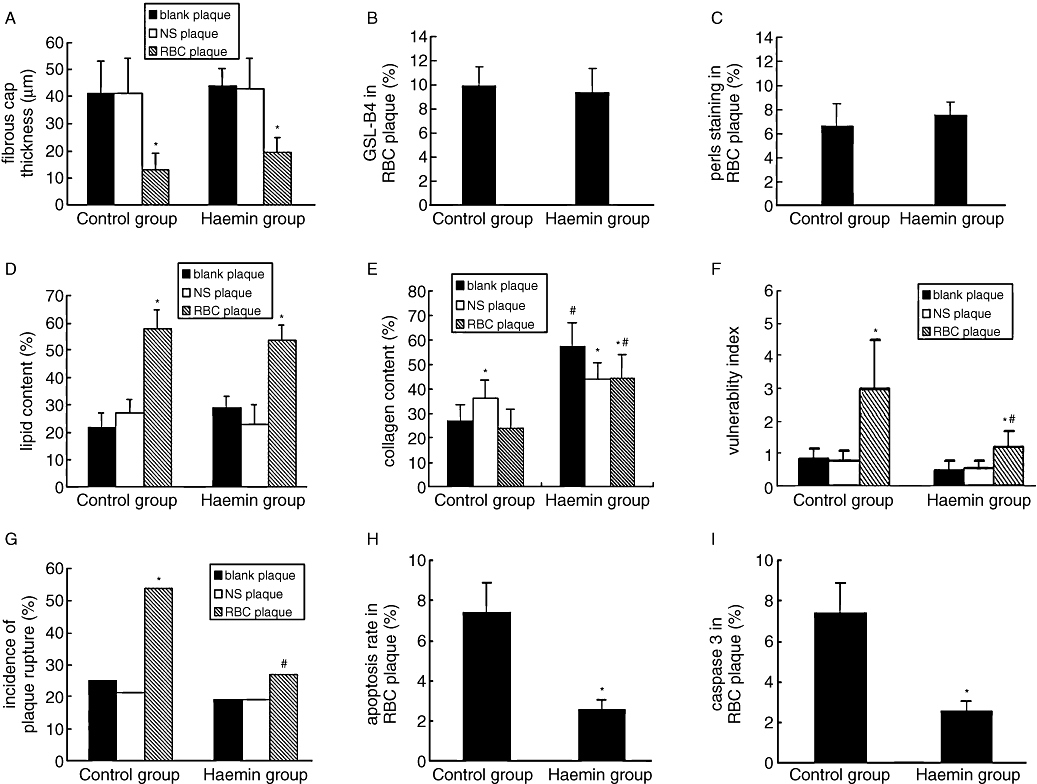
Pathological measurements of atherosclerotic plaques in two groups of rabbits. (A–I) comparison of the fibrous cap thickness (A), relative positive staining of GSL-B4(B), Pearls (C), lipids (D) and collagen (E), vulnerability index (F), incidence of plaque rupture (G), cell apoptosis rate (H) and relative positive staining of caspase 3 (I) among three types of plaques of the control and haemin groups. *P < 0.05 versus blank plaque in the same group; #P < 0.05 versus the same treatment in the control group (control group n = 28, haemin group n = 26).
Figure 4.
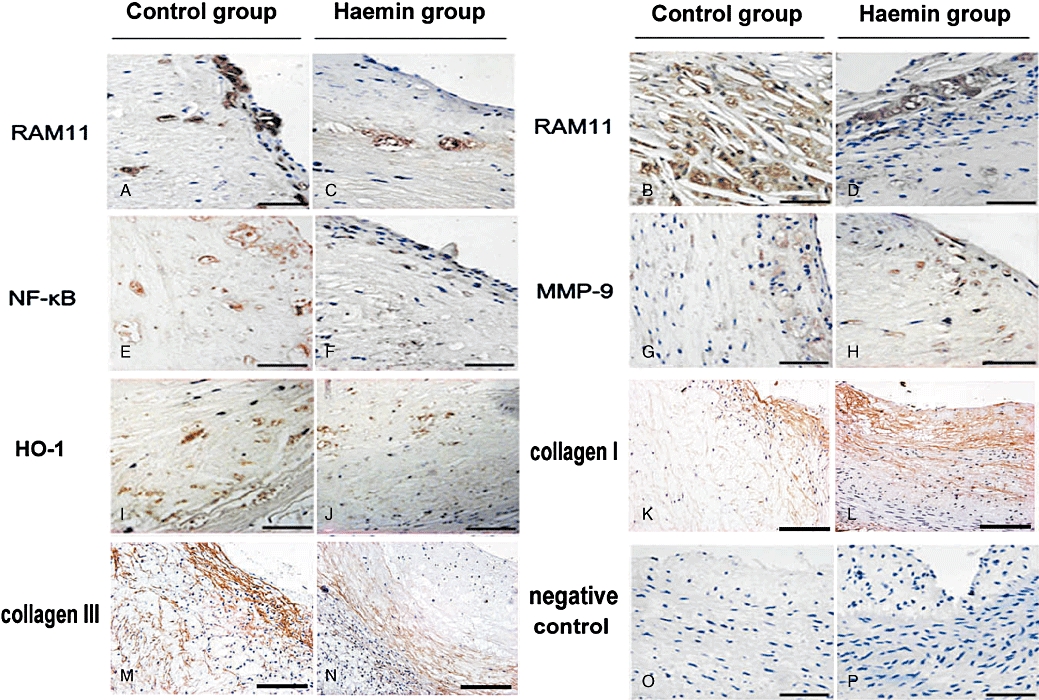
Immunohistochemical staining of plaques in two groups of rabbits. (A–D) RAM11 immunostaining in NS plaques of control group (A), in RBC plaques of control group (B), in NS plaques of haemin group (C) and in RBC plaques of haemin group (D). More extensive macrophage infiltration was seen in RBC plaques than in NS plaques, and less macrophage accumulation was found in the haemin group than the control group (bar = 50 µm); (E–F) NF-κB immunostaining in RBC plaques of the control group (E) and in RBC plaques of the haemin group (F) (bar = 50 µm); (G–H) MMP-9 immunostaining in RBC plaques of the control group (G) and in RBC plaques of the haemin group (H) (bar = 50 µm); (I–J) HO-1 immunostaining in RBC plaques of the control group (I) and in RBC plaques of the haemin group (J) (bar = 50 µm); (K–L) collagen I immunostaining in RBC plaques of the control group (K) and the haemin group (L) (bar = 50 µm); (M–N) collagen III immunostaining in RBC plaques of the control group (M) and the haemin group (N) (bar = 50 µm); (O–P) Mouse IgG negative controls in the control group (K) and the haemin group (L) (bar = 50 µm) (control group n = 28, haemin group n = 26). HO-1, haem oxygenase-1; NS, normal saline; RBC, erythrocyte.
