

Abstract
Background and purpose:
Although trace amines (TAs) are historically considered ‘false neurotransmitters’ on the basis of their ability to induce catecholamine release, there is evidence that they directly affect neuronal activity via TA receptors, ligand-gated receptor channels and/or σ receptors. Here, we have investigated the effects of two TAs, tyramine (TYR) and β-phenylethylamine (β-PEA), on electrophysiological responses of substantia nigra pars compacta (SNpc) dopaminergic cells to the D2 receptor agonist, quinpirole.
Experimental approach:
Electrophysiological recordings of D2 receptor-activated G-protein-gated inward rectifier K+ channel (GIRK) currents were performed on dopaminergic cells from midbrain slices of mice and on Xenopus oocytes expressing D2 receptors and GIRK channels.
Key results:
TYR and β-PEA reversibly reduced D2 receptor-activated GIRK currents in a concentration-dependent manner on SNpc neurones. The inhibitory effect of TAs was still present in transgenic mice with genetically deleted TA1 receptors and they could not be reproduced by the selective TA1 agonist, o-phenyl-3-iodotyramine (O-PIT). Pretreatment with antagonists of σ1 and σ2 receptors did not block TA-induced effects. In GTPγS-loaded neurones, the irreversibly-activated GIRK-current was still reversibly reduced by β-PEA. Moreover, β-PEA did not affect basal or dopamine-evoked GIRK-currents in Xenopus oocytes.
Conclusions and implications:
TAs reduced dopamine-induced responses on SNpc neurones by acting at sites different from TA1, σ-receptors, D2 receptors or GIRK channels. Although their precise mechanism of action remains to be identified, TAs, by antagonizing the inhibitory effects of dopamine, may render dopaminergic neurones less sensitive to autoreceptor feedback inhibition and hence enhance their sensitivity to stimulation.
Keywords: dopaminergic neurone, dopamine D2 receptors, trace amine associated receptor 1, σ receptors, tyramine, β-phenylethylamine
Introduction
The trace amines (TAs), tyramine, β-phenylethylamine (β-PEA), octopamine and tryptamine, are a class of endogenous compounds similar to classical monoamine neurotransmitters in their structure, metabolism and tissue distribution (Berry, 2004). Several lines of evidence suggest a link between TAs and the mesencephalic dopaminergic system. Thus, a relatively high level of tyramine (TYR) and β-PEA is present in nigrostriatal and mesolimbic regions of the brain (Boulton, 1976; Paterson et al., 1990). Moreover, β-PEA is synthesized by dopaminergic neuron (Greenshaw et al., 1986) and specific high affinity binding sites for β-PEA (Richard et al., 1982) and TYR (Vaccari, 1986) have been described in the striatum and other dopamine-rich areas. These observations are consistent with the notion that TAs might play an important role in the control of movement, mood, reward and cognition (Boulton, 1980; Davis and Boulton, 1994; Bergman et al., 2001; Sotnikova et al., 2008). Furthermore, a dysregulation in the actions of TAs has been linked to various neuropsychiatric disorders including schizophrenia, attention-deficit/hyperactivity disorders, depression and Parkinson's disease, all of which are characterized by altered activity of monoaminergic networks (Boulton, 1980; Davis and Boulton, 1994; Branchek and Blackburn, 2003; Burchett and Hicks, 2006; Millan, 2006)
Although TAs are phylogenetically older than classical biogenic amines, as mentioned earlier, they are structurally related and share metabolic pathways (Paterson et al., 1990). In fact, TAs are primarily synthesized by the enzyme L-amino acid decarboxylase (AADC) and levels in the brain depend on the activity of monoamine oxidase (MAO). Interestingly, TAs are stored in nerve terminals with classical biogenic amines and can be co-released in a vesicular and non-vesicular manner (Kosa et al., 2000). In spite of these intriguing observations, the physiological roles and mechanisms of action of TAs in the mammalian brain remain poorly understood (Borowsky et al., 2001; Bunzow et al., 2001; Premont et al., 2001; Branchek and Blackburn, 2003; Burchett and Hicks, 2006; Millan, 2006).
In fact, the best-characterized effect of TAs consists of their ability to release, in an amphetamine-like manner, catecholamines via their displacement from synaptic vesicles and reversal of plasma membrane transporters. Furthermore, of particular pertinence to dopaminergic transmission, it has been demonstrated that TAs are able to release dopamine from synaptosomes (Raiteri et al., 1977) and striatal brain slices (Dyck, 1983). Moreover, in vivo studies (Philips and Robson, 1983; Bailey et al., 1987) have shown that β-PEA releases dopamine in the striatum (Philips and Robson, 1983) and nucleus accumbens (Nakamura et al., 1998) and induces ipsilateral rotation in unilateral 6-hydroxydopamine-lesioned animals (Barroso and Rodriguez, 1996). Consistent with these findings, we have observed inhibitory effects of TYR and β-PEA on dopaminergic perikarya that appear to be mediated by the extracellular release of dopamine (Geracitano et al., 2004). Moreover, indirect effects of TYR, mediated by the release of dopamine acting at D2-like receptors, depolarize subthalamic neurones and depress GABAergic transmission (Zhu et al., 2007).
An additional interesting dimension to the physiology of TAs is the possibility that they may exert direct actions independently from their neurotransmitter releasing properties. Their modulation of neuronal activity could be related to the activation of G protein-coupled receptors (GPCRs) and/or ligand-gated ion channels. Indeed a novel family of TA receptors was recently identified of which two, TA1 and TA2 (Maguire et al., 2009), were found to be sensitive to TAs (Borowsky et al., 2001; Bunzow et al., 2001). TA1 are positively coupled to Gs protein, leading to the intracellular accumulation of cAMP upon activation of adenylyl cyclase. Interestingly, recent experimental evidence suggests that TA1 (also called ‘TA associated receptor 1’, TAAR1) modulate monoaminergic transmission (Sotnikova et al., 2008; Lindemann et al., 2008; Xie and Miller, 2009) by an effect on monoamine transporters (Xie et al., 2007, 2008; Xie and Miller, 2009) and the firing activity of mesencephalic dopaminergic neurones (Lindemann et al., 2008).
Apart from TA1 receptors coupled to G-proteins, it is conceivable that ionotropic receptors are activated by TAs as, in invertebrates, ligand-gated chloride channels are opened by TAs (Pirri et al., 2009; Ringstad et al., 2009). Moreover, it has been demonstrated that both β-PEA and TYR activate σ receptors (Fontanilla et al., 2009). σ-Receptors were initially described as a subtype of opioid receptor (Martin et al., 1976) but are now recognized to be a distinct family comprising two subtypes (σ1 and σ2) possessing distinct biochemical and pharmacological profiles (Itzhak et al., 1991; Monnet and Maurice, 2006). Therefore, TAs may fine-tune neuronal activity by directly acting on σ receptors. In fact, the activation of σ1 and σ2 receptors has been reported to inhibit various types of K+ and Ca2+ ion channels (Nguyen et al., 1998; Zhang and Cuevas, 2002) through direct protein-protein interactions in a G-protein-independent manner unrelated to protein kinases (Lupardus et al., 2000; Zhang and Cuevas, 2005).
From the comments mentioned earlier, it appears that TAs may exert a direct influence upon neuronal activity. In the present manuscript, we present evidence that both TYR and β-PEA reduce dopamine-induced responses in midbrain dopaminergic neurones via a G protein-independent signalling mechanisms mediated neither by TA1 nor by σ receptors.
Methods
Slice preparations
The electrophysiological recordings were made from dopaminergic neuron of SNpc in acute slices of ventral midbrain obtained using standard procedures (Mercuri et al., 1995). C57BL6 mice and TA1 KO mice (background C57BL6; 18–25 days old) were anaesthetized with halothane and decapitated. All experiments were carried out in accordance with the international guidelines on the ethical use of animals from the European Communities Council Directive of 24 November 1986 (86/609/EEC) and Comitato Etico of the Tor Vergata University, Rome. The brain were rapidly removed from the skull and a tissue block containing the midbrain was mounted on an agar block and immerged in cold artificial cerebrospinal fluid (aCSF) at 8–10 C°. The aCSF contained 126 mM NaCl, 2.5 mM KCl, 1.2 mM MgCl2, 2.4 mM CaCl2, 1.2 mM NaH2PO4, 24 mM NaHCO3 and 10 mM glucose and was saturated with 95% O2–5% CO2 (pH 7.4). Horizontal slices (250 µm) of the ventral midbrain, containing the substantia nigra, were cut using a vibratome (Leica VT1000S, Leica Microsystems, Wetzlar, Germany). Slices were maintained in aCSF at 33.0 ± 0.5 C° for 45 min before being transferred to the recording chamber.
Electrophysiology
Intracellular recordings from midbrain SNpc dopaminergic neurones were made with sharp microelectrodes. The experiments were performed at 33.0 ± 0.5 C° in a recording chamber, containing aCSF flowing at a rate of 2.5–3 mL·min−1 and continuously oxygenated, on the stage of an upright microscope (Axioscope FS, Zeiss, Gottingen, Germany), equipped for infrared video microscopy (Hamamatsu, Tokyo, Japan) in order to allow the direct visualization of the recorded neuron. Neurones, selected for their location and morphology, were identified as dopaminergic by their electrophysiological properties, such as the presence of a regular spontaneous firing activity (0.5–4 Hz), a large inward current (Ih) in response to hyperpolarizing voltages and a membrane hyperpolarization/outward current caused by the application of dopamine (10–30 µM) or quinpirole (10–300 nM) (Mercuri et al., 1995). The electrodes used were filled with 2 M KCl and had a tip resistance of 30–80 MΩ. Recordings were made with an Axoclamp 2B amplifier (Axon instruments, Foster City, CA, USA) using Clampex software and a Digidata 1200B connected to a PC.
