

Abstract
Background and purpose:
We investigated how McN-A-343 inhibited the alkylation of the M1 muscarinic receptor by its nitrogen mustard derivative and that of ACh to identify whether it interacts allosterically or orthosterically.
Experimental approach:
We incubated the M1 muscarinic receptor expressed in Chinese hamster ovary cells with ACh mustard for various periods of time in the presence of McN-A-343 or known allosteric and orthosteric ligands. After stopping the reaction and removing unreacted ligands, unalkylated receptors were measured using [3H]N-methylscopolamine. Analogous experiments were done using a nitrogen mustard analog of McN-A-343. Affinity constants, cooperativity values for allosteric interactions and rate constants for receptor alkylation were estimated using a mathematical model.
Key results:
The kinetics of receptor alkylation by the nitrogen mustard derivatives of ACh and McN-A-343 were consistent with a two-step model in which the aziridinium ion rapidly forms a reversible receptor complex, which converts to a covalent complex at a slower rate. The inhibition of receptor alkylation by acetycholine, N-methylscopolamine and McN-A-343 was consistent with competitive inhibition, whereas that caused by gallamine was consistent with allosterism. Affinity constants estimated from alkylation kinetics agreed with those measured by displacement of [3H]N-methylscopolamine binding.
Conclusions and implications:
Our results suggest that McN-A-343 and its nitrogen mustard derivative interact competitively with ACh and N-methylscopolamine at the orthosteric site on the M1 muscarinic receptor. Measuring how drugs modulate the kinetics of receptor alkylation by an irreversible ligand is a powerful approach for distinguishing between negative allosteric modulators and competitive inhibitors.
Keywords: M1 muscarinic receptor, ACh mustard, McN-A-343, nitrogen mustard derivative, allosterism, competitive inhibition, irreversible binding
Introduction
The compound, [4-[[N-(3-chlorophenyl)carbamoyl]oxy]-2-butynyl] trimethylammonium chloride (McN-A-343), was first shown by Roszkowski (1961) to stimulate sympathetic ganglia. When administered to cats and dogs, it caused an increase in blood pressure, and this action was blocked by atropine, adrenergic antagonism and bilateral adrenalectomy and sympathectomy, but not by hexamethonium. The pressor response occurred rapidly upon arterial injection of McN-A-343 close to the superior sympathetic ganglia, and it was later shown that the compound caused a release of catecholeamines in the rat (Martin, 1996). Thus, McN-A-343 behaves as a novel muscarinic ganglionic stimulant. In contrast, most non-selective muscarinic agonists decrease blood pressure by activating the M3 muscarinic receptor (receptor nomenclature follows Alexander et al., 2009) on the endothelium of peripheral blood vessels. The pressor response to McN-A-343 in vivo is antagonized by the M1 selective antagonist pirenzepine with high potency (Hammer and Giachetti 1982), and McN-A-343 behaves as an M1/M4 selective muscarinic receptor agonist in signalling assays on cell lines expressing muscarinic receptors (McKinney et al., 1991; Lazareno et al., 1993; Figueroa et al., 2008).
The selective action of McN-A-343 raises the question of how it binds differentially with muscarinic receptor subtypes. The interaction between the binding of McN-A-343 and the orthosteric, muscarinic antagonist [3H]N-methylscopolamine ([3H]NMS) in the cerebral cortex and heart has been interpreted as competitive and allosteric, respectively (Birdsall et al., 1983). Given the abundance of M1 and M4 receptors in the cortex, and M2 receptors in the heart, this conclusion suggests that McN-A-343 interacts competitively with [3H]NMS at M1 and M4 receptors, and allosterically with [3H]NMS at the M2 receptor. Site-directed mutagenesis studies have led to speculation that McN-A-343 (May et al., 2007) and desmethylclozapine (Sur et al., 2003) interact allosterically with [3H]NMS at the M2 muscarinic receptor. More recently, it has been suggested that the binding site for McN-A-343 on the M2 receptor shares a common region with that of orthosteric ligands, indicating a competitive interaction of McN-A-343 with [3H]NMS and ACh even though part of McN-A-343 may interact with an allosteric site (Valant et al., 2008). In a study on the interaction between McN-A-343 and [3H]NMS at the M2 receptor in the presence of d-tubocurarine, the data were interpreted according to the postulate that McN-A-343 competitively inhibits the binding of [3H]NMS at relatively low concentrations, and at higher concentrations it binds to the allosteric site (Waelbroeck, 1994). How this mechanism can account for the incomplete inhibition of [3H]NMS binding by McN-A-343 is unclear, although it is interesting that McN-A-343 causes complete inhibition of [3H]NMS binding to the M2 receptor in the presence of d-tubocurarine (Waelbroeck, 1994).