GSL-B4 and Perls staining was positive only in RBC plaques but not in NS or blank plaques in either haemin group or control group, although the positive staining in RBC plaques showed no difference between the haemin group and control group (Figures 2C,D, 3B,C). Movat pentachrome staining revealed few cholesterol crystals in NS or blank plaques but a large amount of cholesterol crystals or lipids and foam cells in RBC plaques (Figure 2E,F).
The relative content of lipids in RBC plaques was markedly higher than that in NS or blank plaques (all P < 0.05) in either haemin group or control group; however, the lipid content showed no significant difference between the NS plaques and blank plaques in both haemin and control groups (Figures 2G,H, 3D).
As revealed by RAM11 immunohistochemistry, macrophage content was higher in RBC plaques than in NS or blank plaques for both groups (P < 0.05) and higher in the control group than in the haemin group (P < 0.05). The results of anti-NF-κB and anti-MMP-9 immunostaining were similar to that of macrophage staining except that anti-MMP-9 staining did not differ between the haemin group and the control group. The positive staining for HO-1 and CD163 was higher in RBC plaques than in NS or blank plaques (P < 0.05) and was much higher in the haemin than the control group (Figures 4A–J, 5, 6A,B).
Figure 5.
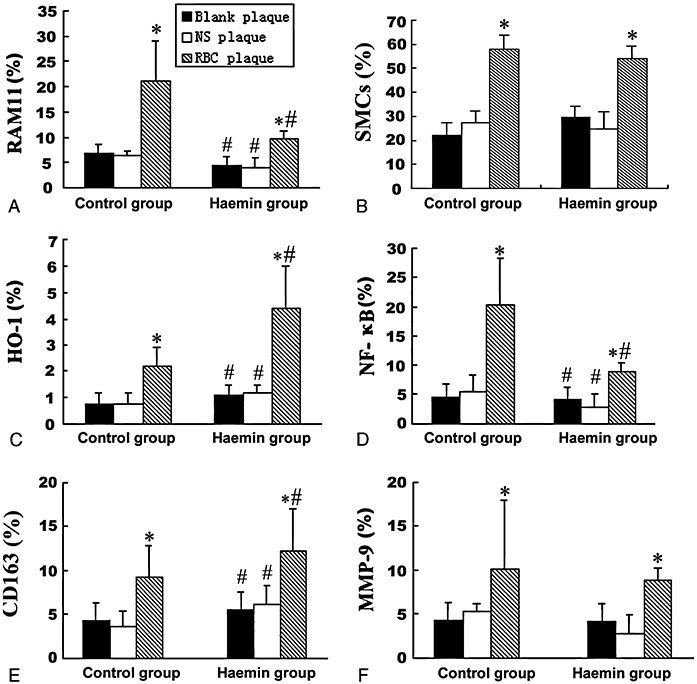
Quantification of immunohistochemical staining in three types of plaques. The score in each diagram was calculated as the percentage of the immunoreactive area to the total cross sectional area. Measurements for RAM11 (A), SMCs (B), HO-1(C), NF-κB (D), CD163 (E) and MMP-9 (F) in three types of plaques of the control and haemin groups were shown. *P < 0.05 versus blank plaque in the same group, #P < 0.05 versus the same treatment in the control group. HO-1, haem oxygenase-1; MMP, matrix metalloproteinase; NF-κB, nuclear transcription factor κB.
Figure 6.