Voltage clamp experiments were performed while maintaining the membrane voltage between –60 and –65 mV by hyperpolarizing current injection in order to avoid spontaneous spike triggering. The single-electrode voltage-clamp technique (Lacey et al., 1987) was used in preference to the whole-cell patch-clamp technique because the D2 receptor-mediated outward current could be stably followed for more then 1 h with little fading with the former method. In some experiments, current–voltage (I/V) relationships were constructed by applying voltage pulses of 14–16 mV increments from about −45 to −130 mV in voltage-clamped neurones in control conditions, during the application of quinpirole and the co-application of quinpirole plus β-PEA.
For the experiments performed to verify the potential involvement of G protein, the dopaminergic neurones were loaded with GTPγS (1 mM) dissolved in the microelectrode pipette filling solution. To inhibit dopa decarboxylase and MAO B, midbrain slices were incubated in aCSF (at 33.0 ± 0.5 C°) containing carbidopa (300 µM) and selegiline (30–100 nM), respectively, for more than an hour to ensure complete enzymatic inhibition and then transferred to the recording chamber.
Oocytes preparation and RNA injection
Xenopus laevis care and handling were in accordance with the highest standards of institutional guidelines in compliance with both national and international laws and policies. Frogs underwent no more than two surgical procedures, separated by at least 3 weeks. Frogs were anaesthetized with an aerated solution of 3-aminobenzoic acid ethyl ester. Stages V–VI oocytes were isolated and stored at 16°C in fresh ND96 medium (NaCl 96 mmol·L−1, KCl 2 mmol·L−1, MgCl2 1 mmol·L−1, CaCl2 1.8 mmol·L−1, HEPES 5 mmol·L−1, gentamicin 50 µg·mL−1). D2 receptors and Kir 3.2 channel subunits composing GIRK2 channels the same as those expressed in SNpc neurones of adult mice were subcloned into the oocyte expression vector pBF, which provides 5’ and 3’ untranslated regions from the Xenopusβ-globin gene flanking a polylinker containing multiple restriction sites. Capped mRNAs were synthesized in vitro by using the SP6 mMESSAGE mMACHINE kit (Ambion). Kir3.2 mRNA (50 nL) and D2 receptor mRNA (50 nL) were injected into the oocytes (Nanoject, Drummond, Broomall, PA, USA). All the reagents were supplied by Sigma (Milan, Italy).
Electrophysiology
Electrophysiological recordings and data analysis were performed as previously described (Pessia et al., 1996). Briefly, two-electrode voltage-clamp recordings (TEVC) were obtained from Xenopus oocytes at ∼22°C, 2–8 days after injection. A GeneClamp 500 amplifier (Axon Instruments) interfaced to a PC computer with an ITC-16 interface (Instrutech Corp., Port Washington, NY USA) was used. Microelectrodes were filled with KCl 3 M and had resistances of 0.1–0.5 MΩ. Standard recording solution contained 90 mM KCl, 3 mM MgCl2, 10 mM Hepes (pH 7.4) was used, unless otherwise stated. Currents were evoked by voltage commands from a holding potential of –10 mV, delivered in –10 mV increments from +50 to –120 mV, unless otherwise stated. The recordings were filtered at 2 kHz and acquired at 5 kHz with Pulse software (HEKA elektronik GmbH, Lambrecht, Germany). Data analysis was performed by using IGOR (Wavemetrics, Lake Oswego, OR, USA), PulseFit (HEKA elektronik GmbH) and KaleidaGraph (Synergy Software, Reading, PA, USA). Leak and capacitative currents were subtracted using a P/4 protocol.
Mice genetically lacking TA1 receptors
Mice genetically deleted of TA1 receptors were custom-generated by GenOway (Lyon, France) employing a conventional beta-lac strategy, as employed by others (Lindemann et al., 2008). Briefly, the TA1 receptor coding sequence was replaced by a cDNA encoding IRES-LacZ-NeoTk cassette. 129SvPas ES cells were transfected and chimeras obtained from C57BL/6 blastocysts. Homozygous mice showing a pure C57BL/6J genetic background were bred and back-crossed for 8 generations prior to utilization of congenic knockout and wild-type mice (De Groote et al., 2007; Millan et al., 2008). Adult knockout mice were healthy and normal with no apparent phenotype, hence indistinguishable from their wild-type counterparts. The effectiveness of gene deletion was validated by employing standard histoenzymological detection of b-galactosidase on 30 µm saggital brain sections of knockout mice, as well as real-time polymerase chain reaction (PCR) quantification of TA1 receptor transcripts in defined (micropunched) cerebral regions, including the SNpc (Millan et al., 2008).
Drugs
All drugs were prepared in stock solutions and bath-applied at known concentrations via a three-way tap system. A complete exchange of the solution in the recording chamber occurred in about 1 min. In some experiments GTPγS (1 mM) was infused into the cytosol through the recording pipettes. The following substances were used: dopamine hydrochloride, β-phenylethylamine (β-PEA), tyramine (TYR), d-amphetamine sulphate (AMPH), cocaine hydrochloride, S-(–)-carbidopa, R-(–)-selegiline hydrochloride, GTPγS tetralithium salt were purchased from Sigma. (–)-Quinpirole hydrochloride, SM-21 maleate, BD1047 dihydrobromide were obtained from Tocris Cookson Inc. (Bristol, UK). o-Phenyl-3-iodotyramine (O-PIT) was synthesized by Servier chemists.
Data analysis
Numerical data are expressed as mean ± SEM. Student's t-test for paired and unpaired observations was used to compare data. The threshold level of significance for all analyses was P < 0.05. To estimate the IC50 and maximal response, concentration-response curves were fitted using the logistic equation Y = a/{1 + exp[–(x–x0)/b]} where a is maximal effect, b is minimum effect, x is drug concentration, x0 is the concentration attaining half-maximal effect.
Nomenclature
The nomenclature of all molecular targets (receptors, ion channels, enzymes, etc.) cited in this work conforms to the British Journal of Pharmacology's Guide to Receptors and Channels (Alexander et al., 2009).
Results
TAs reduce D2-activated responses on dopaminergic neurones
Intracellular recordings with sharp microelectrodes were made from dopaminergic neurones in the mice SNpc (n = 92). The electrophysiological and pharmacological characteristics of these cells have been described previously (Lacey et al., 1989; Mercuri et al., 1995).
Bath application of dopamine (10–30 µM) and the selective D2 receptor agonist, quinpirole (0.01–1 µM), activated the somatodendritic receptors, which led to a membrane hyperpolarization/outward current because of opening of Gi/o–gated K+ channels (Lacey et al., 1987). In the present study, we routinely perfused quinpirole (300 nM) to induce a maximal and constant GIRK activation in single-electrode voltage-clamped neurones held at –60 mV, and evaluated whether the co-application of TAs modulated the quinpirole-induced outward current. Both β-PEA and TYR reversibly reduced the amplitude of the sustained outward current (78.9 ± 3.6 pA) caused by the continuous perfusion of quinpirole (300 nM) (n = 59). In particular, co-application (4–7 min) of β-PEA (10–300 µM) or TYR (30–300 µM) inhibited the D2 receptor-activated GIRK current (Figure 1A, D). The effect of the TAs peaked at 3–5 min and washed out in 8–15 min. It was reproducible and concentration-dependent (Figure 1A, B, E). The IC50 value for β-PEA was 82.9 ± 9.6 µM and the maximal inhibition produced by this drug was obtained at a concentration of 300 µM (84.2 ± 8.3%, n = 8). Interestingly, an inhibitory effect similar to that of TAs was produced by the dopamine releaser AMPH (30 µM; Figure 1C, F) but not by the dopamine reuptake blocker cocaine (10 µM; Figure 1D). In particular AMPH perfusion (30 µM, n = 7) reduced the D2 receptor-activated GIRK currents by 41.2 ± 2.1%, whereas cocaine (10 µM, n = 4) was unable to inhibit quinpirole-induced currents and to prevent the depressant effects produced by TAs (Figure 1D, F).
Figure 1.