Measuring how a test drug interferes with the covalent binding of a reactive orthosteric ligand to a receptor is a powerful means of detecting allosteric interactions (Suga et al., 2008). Because neither the test drug nor reactive ligand is radioactive, they can be used over a range of concentrations much wider than that typically used in experiments investigating the modulation of radioligand binding by a putative negative allosteric modulator, for example. When used with site-directed mutagenesis, either to introduce or to remove accessible reactive amino acids lining the binding pocket, this approach enables the identification of the binding site for the irreversible ligand and its competitive inhibitors.
In the present report, we have utilized both ACh mustard (AChM) and a nitrogen mustard derivative of McN-A-343, 4-[(2-bromoethyl)methyl-amino]-2-butynyl N-(3-chlorophenyl)carbamate (BR384, see Figure 1) (Ringdahl et al., 1990), to explore the interactions of orthosteric and allosteric ligands with the human M1 receptor. We find that McN-A-343, as well as ACh and NMS, competitively protects the M1 receptor from irreversible alkylation by both AChM and BR348, whereas 17-β-hydroxy-17-α-ethynyl-5-α-androstano[3,2-β]pyrimido[1,2-α]benzimidazole (WIN 51708) has little effect, and gallamine modulates alkylation through an allosteric mechanism. Our approach has widespread application to the study of allosterism at subtypes of the muscarinic receptor and other receptors.
Figure 1.
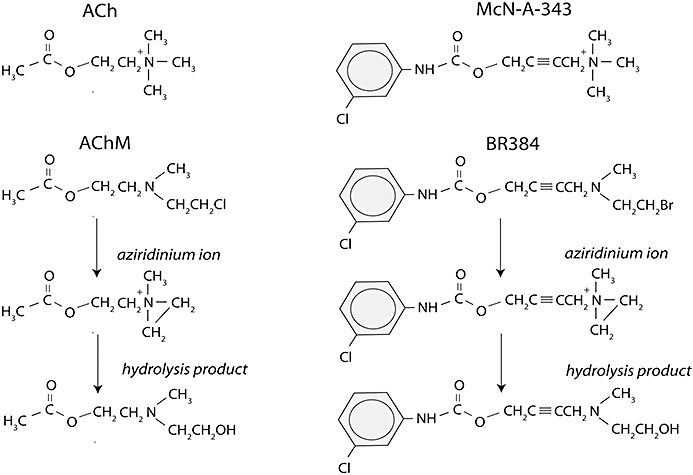
Structures of AChM, BR384, their aziridinium ions and alcoholic hydrolysis products. Also shown for comparison are ACh and McN-A-343.
Methods
Cell culture
Chinese hamster ovary (CHO) cells stably expressing the human muscarinic M1 receptor (CHO hM1) were obtained from Acadia Pharmaceuticals (San Diego, CA, USA). CHO hM1 cells were cultured in F-12 media Kaighn's modification supplemented with 10% fetal calf serum, penicillin–streptomycin (100 units·mL−1 and 100 µg·mL−1) and G418 (0.4 mg·mL−1) at 37°C with 5% CO2.
Animals
All animal care and experimental procedures were approved by the Institutional Animal Care and Use Committee at the University of California, Irvine. Four male Sprague Dawley rats (200–250 g) were used as a source of cerebral cortex for binding experiments. The animals were maintained in the university vivarium on a 12 h light–dark cycle with free access to food and water.
Receptor alkylation experiments in intact cells and homogenates
The strategy for measuring the rate of inactivation of the M1 receptor by AChM and BR384 involved: (i) incubating the M1 receptor preparation with the mustard; (ii) stopping the reaction with thiosulphate and a competitive inhibitor; (iii) washing the preparation to remove the stopping reagents; and (iv) measuring the specific binding of [3H]NMS to estimate the residual, unalkylated receptors. Experiments on intact cells were carried out as described previously (Suga et al., 2008). CHO hM1 cells incubated with the aziridinium ion of the mustard analogs at 37°C for various times in a final volume of 0.6 mL of Krebs–Ringer bicarbonate buffer (KRB buffer; 26 mM NaHCO3, 1.2 mM KH2PO4, 124 mM NaCl, 5 mM KCl, 1.8 mM CaCl2, 1.3 mM MgCl2, 10 mM glucose, pH 7.4). The reaction was stopped at the indicated times by the addition of a mixture of thiosulphate (10 mM) and N-methyl-amitriptyline (10 µM), which completely prevented receptor alkylation by AChM (1 mM) and BR384 (0.1 mM). The stopping solution was left on the cells for a minimum of 15 min. The adherent cells were washed five times, and residual muscarinic receptors were estimated using an intact cell [3H]NMS binding assay at room temperature.