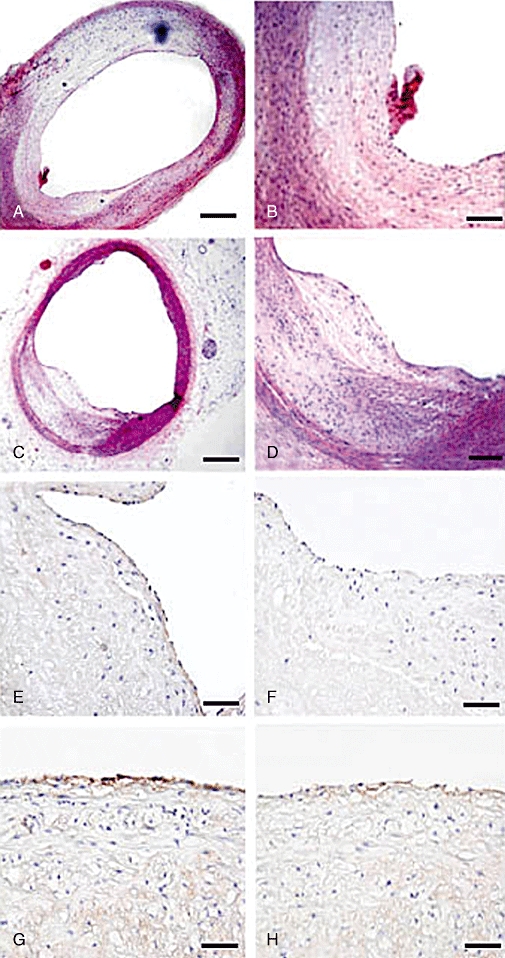
CD163 immunostaining, Masson's trichome staining, TUNEL staining and caspase 3 immunostaining of plaques in two groups of rabbits. (A,B) CD163 staining of RBC plaques in the control (A) and the haemin (B) groups of rabbits (bar = 50 µm); (C) Masson's trichome staining of a RBC plaque in the control group showing an intraluminal thrombus (arrow) overlying a fissured plaque (bar = 100 µm); (D) Masson's trichome staining showing a buried fibrous cap (arrow) of a RBC plaque in the control group (bar = 200 µm); (E–F) TUNEL staining of RBC plaques in the control (E) and the haemin (F) groups of rabbits (bar = 50 µm); (G–H) Caspase 3 immunostaining of RBC plaques in the control (G) and the haemin (H) groups of rabbits (bar = 50 µm). RBC, erythrocyte.
The vulnerability index was higher in RBC plaques than in NS or blank plaques (P < 0.05). However, the vulnerability index in RBC plaques of the haemin group was lower than that of the control group (P < 0.05) (Figure 3E). In the control group but not in the haemin group, the incidence of RBC plaque rupture was higher than that of NS or blank plaques. The incidence of RBC plaque rupture was higher in the control than in the haemin group, but the groups did not differ in the incidence of NS or blank plaque rupture (Figures 3F, 6C,D).
Cell apoptosis shown by TUNEL and positive caspase 3 staining in RBC plaques of the control group was more significant than in RBC plaques of the haemin group (Figures 3G,H, 6E–H), whereas no apoptotic cells were detected in NS or blank plaques of the control or haemin group. Most apoptotic cells were located in the endothelium with their nuclei in a fusiform shape, suggesting they were endothelial cells.
MMP-9 activity measured by gelatin zymography
Gelatin zymography revealed that MMP-9 activity was higher in RBC plaques than in NS or blank plaques for both groups (P < 0.01) and higher in the control group than in the haemin group (P < 0.01) (Figure 7).
Figure 7.
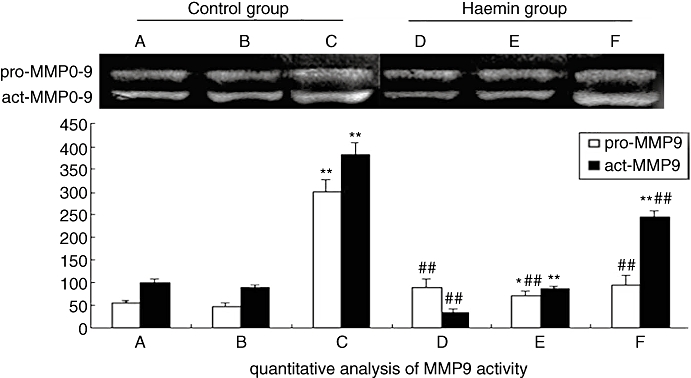
Zymograms of plaque tissue samples in two groups of rabbits. (A) blank plaque in the control group; (B) NS plaque in the control group; (C) RBC plaque in the control group; (D) blank plaque in the haemin group; (E) NS plaque in the haemin group; (F) RBC plaque in the haemin group. *P < 0.05, **P < 0.01 versus blank plaque in the same group; ##P < 0.01 versus the same treatment in the control group (control group n = 28, haemin group n = 26). NS, normal saline; RBC, erythrocyte.
Changes in mRNA expression of the target genes
The level of HO-1 mRNA expression was significantly higher in RBC plaques than in NS or blank plaques and was much higher in the haemin than in the control group (all P < 0.05) (Figure 8A). The relative mRNA expression level of MCP-1, MMP-9 and VCAM-1 in RBC plaques was higher than that in NS or blank plaques in both groups (all P < 0.05), and that of MCP-1 and VCAM-1 was lower in the haemin group than in the control group (all P < 0.05). Although the mRNA expression level of MMP-9 tended to be reduced in RBC plaques of the haemin group compared with that of the control group, the difference did not reach a statistical significance. On the contrary, the TIMP-1 mRNA expression level was lower in RBC plaques than in NS and blank plaques in both groups and was higher in the haemin group than in the control group (all P < 0.05) (Figure 8B–E).
Figure 8.