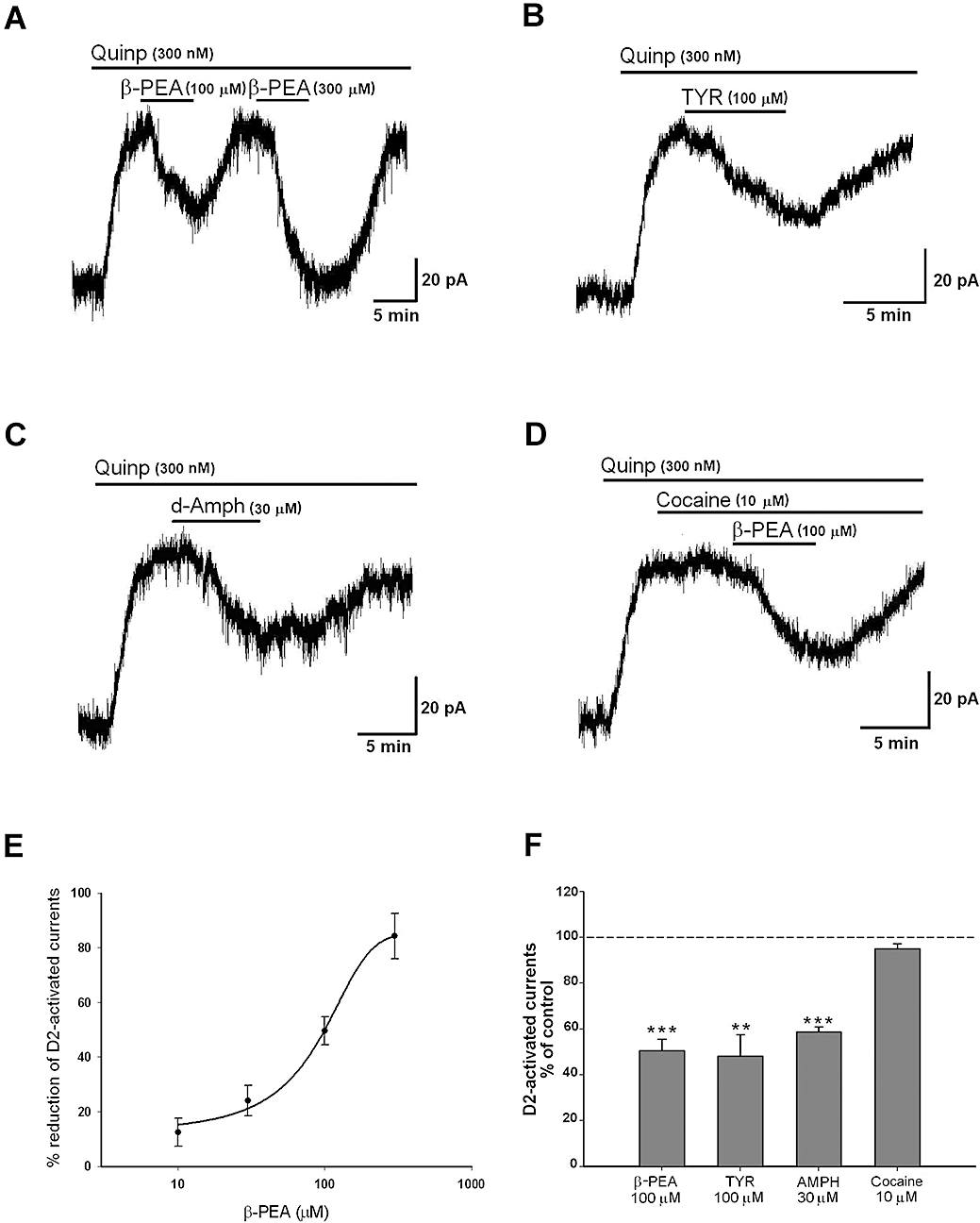
(A) Representative recordings showing the inhibitory effect of β-phenylethylamine (β-PEA) (A) and tyramine (TYR) (B) on the quinpirole-induced current. Effects were mimicked by d-amphetamine (AMPH) (C) but not by cocaine (D). Of note, cocaine does not modify the action of β-PEA. From the concentration-response curve shown in (E), the value of the IC50 was 82.9 ± 9.6 µM for β-PEA (n = 6–8 cells for each concentration). (F) The columns indicate the normalized residual quinpirole-induced current (% of control) observed in the presence of β-PEA (100 µM, n = 17, P < 0.001), TYR (100 µM, n = 10, P < 0.005), AMPH (30 µM, n = 7, P < 0.001) and cocaine (10 µM, n = 4, P > 0.05).
In order to verify whether a reduction of TA catabolism could influence their inhibitory effect on the quinpirole-activated GIRK current, we performed recordings in midbrain slices preincubated with the MAO B inhibitor, selegiline. Indeed, in slices preincubated with selegiline (30–100 nM for more then 1 h) the inhibitory effect on GIRK currents was achieved at lower concentrations of the TAs. The concentration-response curve for β-PEA was shifted to the left (Figure 2A) and the IC50 value reduced to 19.7 ± 8.5 µM (n = 7) indicating that inhibition of MAOs reduces the effective doses of the TAs.
Figure 2.
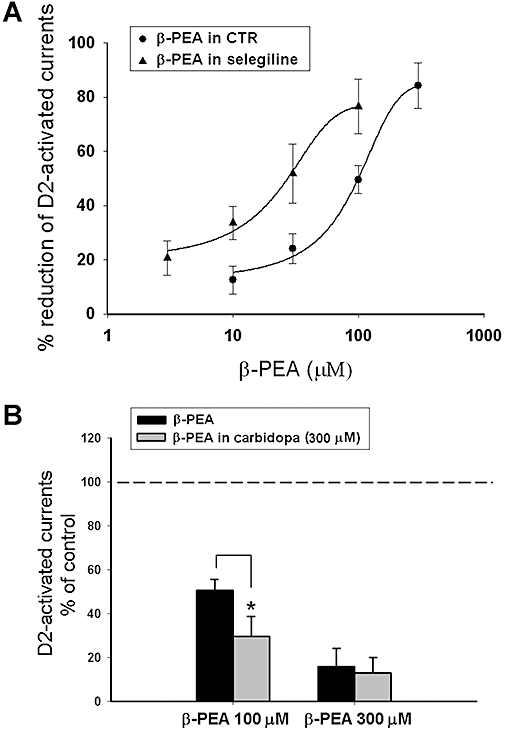
Effects of β-phenylethylamine (β-PEA) in the presence of enzymatic inhibition of catabolic and synthesizing pathways. (A) The concentration-response curve of β-PEA was shifted to the left in the presence of the monoamine oxidase (MAO) B inhibitor selegiline (100 nM) (n = 7 cells per concentration). (B) The L-amino acid decarboxylase (AADC) inhibitor carbidopa did not modify the effect of 300 µM β-PEA but increased the responses to 100 µM β-PEA. The enhancing effect of carbidopa at the lower concentration of β-PEA (100 µM) could be because of a drop in the endogenous level of amines caused by this AADC inhibitor and thus, a more marked effect of the exogenously applied trace amine is detected (*P < 0.05; the results depicted by the columns were obtained from n = 7 experiments).
In another set of experiments, the irreversible AADC inhibitor carbidopa was used to reduce the synthesis of both dopamine and TAs. Carbidopa, which abolishes the indirect dopamine-mediated effects of TAs on dopaminergic neurones (Geracitano et al., 2004), by reducing the rate of synthesis of TAs, might unmask a greater effect of exogenously applied TAs. In fact, under these conditions the TAs still inhibited the D2 receptor-activated GIRK current and the percentage reduction was slightly higher compared with control conditions (Figure 2B). In particular β-PEA (100 µM) produced an inhibition of 73.3 ± 10.4% (n = 7) in carbidopa-treated cells versus 49.5 ± 5.1% (n = 18; P < 0.05) in control cells.
The effects of TYR and β-PEA are not caused by the activation of TA1 receptors
It was possible that the effects of TAs on D2 receptor-induced outward current were the result of the activation of TA1 receptors. We addressed this issue by utilizing mice genetically knocked-out for TA1, as verified both by PCR quantification of cerebral levels of TA1 (complexly absent in the SNpc and other CNS regions of knockout vs. wild-type mice) and of beta-lac (present in knockout vs. wild-type mice) (Millan et al., 2008). The depressant effects of TYR (n = 5), β-PEA (n = 7) and AMPH (n = 4) were still present in these mice (Figure 3Aa, b). The TAs-induced depression was similar to that observed in C57BL6 wild-type mice both for the kinetics and the percentages of reduction obtained (Figure 3Ab). Specifically in TA1 KO mice, β-PEA (100 µM) and TYR (100 µM) reduced the quinpirole-induced current to 25.5 ± 9.8% (n = 7) and to 56.6 ± 11.8% (n = 5) of control values, respectively, whereas in wild-type mice corresponding doses inhibited GIRK currents to 49.5 ± 5.1% (n = 18, P < 0.05) and 48.1 ± 9.3% (n = 10, P > 0.05) of control.
Figure 3.