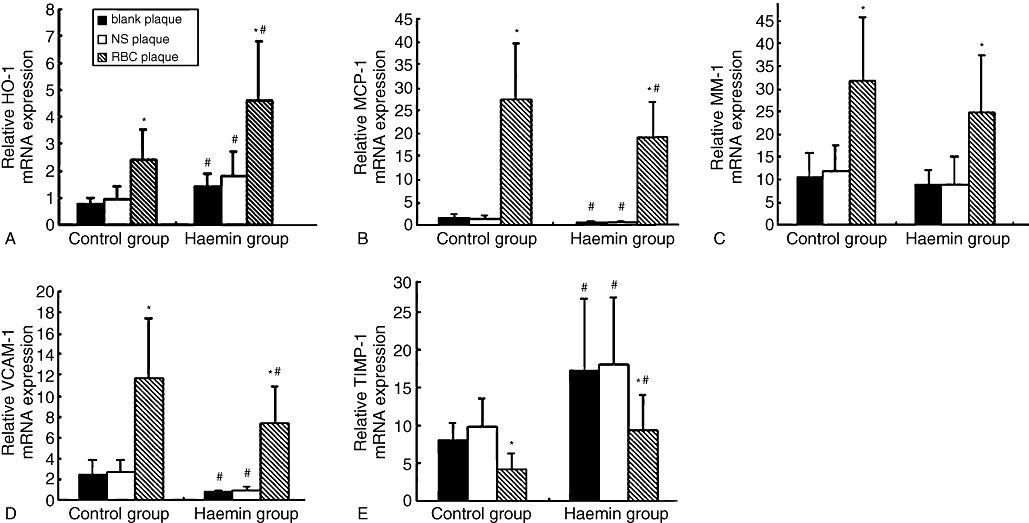
Quantitative real-time RT-PCR in plaques of two groups of rabbits. The relative mRNA expression of HO-1(A), VCAM-1(B), MMP-9(C), TIMP-1(D) and MCP-1(E) in blank, NS and RBC plaques in the control and haemin groups were shown. *P < 0.05 versus blank plaque in the same group; #P < 0.05 versus the same treatment in the control group. (control group n = 28, haemin group n = 26). HO-1, haem oxygenase-1; MCP-1, monocyte chemoattractant protein-1; MMP, matrix metalloproteinase; NS, normal saline; RBC, erythrocyte; TIMP-1, tissue inhibitor of metalloproteinase 1; VCAM-1, vascular cell adhesion molecule-1.
Discussion
A major finding of the present study is that RBC plaques showed greater expression of inflammatory factors and macrophage infiltration, thinner fibrous caps, more lipid content and higher incidence of plaque rupture than blank or NS plaques. In contrast, haemin treatment resulted in higher HO-1 and CD163 expression, more VSMCs and collagen content, less macrophage infiltration, cell apoptosis and PB%, decreased MMP-9 activity, and lower vulnerability index and incidence of plaque rupture than those in the control group, which led to a stabilized phenotype of atherosclerotic plaques. To the best of our knowledge, this is the first study to report the beneficial effects of HO-1 in stabilizing erythrocyte-induced vulnerable plaques.
Another interesting finding of this study was that the plaque burden (PB%) measured by IVUS was substantially lower in blank, NS and RBC plaques of the haemin group than that in the corresponding plaques of the control group (Figure 1D). Although haemin treatment led to a significant improvement of many parameters measured in this study in the RBC plaque, there were still significant differences in these parameters between the RBC plaque and the blank or NS plaque in the haemin group. By comparison, only PB% was similar among the three types of plaques after haemin treatment. These results suggest that enhanced HO-I expression was able to attenuate plaque progression, regardless of plaque type. The mechanism underlying this effect is unclear but may be related to the reduced macrophage infiltration and inflammatory cytokine expression in plaques after haemin treatment as demonstrated in this study.
In the present study, erythrocyte membrane cholesterol and oxidant status increased along with elevated plasma cholesterol level, which was maintained at a high level even after the plasma cholesterol level had decreased to the baseline level. Therefore, once erythrocyte membrane cholesterol and oxidant status increases, it is difficult for these abnormalities to normalize. As erythrocytes were injected into plaques, the lipids derived from the erythrocyte membrane would increase the lipid content of plaques, as shown by oil red O staining. At the same time, the oxidative stress in plaques would be aggravated. Moreover, the breakdown of erythrocytes may result in accumulation of other substances, such as hemoglobin, haem, iron and phospholipids. Haemoglobin, when oxidized, may produce a cytotoxic effect through the generation of oxidized LDL by releasing haem or combining with LDL to form a haemoglobin-modified LDL (HbLDL), which is highly susceptible to oxidation (Asatryan et al., 2003). Haem is a strong pro-oxidant that can trigger peroxidization and induce atherosclerosis (Fernandez et al., 2001). Moreover, iron derived from haem can act as a catalyst in the formation of free radicals, which may contribute to the modification of LDL (Yuan et al., 2004). In addition, phospholipids, once oxidized, can induce expression of genes such as tissue factor, MCP-1, and interleukin-8 via transcription factors such as peroxisome proliferator-activated receptor (PPAR)-α, the nuclear factor of activated T cells (Vila et al., 2002; Kronke et al., 2003). Furthermore, with a large amount of cholesterol and oxidant accumulation in plaques at the same time, a large pool of oxidized-LDL would be available and result in macrophage infiltration and abundant inflammatory factors.
An important pathological feature of vulnerable plaques is intensive inflammation and cellular apoptosis which may involve endothelial cells, SMCs and macrophages. In the current animal model, intraplaque injection of RBC enhanced oxidative stress and macrophage infiltration, which induced cellular apoptosis. The endothelial location and the fusiform nuclei of apoptotic cells suggested that they were likely to originate from endothelial cells. Our recent studies in p53 gene-induced vulnerable plaques showed that cell apoptosis may inversely aggravate the inflammatory process by increasing the production and release of inflammatory molecules including VCAM-1, MCP-1 and proteolytic enzymes (Chen et al., 2007; Zhang et al., 2009). Acting synergistically, these cytokines may stimulate macrophage recruitment and production of many inflammatory and cytotoxic molecules from macrophages. In contrast, haemin treatment substantially attenuated oxidative stress, inflammation and cellular apoptosis, which may in turn inhibit macrophage production.
Using our intraplaque haemorrhage model, we demonstrated the presence of erythrocyte membrane and iron deposition in RBC plaques which paralleled the extent of macrophage and inflammatory cytokine staining. The mRNA expression level of VCAM-1, MCP-1 and MMP-9 was increased and that of TIMP-1 decreased, which confirmed that erythrocytes acted as a source of inflammation in the plaque. Our zymogram data demonstrated that the expression levels of both pro-MMP-9 and active-MMP-9 were significantly elevated in RBC plaques of the control group and after haemin treatment, the pro-MMP-9 level returned to normal and the active-MMP-9 level was significantly lowered in RBC plaques of the haemin group. These data indicated that the synthesis and activity of MMP-9 were strikingly increased in RBC plaques of the control group but were markedly attenuated in RBC plaques of the haemin group. With the increased expression of inflammatory cytokines, more inflammatory cells would be recruited in the plaque and more toxic materials such as MMPs would be excreted to degrade the matrix and attenuate the fibrous caps (de Nooijer et al., 2006), leading to a thin fibrous cap in RBC plaques.
In this study, HO-1 expression was higher in RBC plaque than NS and blank plaque in the control group. CD163 as a receptor binds haptoglobin and haemoglobin in complex and scavenges haemoglobin by mediating endocytosis of the haptoglobin-haemoglobin complex. Such a receptor-ligand interaction activates intracellular signalling pathways and up-regulates HO-1 expression (Kristiansen et al., 2001; Abraham and Drummond, 2006). Despite the increased HO-1 and CD163 expression, macrophage infiltration, expression of other inflammatory factors and the incidence of plaque rupture in RBC plaques were still greater than that of control plaques. Similar results were obtained in previous studies. Ijäs et al. (2007) studied symptomatic and asymptomatic carotid plaques from the same patient and found that symptomatic plaques show a more pronounced induction of HO-1 and CD163 in response to plaque haemorrhages. In contrast, when a competitive inhibitor of HO-1 was introduced to Watanable heritable hyperlipidemic rabbits, the aortic atherosclerotic lesions were markedly aggravated when compared with a control group, proving that HO-1 has anti-atherogenic properties in vivo (Ishikawa et al., 2001a). Taken together, these results indicate that physiologically up-regulated HO-1 expression in atherosclerotic plaques is not sufficient to counteract the adverse effects of enhanced oxidative stress induced by intraplaque hemorrhage. In contrast, only pharmacologically enhanced HO-1 expression can effectively attenuate local oxidative stress and stabilize vulnerable plaques as demonstrated in the present study.
After treatment with haemin, an exogenous inducer of HO-1, the animals exhibited a markedly increased level of HO-1 and CD163 expression in comparison with the control group. The extent of macrophage infiltration, the expression level of VCAM-1, MCP-1 and NF-κB were significantly reduced while that of TIMP-1 was significantly increased in the haemin group compared with the control group. In addition, the activity of MMP-9 was decreased in the haemin group as demonstrated by zymography. These results were found not only in plaques without erythrocytes, as was previously reported (Goto et al., 2002; Kadl et al., 2002; Asatryan et al., 2003; Yet et al., 2003), but also in RBC plaques, which suggests that HO-1 exerts an anti-inflammatory property in plaques. More importantly, the RBC plaques of the haemin group had a much lower vulnerability index and incidence of plaque rupture than that of the control group. However, the decreased vulnerability index and incidence of plaque rupture were not found in the NS or control plaques in the two groups. The attenuated RBC plaque vulnerability may be due to the effect of HO-1 in eliminating more haem, oxidized phospholipids or other oxidants, and producing more biliverdin and bilirubin, which may inhibit LDL oxidation and scavenge oxygen radicals from phospholipids, triglycerides and cholesterol esters (Clark et al., 2000; Stocker, 2004). They suppress the oxygen radicals to a greater extent than α-tocopherol, which provides powerful protection against lipid peroxidation (Stocker et al., 1987; Ishikawa et al., 2001b; Jeney et al., 2002; Liu et al., 2002). The changes of toxic substances into beneficial factors may prevent the progression of atherosclerosis and increase the stability of plaques, leading to a decreased incidence of plaque rupture. Unfortunately, however, the activity of HO-1 in plaques was not measured in the present study. Moreover, haemin as a non-specific inducer of HO-1 may cause other gene changes. Because of these limitations, further studies are required to specifically up-regulate or down-regulate the expression of HO-1 and to attest the causal relationship between HO-1 expression and plaque stability.