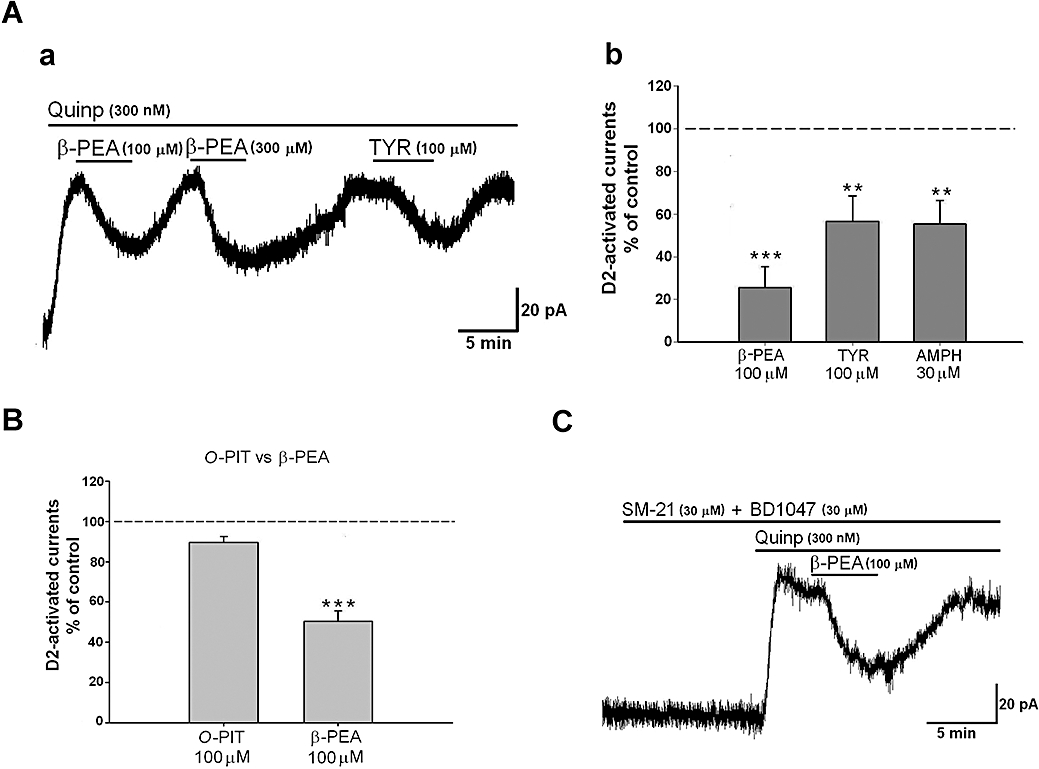
(A) (a) Representative trace showing that a consistent and reproducible inhibition of the quinpirole-induced outward current was caused by β-phenylethylamine (β-PEA; 100–300 µM) and tyramine (TYR; 100 µM) in a dopaminergic neurone obtained from a TA1 knockout mice. (b) A similar depression was caused by d-amphetamine (AMPH) (30 µM). (B) The histogram shows that the specific TA1 agonist o-phenyl-3-iodotyramine (O-PIT; 100 µM) had no effect on the D2 receptor-mediated outward response. In contrast to β-PEA (P < 0.001 for O-PIT vs. β-PEA), an infusion of O-PIT (100 µM) induced only a small but non-significant reduction of G protein-gated inward rectifier K+ channel (GIRK) currents (n = 6, P > 0.05). (C) Representative trace showing that β-PEA still inhibits the quinpirole-induced outward current in the presence of the σ2 antagonist, SM-21 (30 µM) and the σ1 antagonist, BD1047 (30 µM).
In addition, in normal mice, the selective agonist of TA1O-PIT (Hart et al., 2006), bath applied at a concentration of 100 µM (n = 6) for more than 5 min, was not able to mimic the reduction of the D2 receptor-mediated responses induced by the TAs (Figure 3B). β-PEA 100 µM induced a reduction of 49.5 ± 5.1% whereas that to O-PIT 100 µM was 10.3 ± 2.9% (Figure 3B).
The effects of TYR and β-PEA are not caused by activation of σ receptors
The next step was to investigate whether the inhibitory effect induced by TAs on quinpirole-activated GIRK currents was caused by the activation of σ receptors. Recently, it was shown that both β-PEA and TYR can bind and activate σ receptors (σ1 and σ2), displaying the highest affinities for the σ2 receptors (Fontanilla et al., 2009). We tested this possibility by superfusing β-PEA with a cocktail of σ1 and σ2 antagonists. The concomitant perfusion of the σ2 antagonist, SM-21 (30 µM for 10 min) with the selective σ1 antagonist, BD1047 (30 µM for 10 min) had no effect on the depressant effect of β-PEA (Figure 3C) on the quinpirole-induced current (n = 4, 46.3 ± 6.1% in presence of antagonists vs. 49.5 ± 5.1% in control conditions, P > 0.05).
TAs reduce D2-activated GIRK current through a G-protein independent mechanism
In order to ascertain whether the D2 receptor-mediated outward current is negatively modulated by TAs, we generated current–voltage plots in control conditions during the perfusion of quinpirole and quinpirole plus β-PEA. As expected for an involvement of GIRK the crossing of the resultant net currents was –101 mV (Figure 4A) (Lacey et al., 1987).
Figure 4.
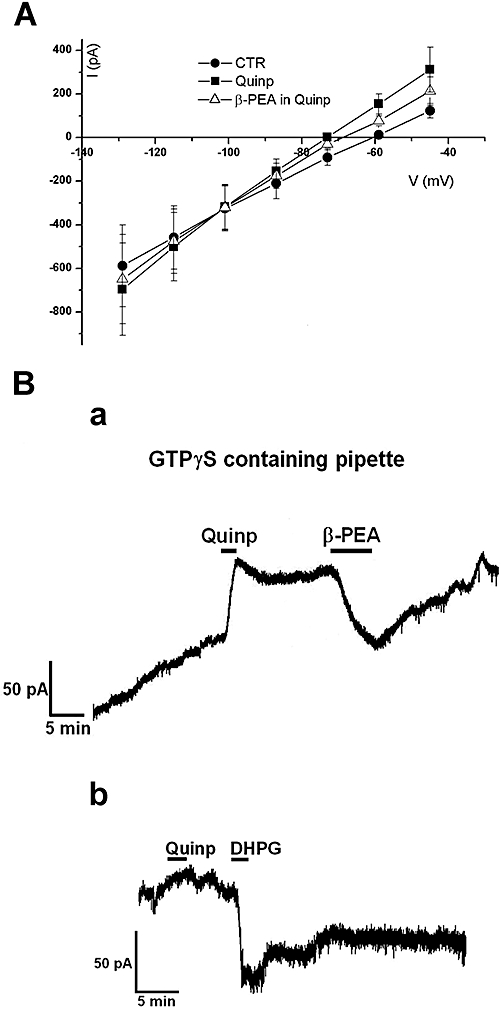
Trace amines reduced the D2 receptor-activated G protein-gated inward rectifier K+ channel (GIRK) current through a G-protein independent mechanism. (A) The current–voltage relationships performed by voltage steps in control conditions, in quinpirole and in quinpirole plus β-phenylethylamine (β-PEA; 100 µM) demonstrate that the quinpirole-induced outward currents and the β-PEA-induced inward current (quinp plus β-PEA) all reversed at –101 mV. Note that the point of intersection for the currents in the three different conditions above is close to K+ reversal potentials. Each point represents the mean ± SEM (n = 4). (B) (a) Representative current trace showing that the intracellular diffusion of GTPγS in a dopaminergic neurone caused a progressive outward current that was augmented by quinpirole (300 nM). Under this condition a relative short application of the D2 receptor agonist caused a sustained outward current that was transiently depressed by β-PEA (100 µM). (b) Noteworthy, whereas the inward current caused by β-PEA washed-out, the one induced by the mGluR 1 agonist DHPG (10 µM) was sustained indicating the involvement of G-protein in the latter case.
To further characterize the mechanism by which TAs depress the D2 receptor-activated GIRK current we analysed the potential involvement of G-protein signalling pathways. Hence, we loaded dopaminergic neurones with the non-hydrolyzable GTP analogue, GTP-γ-S trilithium salt (GTPγS, 1 mM). It has been demonstrated previously that intracellular GTPγS diffusion in dopaminergic neurones leads to activation of a sustained outward current because of progressive and permanent activation of Gi/o proteins (Lacey et al., 1987). Under these conditions, the stimulation of D2 receptors results in an irreversible outward response suggesting a persistent GIRK opening (Figure 4Ba). The mean D2 receptor-mediated outward current was 183 ± 32 pA (n = 5) whereas the subsequent application of β-PEA (100 µM, 5 min) induced a sustained inward current of 95 ± 18 pA (n = 5), which was reversible (wash out 15 min). Additional experiments were performed in GTPγS-treated neurones using the GABAB agonist, baclofen (10 µM), which activates the same current as dopamine (Lacey et al., 1988). Under these conditions, the outward current of 187 ± 22 pA (n = 3), irreversibly activated by baclofen, was transiently depressed by β-PEA (100 µM, 5 min; not illustrated).
In contrast, the superfusion of the mGluR agonist DHPG (10 µM), superimposed on the quinpirole-induced outward current, caused an inward current (Guatteo et al., 1999) that did not wash out, implying that other G-protein-mediated events (presumably Gq, see Discussion, Cho et al., 2005) could be still irreversibly activated when GTPγS is infused into the cells (Figure 4Bb).
TAs do not affect GIRK currents expressed in oocytes
In order to test the effect of β-PEA on Kir3.2 potassium channels (GIRK2) activity, RNA coding for Kir3.2 channels was injected into Xenopus laevis oocytes and the resulting currents were recorded under TEVC configuration. Figure 5A shows representative current traces recorded from an oocyte expressing Kir3.2 channels before (CTRL) and after the superfusion with a solution containing β-PEA (200 µM). The application of β-PEA did not affect the Kir3.2 potassium current recorded at –120 mV. (Figure 5A, C). In particular, the Kir3.2 potassium current in control conditions was 580 ± 140 nA (n = 12), whereas after β-PEA application it was 610 ± 210 nA (n = 12, P > 0.05). In contrast, the application of Ba2+ (200 µM), a Kir channel blocker, inhibited the current amplitudes readily (Figure 5B, C). Therefore, to determine whether β-PEA only modulates the dopamine-evoked potassium current, Kir3.2 were co-expressed with D2 receptors and the effect of β-PEA on the dopamine-evoked Kir3.2 potassium current was tested. Figure 5D shows representative whole-cell current traces recorded from an oocyte co-expressing Kir3.2 and D2 receptors under basal condition (CTRL), during the superfusion of dopamine (20 µM), β-PEA (200 µM) and after wash-out (W/O). Dopamine (20 µM) increased basal Kir3.2 potassium currents (1130 ± 300 nA, n = 10) ∼1.5-fold (1700 ± 300 nA, n = 10, P < 0.001) in a reversible manner (Werner et al., 1996), whereas β-PEA did not affect dopamine-evoked currents (Figure 5D, F). Indeed, after the application of β-PEA, the dopamine-evoked current was 1500 ± 320 nA (n = 10). Similar negative effects were obtained with a superfusion of TYR (200 µM; not shown).
Figure 5.
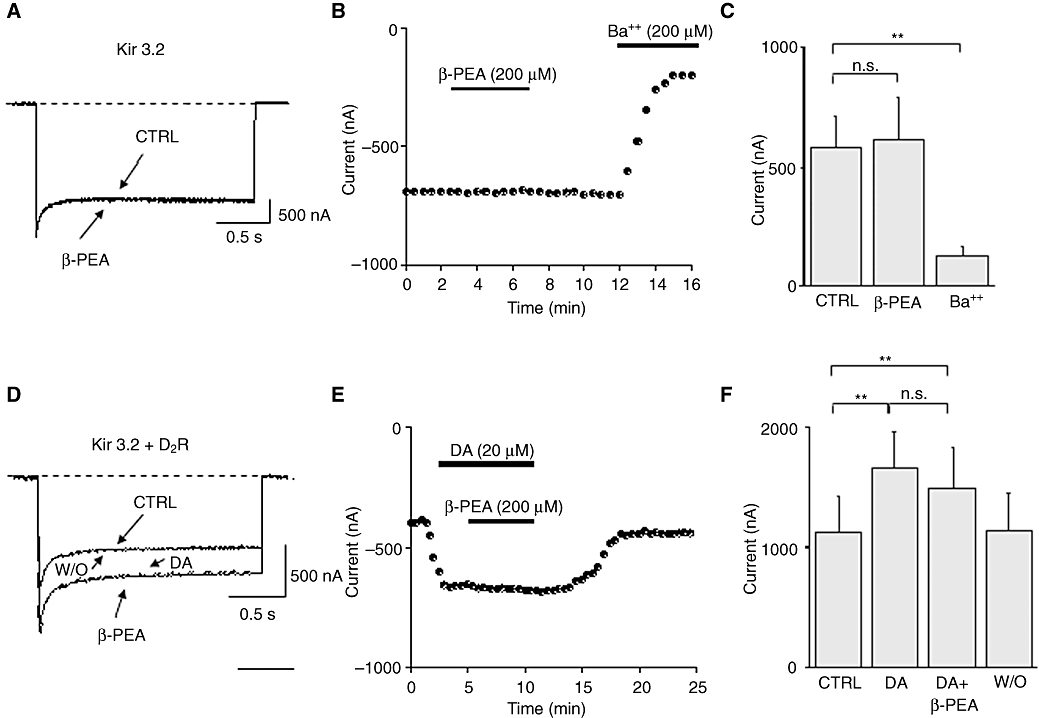
Effect of β-phenylethylamine (β-PEA) on both the basal and dopamine-evoked Kir3.2 potassium currents in heterologous expression systems. (A) Representative whole-cell current traces recorded from a Xenopus oocyte expressing Kir3.2 channels before (CTRL) and after superfusion of β-PEA (200 µM). Currents were elicited by hyperpolarizing steps to –120 mV for 2 s from a holding potential of –10 mV. The dashed line indicates zero current. (B) Time-course for the effect of β-PEA (200 µM) and Ba2+ (200 µM) on current amplitudes recorded from oocytes expressing Kir3.2 channels. The horizontal bar indicates the time of β-PEA and Ba2+ application. (C) Histogram showing the mean current amplitudes recorded at –120 mV from oocytes expressing Kir3.2 channels before and after the superfusion of β-PEA and Ba2+. (D) Representative whole-cell current traces recorded from an oocyte co-expressing Kir3.2 and D2 receptors showing the basal Kir3.2 current (CTRL), the dopamine-evoked Kir3.2 current, the effect of β-PEA (200 µM) application and wash-out (W/O). (E) Time-course of current amplitudes recorded before and after the application of dopamine (20 µM) and β-PEA (200 µM), from an oocyte co-expressing Kir3.2 and D2 receptors. The horizontal bars indicate the time of the dopamine and β-PEA application. (F) Histogram showing the mean Kir3.2 current amplitudes recorded at –120 mV under control conditions, during the application of dopamine (20 µM), β-PEA (200 µM) and after wash-out of dopamine (W/O). These results indicate that β-PEA does not affect either basal or dopamine-evoked Kir3.2 channel activity. The data are mean ± SEM of six to eight cells. (***P < 0.001, Student's t-test).
Discussion
Depressant effects of TAs on D2 receptor-mediated responses
The present results demonstrate that the prototypical TAs, β-PEA and TYR, both concentration-dependently depressed the D2 receptor-mediated outward current in dopaminergic neurones. Inasmuch as the effects of β-PEA were particularly robust and consistent, we focused our attention on this agent which expressed its actions with an IC50 of around 80 µM. This concentration is higher than that which has generally been detected in the brain tissue homogenates (Boulton, 1976; Berry, 2004; Grandy, 2007), but is still likely to be physiologically relevant in view of: (i) the extremely rapid turnover of β-PEA and other TAs (which have a half-life of approximately 30 s); and (ii), the likelihood that free, synaptic, extracellular levels of TA are, at least transiently, much higher upon their release (Durden and Philips, 1980). Moreover, it must be emphasized that the penetration of TAs into tissue slices is limited by the diffusion barrier and degradation/sequestering mechanisms: hence, a high amount needs to be applied in in vitro studies in order to achieve a sufficient concentration at the effective site of action. By analogy, dopamine only affects neuronal activity when superfused in the micromolar range (Lacey et al., 1987; Suppes and Pinnock, 1987) at concentrations far higher than those found in tissue or in the extracellular fluid, but corresponding to concentrations present and active at the synaptic level (Ford et al., 2009). It should also be pointed out that cerebral levels of TAs are not constant inasmuch as pathological states such as Rett syndrome and phenylketonuria are characterized by low (Satoi et al., 2000) and high (Perry, 1962) brain levels of β-PEA, respectively. Furthermore, the dose-response curve for β-PEA was shifted to the left (4.2-fold) in the presence of the MAO B inhibitor, selegiline, consistent with the notion that the functional activity of this catabolic enzyme is important for regulating the turnover and, hence, the effects of TAs on neurones. Accordingly, in certain pathophysiological states, and following functional, genetic or pharmacological manipulations that reduce MAO activity, the availability of TAs will be enhanced and their neuronal actions amplified.
Based on experiments with the AADC inhibitor, carbidopa, which by reducing the formation of dopamine and noradrenaline blunts their TA-induced release (Geracitano et al., 2004), it appears that the effects of β-PEA and TYR were not indirectly mediated via extracellular increases in levels of these two catecholamines. In line with these findings, it has been shown that an intraventricular injection of TYR elicits locomotor activity in rats that had been pretreated with reserpine or α-methyl-p-tyrosine to exclude the possibility of indirect receptor stimulation following catecholamine release (Stoof et al., 1976). Moreover, other effects of TA exerted independently of dopamine and noradrenaline (Paterson, 1993) have also been described.
Noteworthy, in the present study, the inhibitory effect of TAs on the D2 receptor-mediated outward current was mimicked by the dopamine-releasing drug AMPH but not by cocaine, a dopamine uptake blocker. Thus, the persistence of the modulatory effect induced by the TAs in the presence cocaine suggests that dopamine transporters (DATs) are not involved in this effect.
Absence of mechanisms mediated by TA1 receptors
Interestingly, the depressant actions of β-PEA and TYR were present in transgenic mice in which the TA1 receptor had been genetically deleted (TA1 null mice) (De Groote et al., 2007; Millan et al., 2008). Underpinning these observations, the selective TA1 agonist, O-PIT (Hart et al., 2006) proved to be ineffective in depressing D2 receptor-mediated responses, despite the fact that several other functional responses to this agent, like the induction of hypothermia, are abolished in TA1 knock-out mice (above citations).
Collectively, these data support the idea that TA1 receptors are not involved in the depression of the D2 receptor-mediated responses of SNpc dopaminergic neurones induced by β-PEA and TYR observed in the present study.