Our study has important clinical implications. Previous studies revealed that plaques with intraplaque haemorrhage at baseline are more likely to have new plaque haemorrhages at 18 months and repeated bleeding into the plaque may produce a stimulus for the progression of atherosclerosis by enlarging the lipid core and creating new destabilizing factors (Takaya et al., 2005). Therefore, once intraplaque haemorrhage occurs, the plaque would be involved in a vicious cycle, finally resulting in plaque rupture. Consequently, finding an efficient way to interrupt the pathological progress is imperative and HO-1, as revealed in our study, appears to be promising therapeutic target for preventing the intraplaque hemorrhage-induced progression of atherosclerosis.
In conclusion, using a modified intraplaque hemorrhage model, we found that erythrocytes could enhance the instability of plaques by increasing the level of cholesterol and oxidants in plaques. Once highly expressed, HO-1 may eliminate haem or other oxidants, exert unexpected anti-oxidative and anti-inflammatory effects and serve as a promising approach to the direct inhibition of erythrocyte-induced plaque instability.
Acknowledgments
We gratefully acknowledge the assistance of Professor Jie Zhen Wang in statistical analysis. The study was supported by the National 973 Basic Research Program of China (No. 2006CB503803), the National High-tech Research and Development Program of China (No. 2006AA02A406), the Program of Introducing Talents of Discipline to Universities (No. B07035), the State Key Program of National Natural Science of China (No. 60831003, No. 60971023), grant of Natural Science Foundation of Shandong Province (No. Y2007C064, No. ZR2009CM021), the Specialized Research Fund for the Doctoral Program of Higher Education of China (Grant No. 20090131120066) and the Cultivation Fund of the Key Scientific and Technical Innovation Project,Ministry of Education of China (No. 704030).
Glossary
Abbreviations:
- EEMA
external elastic membrane area
- GAPDH
glyceraldehyde 3-phosphate dehydrogenase
- HDL
high-density lipoprotein
- HO-1
haem oxygenase-1
- IVUS
intravascular ultrasound
- LA
lumen area
- LDL
low-density lipoprotein
- MCP-1
monocyte chemoattractant protein-1
- MDA
malondialdehyde
- MMP
matrix metalloproteinase
- NF-κB
nuclear transcription factor κB
- NS
normal saline
- PB
plaque burden
- PBS
phosphate buffered saline
- RBC
erythrocyte
- SDS-PAGE
sodium dodecyl sulphate-polyacrylamide gel electrophoresis
- SOD
superoxide dismutase
- TC
total cholesterol
- TG
triglycerides
- TIMP-1
tissue inhibitor of metalloproteinase 1
- VCAM-1
vascular cell adhesion molecule-1
Conflict of interest
The authors declare no conflict of interest.
Supporting information
Additional Supporting Information may be found in the online version of this article:
Figure S1 Measurements of biochemical assay and erythrocyte oxidative status. Shown in the diagrams were serum levels of TC (A), TG (B), HDL(C) and LDL (D), erythrocyte membrane cholesterol level (E), and erythrocyte MDA level (F) and SOD (G) activities at week 0, 10, 18 and 24 in all rabbits. *P < 0.05, **P < 0.01. (control group n = 28, haemin group n = 26).
{kind=link}
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Abraham NG, Drummond G. CD163-Mediated hemoglobin-heme uptake activates macrophage HO-1, providing an antiinflammatory function. Circ Res. 2006;99:911–914. doi: 10.1161/01.RES.0000249616.10603.d6. [DOI] [PubMed] [Google Scholar]
- Asatryan L, Ziouzenkova O, Duncan R, Sevanian A. Heme and lipid peroxides in hemoglobin-modified low-density lipoprotein mediate cell survival and adaptation to oxidative stress. Blood. 2003;102:1732–1739. doi: 10.1182/blood-2003-01-0293. [DOI] [PubMed] [Google Scholar]
- Burke AP, Farb A, Malcom GT, Liang Y, Smialek JE, Virmani R. Plaque rupture and sudden death related to exertion in men with coronary artery disease. J Am Med Assoc. 1999;281:921–926. doi: 10.1001/jama.281.10.921. [DOI] [PubMed] [Google Scholar]
- Chen WQ, Zhang L, Liu YF, Chen L, Ji XP, Zhang M, et al. Prediction of atherosclerotic plaque ruptures with high-frequency ultrasound imaging and serum inflammatory markers. Am J Physiol Heart Circ Physiol. 2007;293:H2836–H2844. doi: 10.1152/ajpheart.00472.2007. [DOI] [PubMed] [Google Scholar]
- Clark JE, Foresti R, Green CJ, Motterlini R. Dynamics of haem oxygenase-1 expression and bilirubin production in cellular protection against oxidative stress. Biochem J. 2000;348(Pt 3):615–619. [PMC free article] [PubMed] [Google Scholar]