Of note, TA1 is distributed throughout cerebral populations of monoaminergic neurones and is co-localized with DAT in a subset of dopaminergic neurones in mouse and rhesus monkey SNpc (Xie et al., 2007). A mutual functional interaction between TAs and dopaminergic transmission has been described (Lindemann et al., 2008; Sotnikova et al., 2008; Xie et al., 2008), in that D2-autoreceptor activation appears to reduce TA1 signalling (Xie et al., 2007) that, contrarily, is increased by the co-expression of DAT with TA1 in HEK293 cells (Miller et al., 2005). Reciprocally, both TA1 and D2 receptors modulate DAT activity but exert opposite effects (Xie et al., 2008). Notably, the neuronal hyperpolarization produced in dopaminergic mesencephalic neurones by TA-induced release of DA via DAT was impaired in TA1 KO mice (Lindemann et al., 2008). Moreover, it has been recently shown that TA1 modulates D2 receptor activity, fine-tuning their desensitization rate and the potency of agonists (Bradaia et al., 2009). It should be mentioned that, in our experimental conditions, the outward current induced by long-lasting application of quinpirole (300 nM) did not desensitize, so this phenomenon was not monitored. Recently, it was proposed that TYR inhibits the firing activity of dopaminergic neurones and induces the opening of inwardly rectifying K+ channels through a TA1-dependent mechanism (Bradaia et al., 2009). These data are not consistent with our previous results showing that the inhibitory effects of TYR and β-PEA are indirect (mediated by dopamine); they were shown to be abolished by sulpiride and greatly reduced by depletion of dopamine (carbidopa-induced) (Geracitano et al., 2004). Notably, indirect dopamine-mediated effects of TYR have also been reported in the subthalamic nucleus (Zhu et al., 2007). Although species differences (rat versus mice) could partially account for the divergent results, the clear-cut D2 receptor-mediated effects of β-PEA in TA1 KO mice clearly differ from the recent observations of Bradaia et al. (2009).
Absence of mechanisms mediated by σ receptors
σ Receptors have been reported to inhibit diverse classes of K+ and Ca2+ ion channels through direct protein-protein interactions with a G-protein independent mechanism not related to protein kinases (Lupardus et al., 2000; Fontanilla et al., 2009). A reduction of GIRK-mediated response has been reported with σ agonists (Kobayashi et al., 2006; Monassier et al., 2007) both in the heart and also in catecholaminergic neurones in the CNS (Nguyen et al., 1998). As it was recently shown that both β-PEA and TYR activate σ receptors (Fontanilla et al., 2009), we used selective antagonists of σ1 and σ2 subtypes to verify if they are involved in their depressant effect on the D2 receptor-mediated outward current. Inasmuch as they were ineffective, these results suggest that TAs do not stimulate σ receptors to modulate D2 receptor-activated responses in the dopaminergic cells.
Cellular mechanisms of action
It is known that stimulation of D2 autoreceptors activates Gi/o proteins to open inward rectifier potassium channels. This has again been supported by the experiments in which the reversal potential of the D2 receptor-mediated outward current is compatible with potassium equilibrium (Lacey et al., 1987, 1988). Moreover, the involvement of the Gi/o proteins has been confirmed by single-electrode voltage clamp experiments that permitted a successful intracellular diffusion of the irreversible non-hydrolysable GTP analogue GTPγS. Thus, the dissociated Gβγ subunits permanently engaged the receptor-operated K+ channels (Lacey et al., 1987). It is intriguing, then, that the GTPγS experiments conduced herein suggested a lack of G-protein involvement in the depressant effect of TAs. Based on the action of GTPγS – intracellularly dialysed using a patch-clamp technique – we previously reported that G-proteins are involved in the depression of GABAB-mediated outward currents by TAs (Federici et al., 2005). However, in retrospect, there may have been a misinterpretation of our data because of excessive dialysis of the intracellular milieu during prolonged patch-clamp recording. It is likely that an excessive dialysis of the cytoplasmatic content, because of the use of the patch-clamp technique, caused a subsequent rundown of GABAB-mediated responses, which was erroneously considered to represent an irreversible inhibitory effect of TAs. This is also supported by newer experiments using baclofen in GTPγS-treated neurones (see Results). Hence, we suggest that during prolonged electrophysiological experiments requiring GTPγS dialysis and designed to examine G-protein-mediated effects, it is preferable to use – as herein – sharp microelectrode techniques rather than conventional patch-clamp recordings. The current–voltage plots performed in the presence of quinpirole and β-PEA demonstrated that the TA-reduced current reversed at the potassium equilibrium point, confirming that TAs reduce a GIRK-activated channel. Surprisingly, the experiments performed in oocytes suggest that TAs act neither at D2 receptors nor on GIRK channels themselves leading to the hypothesis that a more complex sequence of events leads to the reduction of the D2 receptor-activated GIRK currents by TAs. However, as the Gβγ subunit combination expressed by oocytes is not known, it is possible that they differ from dopaminergic cells of the substantia nigra and are therefore insensitive to modulation by TAs. Thus, taken together, the present data suggest that TAs diminish quinpirole-induced outward currents by a molecular mechanism that needs to be more fully elucidated in future studies. However, a comparable depressant effect of TA has also been observed on GIRK currents activated by GABAB receptors (Federici et al., 2005 and the present data) and, as the same type of GIRK channels are activated by D2 and GABAB receptors (Lacey et al., 1988), a similar mechanism is probably involved in the modulation, by β-PEA and other TAs, of the GIRKs induced by both D2 and GABAB.
It is known that GIRK currents can be inhibited by the activation of receptors coupled to Gq/11 leading, through the induction of phospholipase C (PLC), to depletion of phosphatidylinositol 2,4-bisphosphate (PIP2) (Cho et al., 2005). However, the experiments performed using GTPγS in which mGluR1 (DHPG)/Gq/11–mediated responses were found to be irreversible, make it unlikely that such a mechanism is involved in the reversible inhibition of D2 receptor-activated GIRK currents induced by TAs.
Finally, based on the lack of effects of TAs in D2 receptor- and GIRK-expressing oocytes there is a real possibility that specific neuronal sites exist at which TA could act to affect D2 receptor/G-protein-operated GIRK channel activity. Hence, TAs could modulate neuronal GIRK activity by altering the efficiency of Gβγ coupling to the channel. In line with this notion, recent evidence indicates that – apart from their cognate heterotrimeric G proteins – D2 receptors interact with a large number of dopamine receptor interacting proteins (‘DRIPs’) involved in scaffolding, trafficking and coupling to intracellular effectors (Kabbani and Levenson, 2007). Therefore, it is conceivable that TAs hamper the interaction between receptors and DRIPs, thereby leading to an inhibition of D2 receptor-activated GIRK currents.
Functional considerations
It is possible that the negative regulation of D2 autoreceptor function by TA contributes to the regulation of the normal basal activity of dopaminergic cells, and it might also render them more excitable. An excitatory effect of TAs on cortical and subthalamic neurones has been described previously (Zhu et al., 2007; Kitamura et al., 2008) and it was demonstrated that β-PEA inhibits K+ currents in neocortical neurones of the rat (Kitamura et al., 2008). TA-induced blunting of D2 autoreceptor inhibition has potentially important implications for the control of movement, reward, mood, motor function and cognition and, consequently, may be pertinent to the aetiology and treatment of diseases in which perturbed dopaminergic transmission has been implicated, such as schizophrenia, drug abuse, depression, Parkinson's disease and attention deficit hyperactive disorders (Boulton, 1980; Davis and Boulton, 1994; Bergman et al., 2001; Millan, 2006; Sotnikova et al., 2008).
Concluding remarks
The present data demonstrate that the prototypical TAs, β-PEA and TYR, both concentration-dependently suppress D2 receptor-mediated GIRK currents in SNpc localized dopaminergic neurones, actions expressed neither via TA1 receptors nor via σ1 receptors, and in a G-protein independent fashion. The underlying molecular substrates remain to be determined. However, they may involve intracellular modulation by TAs of protein-protein interactions downstream of D2 receptors thereby modifying their influence on GIRKs – and other cellular effectors. In any event, these data add further weight to the notion that the actions of TA are by no means limited to modulation of catecholamine release and that they directly influence the activity of monoaminergic and, in particular, dopaminergic pathways.
Acknowledgments
This work was supported by Italian Ministry of Health (grant RF.06.71.3 to N.B.M.). The financial support of MIUR-PRIN 2007 and Compagnia di San Paolo (Turin) ‘Programma Neuroscienze’ to M. Pessia is gratefully acknowledged.
Glossary
Abbreviations:
- AADC
aromatic amino acid decarboxylase
- AMPH
d-amphetamine
- β-PEA
β-phenylethylamine
- DA
dopamine
- GPCRs
G protein–coupled receptors
- GIRK
G protein-gated inward rectifier K+ channel
- MAO B
monoamine oxidase B
- SNpc
substantia nigra pars compacta
- TA1
trace amine 1 receptor
- TAs
trace amines
Conflict of interest
None.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Figure S1 Representative current trace showing that the intracellular diffusion of GTPγS in a dopaminergic neurone caused a progressive outward current. Under this condition, a relative short application of the GABAB receptor agonist, baclofen (10 μM), caused a sustained outward current that was transiently depressed by β-phenylethylamine (β-PEA) (100 μM).