- Dodge JT, Mitchkll C, Hanahan DJ. The preparation and chemical characteristics of hemoglobin-free ghosts of human erythrocytes. Arch Biochem Biophys. 1963;100:119–130. doi: 10.1016/0003-9861(63)90042-0. [DOI] [PubMed] [Google Scholar]
- Fernandez AZ, Lopez F, Tablante A, Romano E, Hurt-Camejo E, Camejo G, et al. Intravascular hemolysis increases atherogenicity of diet-induced hypercholesterolemia in rabbits in spite of heme oxygenase-1 gene and protein induction. Atherosclerosis. 2001;158:103–111. doi: 10.1016/s0021-9150(01)00422-1. [DOI] [PubMed] [Google Scholar]
- Goto J, Ishikawa K, Kawamura K, Watanabe Y, Matumoto H, Sugawara D, et al. Heme oxygenase-1 reduces murine monocrotaline-induced pulmonary inflammatory responses and resultant right ventricular overload. Antioxid Redox Signal. 2002;4:563–568. doi: 10.1089/15230860260220058. [DOI] [PubMed] [Google Scholar]
- Hayashi S, Takamiya R, Yamaguchi T, Matsumoto K, Tojo SJ, Tamatani T, et al. Induction of heme oxygenase-1 suppresses venular leukocyte adhesion elicited by oxidative stress: role of bilirubin generated by the enzyme. Circ Res. 1999;85:663–671. doi: 10.1161/01.res.85.8.663. [DOI] [PubMed] [Google Scholar]
- Ijäs P, Nuotio K, Saksi J, Soinne L, Saimanen E, Karjalainen-Lindsberg ML, et al. Microarray analysis reveals overexpression of CD163 and HO-1 in symptomatic carotid plaques. Arterioscler Thromb Vasc Biol. 2007;27:154–160. doi: 10.1161/01.ATV.0000251991.64617.e7. [DOI] [PubMed] [Google Scholar]
- Ishikawa K, Sugawara D, Goto J, Watanabe Y, Kawamura K, Shiomi M, et al. Heme oxygenase-1 inhibits atherogenesis in Watanabe heritable hyperlipidemic rabbits. Circulation. 2001a;104:1831–1836. doi: 10.1161/hc3901.095897. [DOI] [PubMed] [Google Scholar]
- Ishikawa K, Sugawara D, Wang X, Suzuki K, Itabe H, Maruyama Y, et al. Heme oxygenase-1 inhibits atherosclerotic lesion formation in ldl-receptor knockout mice. Circ Res. 2001b;88:506–512. doi: 10.1161/01.res.88.5.506. [DOI] [PubMed] [Google Scholar]
- Jeney V, Balla J, Yachie A, Varga Z, Vercellotti GM, Eaton JW, et al. Pro-oxidant and cytotoxic effects of circulating heme. Blood. 2002;100:879–887. doi: 10.1182/blood.v100.3.879. [DOI] [PubMed] [Google Scholar]
- Kadl A, Huber J, Gruber F, Bochkov VN, Binder BR, Leitinger N. Analysis of inflammatory gene induction by oxidized phospholipids in vivo by quantitative real-time RT-PCR in comparison with effects of LPS. Vascul Pharmacol. 2002;38:219–227. doi: 10.1016/s1537-1891(02)00172-6. [DOI] [PubMed] [Google Scholar]
- Kockx MM, Cromheeke KM, Knaapen MW, Bosmans JM, De Meyer GR, Herman AG, et al. Phagocytosis and macrophage activation associated with hemorrhagic microvessels in human atherosclerosis. Arterioscler Thromb Vasc Biol. 2003;23:440–446. doi: 10.1161/01.ATV.0000057807.28754.7F. [DOI] [PubMed] [Google Scholar]
- Kolodgie FD, Gold HK, Burke AP, Fowler DR, Kruth HS, Weber DK, et al. Intraplaque hemorrhage and progression of coronary atheroma. N Engl J Med. 2003;349:2316–2325. doi: 10.1056/NEJMoa035655. [DOI] [PubMed] [Google Scholar]
- Kristiansen M, Graversen JH, Jacobsen C, Sonne O, Hoffman HJ, Law SK, et al. Identification of the haemoglobin scavenger receptor. Nature. 2001;409:198–201. doi: 10.1038/35051594. [DOI] [PubMed] [Google Scholar]
- Kronke G, Bochkov VN, Huber J, Gruber F, Blüml S, Fürnkranz A, et al. Oxidized phospholipids induce expression of human heme oxygenase-1 involving activation of cAMP-responsive element-binding protein. J Biol Chem. 2003;278:51006–51014. doi: 10.1074/jbc.M304103200. [DOI] [PubMed] [Google Scholar]
- Liu XM, Chapman GB, Peyton KJ, Schafer AI, Durante W. Carbon monoxide inhibits apoptosis in vascular smooth muscle cells. Cardiovasc Res. 2002;55:396–405. doi: 10.1016/s0008-6363(02)00410-8. [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C (T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Lockwood CJ, Oner C, Uz YH, Kayisli UA, Huang SJ, Buchwalder LF, et al. Matrix metalloproteinase 9 (MMP9) expression in preeclamptic decidua and MMP9 induction by tumor necrosis factor alpha and interleukin 1 beta in human first trimester decidual cells. Biol Reprod. 2008;78:1064–1072. doi: 10.1095/biolreprod.107.063743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macchia T, Mancinelli R, Barbini DA, Taggi F, Avico U, Cantafora A. Determination of membrane cholesterol in normal and pathological red blood cells. Clin Chim Acta. 1991;199:59–67. doi: 10.1016/0009-8981(91)90009-2. [DOI] [PubMed] [Google Scholar]
- Mintz GS, Nissen SE, Anderson WD, Bailey SR, Erbel R, Fitzgerald PJ, et al. American College of Cardiology Clinical Expert Consensus Document on Standards for Acquisition, Measurement and Reporting of Intravascular Ultrasound Studies (IVUS). A report of the American College of Cardiology Task Force on Clinical Expert Consensus Documents. J Am Coll Cardiol. 2001;37:1478–1492. doi: 10.1016/s0735-1097(01)01175-5. [DOI] [PubMed] [Google Scholar]
- Miwa S, Inouye M, Ohmura C, Mitsuhashi N, Onuma T, Kawamori R. Relationship between carotid atherosclerosis and erythrocyte membrane cholesterol oxidation products in type 2 diabetic patients. Diabetes Res Clin Pract. 2003;61:81–88. doi: 10.1016/s0168-8227(03)00108-6. [DOI] [PubMed] [Google Scholar]
- de Nooijer R, Verkleij CJ, von der Thusen JH, Jukema JW, van der Wall EE, van Berkel TJ, et al. Lesional overexpression of matrix metalloproteinase-9 promotes intraplaque hemorrhage in advanced lesions but not at earlier stages of atherogenesis. Arterioscler Thromb Vasc Biol. 2006;26:340–346. doi: 10.1161/01.ATV.0000197795.56960.64. [DOI] [PubMed] [Google Scholar]
- Otterbein LE, Zuckerbraun BS, Haga M, Liu F, Song R, Usheva A, et al. Carbon monoxide suppresses arteriosclerotic lesions associated with chronic graft rejection and with balloon injury. Nat Med. 2003;9:183–190. doi: 10.1038/nm817. [DOI] [PubMed] [Google Scholar]
- Shiomi M, Ito T, Hirouchi Y, Enomoto M. Stability of atheromatous plaque affected by lesional composition: study of WHHL rabbits treated with statins. Ann N Y Acad Sci. 2001;947:419–423. doi: 10.1111/j.1749-6632.2001.tb03977.x. [DOI] [PubMed] [Google Scholar]
- Siow RC, Sato H, Mann GE. Heme oxygenase-carbon monoxide signalling pathway in atherosclerosis: anti-atherogenic actions of bilirubin and carbon monoxide? Cardiovasc Res. 1999;41:385–394. doi: 10.1016/s0008-6363(98)00278-8. [DOI] [PubMed] [Google Scholar]
- Stocker R. Antioxidant activities of bile pigments. Antioxid Redox Signal. 2004;6:841–849. doi: 10.1089/ars.2004.6.841. [DOI] [PubMed] [Google Scholar]
- Stocker R, Yamamoto Y, McDonagh AF, Glazer AN, Ames BN. Bilirubin is an antioxidant of possible physiological importance. Science. 1987;235:1043–1046. doi: 10.1126/science.3029864. [DOI] [PubMed] [Google Scholar]
- Takaya N, Yuan C, Chu B, Saam T, Polissar NL, Jarvik GP, et al. Presence of intraplaque hemorrhage stimulates progression of carotid atherosclerotic plaques: a high-resolution magnetic resonance imaging study. Circulation. 2005;111:2768–2775. doi: 10.1161/CIRCULATIONAHA.104.504167. [DOI] [PubMed] [Google Scholar]
- Torkhovskaia TI, Khodzhakuliev BG, Khalilov EM, Kasatkina LV, Polesskii VA. Na+, K+-ATPase activity and cholesterol content in erythrocyte membranes of patients with coronary atherosclerosis in various forms of dyslipoproteinemia. Vopr Med Khim. 1983;29:69–73. [PubMed] [Google Scholar]
- Torzewski M, Klouche M, Hock J, Messner M, Dorweiler B, Torzewski J, et al. Immunohistochemical demonstration of enzymatically modified human LDL and its colocalization with the terminal complement complex in the early atherosclerotic lesion. Arterioscler Thromb Vasc Biol. 1998;18:369–378. doi: 10.1161/01.atv.18.3.369. [DOI] [PubMed] [Google Scholar]
- Vila A, Korytowski W, Girotti AW. Spontaneous transfer of phospholipid and cholesterol hydroperoxides between cell membranes and low-density lipoprotein: assessment of reaction kinetics and prooxidant effects. Biochemistry. 2002;41:13705–13716. doi: 10.1021/bi026467z. [DOI] [PubMed] [Google Scholar]
- Vincent SH, Grady RW, Shaklai N, Snider JM, Muller-Eberhard U. The influence of heme-binding proteins in heme-catalyzed oxidations. Arch Biochem Biophys. 1988;265:539–550. doi: 10.1016/0003-9861(88)90159-2. [DOI] [PubMed] [Google Scholar]
- Williams H, Johnson JL, Carson KG, Jackson CL. Characteristics of intact and ruptured atherosclerotic plaques in brachiocephalic arteries of apolipoprotein E knockout mice. Arterioscler Thromb Vasc Biol. 2002;22:788–792. doi: 10.1161/01.atv.0000014587.66321.b4. [DOI] [PubMed] [Google Scholar]
- Yet SF, Layne MD, Liu X, Chen YH, Ith B, Sibinga NE, et al. Absence of heme oxygenase-1 exacerbates atherosclerotic lesion formation and vascular remodeling. FASEB J. 2003;17:1759–1761. doi: 10.1096/fj.03-0187fje. [DOI] [PubMed] [Google Scholar]
- Yuan XM, Li W, Baird SK, Carlsson M, Melefors O. Secretion of ferritin by iron-laden macrophages and influence of lipoproteins. Free Radic Res. 2004;38:1133–1142. doi: 10.1080/10715760400011692. [DOI] [PubMed] [Google Scholar]
- Zhang L, Liu Y, Lu X, Xu X, Zhao Y, Ji X, et al. Intraplaque injection of Ad5-CMV.p53 aggravates local inflammation and leads to plaque instability in rabbits. J Cell Mol Med. 2009;13:2713–2723. doi: 10.1111/j.1582-4934.2008.00538.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.