{kind=link}
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Alexander SPH, Mathie A, Peter JA. Guide to receptors and channels (GRAC), 4th edn. Br J Pharmacol. 2009;158(Suppl 1):S1–S254. doi: 10.1111/j.1476-5381.2009.00499.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey BA, Philips SR, Boulton AA. In vivo release of endogenous dopamine, 5-hydroxytryptamine and some of their metabolites from rat caudate nucleus by phenylethylamine. Neurochem Res. 1987;12:173–178. doi: 10.1007/BF00979534. [DOI] [PubMed] [Google Scholar]
- Barroso N, Rodriguez M. Action of β-phenylethylamine and related amines on nigrostriatal dopamine neurotransmission. Eur J Pharmacol. 1996;297:195–203. doi: 10.1016/0014-2999(95)00757-1. [DOI] [PubMed] [Google Scholar]
- Bergman J, Yasar S, Winger G. Psychomotor stimulant effects of beta-phenylethylamine in monkeys treated with MAO-B inhibitors. Psychopharmacology (Berl) 2001;159:21–30. doi: 10.1007/s002130100890. [DOI] [PubMed] [Google Scholar]
- Berry MD. Mammalian central nervous system trace amines. Pharmacological amphetamines, physiological neuromodulators. J Neurochem. 2004;90:257–271. doi: 10.1111/j.1471-4159.2004.02501.x. [DOI] [PubMed] [Google Scholar]
- Borowsky B, Adham N, Jones KA, Raddatz R, Artymyshyn R, Ogozalek KL, et al. Trace amines: identification of a family of mammalian G protein-coupled receptors. Proc Natl Acad Sci USA. 2001;98:8966–8971. doi: 10.1073/pnas.151105198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulton AA. Identification, distribution, metabolism, and function of meta and para tyramine, phenylethylamine and tryptamine in brain. Adv Biochem Psychopharmacol. 1976;15:57–67. [PubMed] [Google Scholar]
- Boulton AA. Trace amines and mental disorders. Can J Neurol Sci. 1980;7:261–263. doi: 10.1017/s0317167100023313. [DOI] [PubMed] [Google Scholar]
- Bradaia A, Trube G, Stalder H, Norcross RD, Ozmen L, Wettstein JG, et al. The selective antagonist EPPTB reveals TAAR1-mediated regulatory mechanisms in dopaminergic neurons of the mesolimbic system. Proc Natl Acad Sci USA. 2009;106:20081–20086. doi: 10.1073/pnas.0906522106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branchek TA, Blackburn TP. Trace amines receptors as target for novel therapeutics: legend, myth and fact. Curr Opin Pharmacol. 2003;3:90–97. doi: 10.1016/s1471-4892(02)00028-0. [DOI] [PubMed] [Google Scholar]
- Bunzow JR, Sonders MS, Arttamangkul S, Harrison LM, Zhang G, Quigley DI, et al. Amphetamine, 3,4-methylenedioxymethamphetamine, lysergic acid diethylamide, and metabolites of the catecholamine neurotransmitters are agonists of a rat trace amine receptor. Mol Pharmacol. 2001;60:1181–1188. doi: 10.1124/mol.60.6.1181. [DOI] [PubMed] [Google Scholar]
- Burchett SA, Hicks TP. The mysterious trace amines: protean neuromodulators of synaptic transmission in mammalian brain. Prog Neurobiol. 2006;79:223–246. doi: 10.1016/j.pneurobio.2006.07.003. [DOI] [PubMed] [Google Scholar]
- Cho H, Lee D, Lee SH, Ho WK. Receptor-induced depletion of phosphatidylinositol 4,5-bisphosphate inhibits inwardly rectifying K+ channels in a receptor-specific manner. Proc Natl Acad Sci USA. 2005;102:4643–4648. doi: 10.1073/pnas.0408844102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis BA, Boulton AA. The trace amines and their acidic metabolites in depression – an overview. Prog Neuropsychopharmacol Biol Psychiatry. 1994;18:17–45. doi: 10.1016/0278-5846(94)90022-1. [DOI] [PubMed] [Google Scholar]
- De Groote L, Gobert A, Brocco M, Rivet JM, Di Cara B, Veiga S, et al. Genetic deletion of Trace Amine Associated Receptor 1 receptors does not compromise the actions of mechanistically diverse antidepressants: a neurochemical and behavioural analysis. Am Soc Neuroscience Abstracts. 2007;807:4. [Google Scholar]
- Durden DA, Philips SR. Kinetic measurements of the turnover rates of phenylethylamine and tryptamine in vivo in the rat brain. J Neurochem. 1980;34:1725–1732. doi: 10.1111/j.1471-4159.1980.tb11267.x. [DOI] [PubMed] [Google Scholar]
- Dyck LE. Release of monoamine from striatal slices by phenelzine and β-phenylethylamine. Prog Neuropsychopharmacol Biol Psychiatry. 1983;7:797–800. doi: 10.1016/0278-5846(83)90069-6. [DOI] [PubMed] [Google Scholar]
- Federici M, Geracitano R, Tozzi A, Longone P, Di Angelantonio S, Bengtson CP, et al. Trace amines depress GABAB response in dopaminergic neurons by inhibiting G-βγ-gated inwardly rectifying potassium channels. Mol Pharmacol. 2005;67:1283–1290. doi: 10.1124/mol.104.007427. [DOI] [PubMed] [Google Scholar]
- Fontanilla D, Johannessen M, Hajipour AR, Cozzi NV, Jackson MB, Ruoho AE. The hallucinogen N,N-dimethyltryptamine (DMT) is an endogenous Sigma-1 receptor regulator. Science. 2009;323:934–937. doi: 10.1126/science.1166127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford CP, Phillips PE, Williams JT. The time course of dopamine transmission in the ventral tegmental area. J Neurosci. 2009;29:13344–13352. doi: 10.1523/JNEUROSCI.3546-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geracitano R, Federici M, Prisco S, Bernardi G, Mercuri NB. Inhibitory effects of trace amines on rat midbrain dopaminergic neurons. Neuropharmacology. 2004;46:807–814. doi: 10.1016/j.neuropharm.2003.11.031. [DOI] [PubMed] [Google Scholar]
- Grandy DK. Trace amine-associated receptor-1 – family archetype or iconoclast? Pharmacol Ther. 2007;116:355–390. doi: 10.1016/j.pharmthera.2007.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenshaw A, Juorio AV, Nguyen TV. Depletion of striatal β-phenylethylamine following dopamine but not 5-HT denervation. Brain Res Bull. 1986;17:477–484. doi: 10.1016/0361-9230(86)90214-5. [DOI] [PubMed] [Google Scholar]
- Guatteo E, Mercuri NB, Bernardi G, Knopfel T. Group I metabotropic glutamate receptors mediate an inward current in rat substantia nigra dopaminergic neurons that is independent from calcium mobilization. J Neurophysiol. 1999;82:1974–1981. doi: 10.1152/jn.1999.82.4.1974. [DOI] [PubMed] [Google Scholar]
- Hart ME, Suchland KL, Miyakawa M, Bunzow JR, Grandy DK, Scanlan TS. Trace amine-associated receptor agonists: synthesis and evalutation of thyronamines and related analogues. J Med Chem. 2006;49:1101–1112. doi: 10.1021/jm0505718. [DOI] [PubMed] [Google Scholar]
- Itzhak Y, Stein I, Zhang SH, Kassim CO, Cristante D. Binding of sigma-ligands to C57BL/6 mouse brain membranes: effects of monoamine oxidase inhibitors and subcellular distribution studies suggest the existence of sigma-receptor subtypes. J Pharmacol Exp Ther. 1991;257:141–148. [PubMed] [Google Scholar]
- Kabbani N, Levenson R. A proteomic approach to receptor signaling: molecular mechanisms and therapeutic implications derived from discovery of the dopamine D2 receptor signalplex. Eur J Pharmacol. 2007;572:83–93. doi: 10.1016/j.ejphar.2007.06.059. [DOI] [PubMed] [Google Scholar]
- Kitamura T, Munikata M, Haginoya K, Tsuchiya S, Iinuma K. β-phenylethylamine inhibits K+ currents in neocortical neurons of the rat: a possible mechanism of β-phenylethylamine-induced seizures. Tohoku J Exp Med. 2008;215:333–340. doi: 10.1620/tjem.215.333. [DOI] [PubMed] [Google Scholar]
- Kobayashi T, Washiyama K, Ikeda K. Inhibition of G Protein-activated inwardly rectifying K+ channels by ifenprodil. Neuropsychopharmacology. 2006;31:516–524. doi: 10.1038/sj.npp.1300844. [DOI] [PubMed] [Google Scholar]
- Kosa E, Marcilhac-Flouriot A, Fache MP, Siaud P. Effects of beta-phenylethylamine on the hypothalamo-pituitary-adrenal axis in the male rat. Pharmacol Biochem Behav. 2000;67:527–535. doi: 10.1016/s0091-3057(00)00383-x. [DOI] [PubMed] [Google Scholar]
- Lacey MG, Mercuri NB, North RA. Dopamine acts on D2 receptors to increase potassium conductance in neurones of rat substantia nigra zona compacta. J Physiol. 1987;392:397–416. doi: 10.1113/jphysiol.1987.sp016787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacey MG, Mercuri NB, North RA. On the potassium conductance increase activated by GABAB and dopamine D2 receptors in the rat substantia nigra neurons. J Physiol. 1988;401:437–453. doi: 10.1113/jphysiol.1988.sp017171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacey MG, Mercuri NB, North RA. Two cell types in rat substantia nigra zona compacta distinguished by membrane properties and the action of dopamine and opioids. J Neurosci. 1989;9:1233–1241. doi: 10.1523/JNEUROSCI.09-04-01233.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindemann L, Meyer CA, Jeanneau K, Bradaia A, Ozmen L, Bluethmann H, et al. Trace amine-associated receptor 1 modulates dopaminergic activity. JPET. 2008;324:948–956. doi: 10.1124/jpet.107.132647. [DOI] [PubMed] [Google Scholar]
- Lupardus PJ, Wilke RA, Aydar E, Palmer CP, Chen Y, Ruoho AE, et al. Membrane-delimited coupling between sigma receptors and K+ channels in rat neurohypophysial terminals requires neither G-protein nor ATP. J Physiol. 2000;526:527–539. doi: 10.1111/j.1469-7793.2000.00527.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maguire JJ, Parker WA, Foord SM, Bonner TA, Neubig RR, Davenport AP. International Union of Pharmacology. LXXII. Recommendations for trace amine receptor nomenclature. Pharmacol Rev. 2009;61:1–8. doi: 10.1124/pr.109.001107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin WR, Eades CG, Thompson JA, Huppler RE, Gilbert PE. The effects of morphine and nalorphine like drugs in the nondependent and morphine-dependent chronic spinal dog. J Pharmacol Exp Ther. 1976;197:517–532. [PubMed] [Google Scholar]
- Mercuri NB, Bonci A, Calabresi P, Stefani A, Bernardi G. Properties of the hyperpolarization-activated cation current Ih in rat midbrain dopaminergic neurons. Eur J Neurosci. 1995;7:462–469. doi: 10.1111/j.1460-9568.1995.tb00342.x. [DOI] [PubMed] [Google Scholar]
- Millan MJ. Multi-target strategies for the improved treatment of depressive states: conceptual foundations and neuronal substrates, drug discovery and therapeutic application. Pharmacol Ther. 2006;110:135–370. doi: 10.1016/j.pharmthera.2005.11.006. [DOI] [PubMed] [Google Scholar]
- Millan MJ, Rivet JM, Brocco M, Zhang X, Svenningsson P, Verdouw PM, et al. Behavioural and neurochemical actions of thyronamines are blunted in Trace-Amine-Associated receptor 1 (TAAR1) receptor knock-out mice. Behav Pharmacol. 2008;20(S1):S112. [Google Scholar]
- Miller GM, Verrico CD, Jassen A, Konar M, Yang H, Panas H, et al. Primate trace amine receptor 1 modulation by the dopamine transporter. J Pharmacol Exp Ther. 2005;313:983–994. doi: 10.1124/jpet.105.084459. [DOI] [PubMed] [Google Scholar]
- Monassier L, Manoury B, Bellocq C, Weiwwenburger J, Greney H, Zimmermann D, et al. σ2-receptor ligand-mediated inhibition of inwardly rectifying K+ channels in the heart. J Pharmacol Exp Ther. 2007;322:341–350. doi: 10.1124/jpet.107.122044. [DOI] [PubMed] [Google Scholar]
- Monnet FP, Maurice T. The sigma-1 protein as a target for the non-genomic effects of neuro(active)steroids: molecular and behavioural aspects. J Pharmacol Sci. 2006;100:93–118. doi: 10.1254/jphs.cr0050032. [DOI] [PubMed] [Google Scholar]
- Nakamura M, Ishii A, Nakahara D. Characterization of β-phenylethylamine induced monoamine release in rat nucleus accumbens: a microdioalysis study. Eur J Pharmacol. 1998;349:163–169. doi: 10.1016/s0014-2999(98)00191-5. [DOI] [PubMed] [Google Scholar]
- Nguyen VH, Ingram SL, Kassiou M, Christie MJ. σ-Binding sites ligands inhibit K+ currents in rat locus coeruleus neurons in vitro. Eur J Pharmacol. 1998;361:157–163. doi: 10.1016/s0014-2999(98)00706-7. [DOI] [PubMed] [Google Scholar]
- Paterson IA. The potentiation of cortical neuron responses to noradrenaline by 2-phenylethylamine is independent of endogenous noradrenaline. Neurochem Res. 1993;18:1329–1336. doi: 10.1007/BF00975055. [DOI] [PubMed] [Google Scholar]
- Paterson IA, Juorio AV, Boulton AA. 2-Phenylethylamine: a modulator of catecholamine transmission in the mammalian central nervous system? J Neurochem. 1990;55:1827–1837. doi: 10.1111/j.1471-4159.1990.tb05764.x. [DOI] [PubMed] [Google Scholar]
- Perry TL. Urinary excretion of amines in phenylketonuria and mongolism. Science. 1962;136:879–880. doi: 10.1126/science.136.3519.879. Pharmacol Ther116: 355–390. [DOI] [PubMed] [Google Scholar]
- Pessia M, Tucker SJ, Lee K, Bond CT, Adelamn JP. Subunit positional effects revealed by novel heteromeric inwardly rectifying K+ channels. EMBO J. 1996;15:2980–2987. [PMC free article] [PubMed] [Google Scholar]
- Philips SR, Robson AM. In vivo release of endogenous dopamine from the rat caudate nucleus by phenylethylamine. Neuropharmacology. 1983;22:1297–1301. doi: 10.1016/0028-3908(83)90203-4. [DOI] [PubMed] [Google Scholar]
- Pirri JK, McPherson AD, Donnelly JM, Francis MM, Alkema MJ. A tyramine-gated chloride channel coordinates distinct motor programs of a Caenorhabditis elegans escape response. Neuron. 2009;62:526–538. doi: 10.1016/j.neuron.2009.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Premont RT, Gainetdinov RR, Caron MG. Following the trace of elusive amines. Proc Natl Acad Sci USA. 2001;98:9474–9475. doi: 10.1073/pnas.181356198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raiteri M, Del Carmen R, Bertollini A, Levi G. Effect of sympathomimetic amines on the synaptosomal transport of noradrenaline, dopamine and 5-hydroxytryptamine. Eur J Pharmacol. 1977;41:133–143. doi: 10.1016/0014-2999(77)90202-3. [DOI] [PubMed] [Google Scholar]
- Richard L, Hauger T, Philskolnick, Steven MP. Specific [3H]IS-Phenylethylamine binding sited in rat brain. Eur J Pharmacol. 1982;83:147–148. doi: 10.1016/0014-2999(82)90301-6. [DOI] [PubMed] [Google Scholar]
- Ringstad N, Abe N, Horvitz R. Ligand gated chloride channels are receptors for biogenic amines in C. elegans. Science. 2009;325:96–100. doi: 10.1126/science.1169243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoi M, Matsuishi T, Yamada S, Yamashita Y, Ohtaki E, Mori K, et al. Decreased cerebrospinal fluid levels of beta-phenylethylamine in patients with Rett syndrome. Ann Neurol. 2000;47:801–803. [PubMed] [Google Scholar]
- Sotnikova TD, Zorina OI, Ghisi V, Caron MG, Gainetdinov RR. Trace amine associated receptor 1 and movement control. Parkinsonism Relat Disord. 2008;14:99–102. doi: 10.1016/j.parkreldis.2008.04.006. [DOI] [PubMed] [Google Scholar]
- Stoof JC, Liem AL, Mulder AH. Release and receptor stimulating properties of p-tyramine in rat brain. Arch Int Pharmacodyn. 1976;220:62–71. [PubMed] [Google Scholar]
- Suppes T, Pinnock RD. Sensitivity of neuronal dopamine response in the substantia nigra and ventral tegmentum to clozapine, metoclopramide and SCH 23390. Neuropharmacology. 1987;26:331–337. doi: 10.1016/0028-3908(87)90185-7. [DOI] [PubMed] [Google Scholar]
- Vaccari A. High affinity binding of [3h]-tyramine in the central nervous system. Br J Pharmacol. 1986;89:15–25. doi: 10.1111/j.1476-5381.1986.tb11116.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werner P, Hussy N, Buell G, Jones KA, North RA. D2, D3, and D4 dopamine receptors couple to G protein-regulated potassium chennels in Xenopus oocytes. Mol Pharmacol. 1996;49:656–661. [PubMed] [Google Scholar]
- Xie Z, Miller GM. Trace amine-associated receptor 1 as a monoaminergic modulator in brain. Biochem Pharmacol. 2009;78:1095–1104. doi: 10.1016/j.bcp.2009.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Z, Westmoreland SV, Bahn ME, Chen G, Yang H, Vallender E, et al. Rhesus monkey trace amine-associated receptor 1 signaling: enhancement by monoamine transporters and attenuation by D2 autoreceptor in vitro. J Pharmacol Exp Ther. 2007;321:116–127. doi: 10.1124/jpet.106.116863. [DOI] [PubMed] [Google Scholar]
- Xie Z, Westmoreland SV, Miller GM. Modulation of monoamine transporters by common biogenic amines via trace amine-associated receptor 1 and monoamine autoreceptors in human embryonic kidney 293 cells and brain synaptosomes. J Pharmacol Exp Ther. 2008;325:629–640. doi: 10.1124/jpet.107.135079. [DOI] [PubMed] [Google Scholar]
- Zhang H, Cuevas J. Sigma receptors inhibit high-voltage-activated calcium channels in rat sympathetic and parasympathetic neurons. J Neurophysiol. 2002;87:2867–2879. doi: 10.1152/jn.2002.87.6.2867. [DOI] [PubMed] [Google Scholar]
- Zhang H, Cuevas J. σ receptor activation blocks potassium channels and depresses neuroexcitability in rat intracardiac neurons. J Pharmacol Exp Ther. 2005;313:1387–1396. doi: 10.1124/jpet.105.084152. [DOI] [PubMed] [Google Scholar]
- Zhu ZT, Munhall AC, Johnson SW. Tyramine excites rat subthalamic neurons in vitro by a dopamine-dependent mechanism. Neuropharmacology. 2007;52:1169–1178. doi: 10.1016/j.neuropharm.2006.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.