In other experiments, the rate of inactivation of muscarinic receptors by AChM and BR384 was measured in homogenates of CHO hM1 cells as described previously (Suga et al., 2008). Cellular homogenate was prepared in 20 mM Na/HEPES buffer, pH 7.4, containing EDTA (10 mM) and NaCl (100 mM). Homogenates were incubated at 37°C with AChM, BR384 and other drugs in microfuge tubes for various periods of time, and the incubations were stopped by the addition of thiosulphate (1 mM) and scopolamine. The concentration of scopolamine was 1 µM for BR384 (1–10 µM) and AChM (1–100 µM). When higher concentrations of each mustard were used (BR384, 0.1–0.3 mM; AChM, 1 mM), the concentration of scopolamine was 10 µM. These reagents completely prevented receptor alkylation by AChM (1 mM) and BR384 (0.3 mM). The homogenates were washed twice (1 µM scopolamine) or three times (10 µM scopolamine) by centrifugation, and residual muscarinic receptors were measured using [3H]NMS at 23°C.
Experiments investigating the alkylation of muscarinic receptors by BR384 in rat brain homogenate were carried out using the same procedures as that described previously in a study on an alkylating derivative of oxotremorine-M (Ehlert and Jenden, 1985).
Competitive [3H]NMS binding experiments at 0°C
The competitive binding experiments with the nitrogen mustard analogs and their transformation product were carried out at 0°C as described by Suga et al. (2008).
Data analysis
The competitive and saturation binding data for [3H]NMS were analysed with GraphPad Prism using the variable slope dose–response and one-site binding functions respectively.
We derived a mathematical model to describe the kinetics of receptor alkylation by BR384 under conditions where the concentration of BR384 decays with time. Our approach is similar to that described previously (Ehlert and Jenden, 1985). The model for the interaction of the aziridinium ion of BR384 (X) with the M1 muscarinic receptor (R) in the presence of an allosteric modulator (A) is shown in Scheme 1. We assume that the aziridinium ion of BR384 rapidly equilibrates with the receptor to form a reversible complex (XR) that converts to a covalent complex (X-R) at a slower rate. K1 and K2 denote the affinity constants (inverse molar units; reciprocal of dissociation constant) of the aziridinium ion of BR384 and the allosteric modulator for the free receptor, respectively; α denotes the cooperativity factor for their interaction; and k1 denotes the rate constant for alkylation of the receptor by the aziridinium ion. If the rate constants describing the reversible binding of X and A are fast relative to k1, then the rate of loss of unalkylated receptors (y) can be described by the following differential equation as described previously (Suga et al., 2008):
Scheme 1.
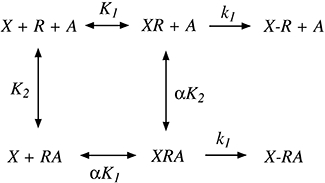
Model for the interaction of the aziridinium ion (X) with the M1 receptor (R) in the presence of an allosteric modulator (A). The aziridinium ion first forms a reversible complex with the receptor (XR), and then forms a covalent complex (X-R). K1 and K2 denote the affinity constants of the reversible complexes of XR and RA, α denotes the cooperativity factor for the allosteric interaction and k1 denotes the rate constant for alkylation of the receptor.
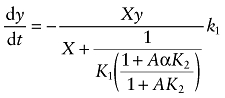 |
(1) |
The concentration of the aziridinium ion is not constant during the incubation, but decays from its peak concentration according to a first-order decay process approximated by the following equation:
| (2) |
in which X0 denotes the concentration of the aziridinium ion at the start of the incubation, and τx denotes the time constant for the decay of the aziridinium ion from its peak concentration. The value of τx was estimated at 14.3 min by regression analysis of the data reported by Ringdahl et al. (1990) for the stability of BR384 in aqueous solution at pH 7.4. We use a time constant to describe the first-order decay of the aziridinium ion from its peak concentration in solution to distinguish this process from the two microscopic rate constants describing the formation and decay of the aziridinium ion (k1 and k2 described by Ringdahl et al., 1990). Substituting in Equation (3) for X in Equation (2) yields:
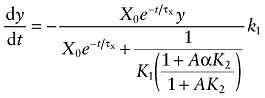 |
(3) |
Dividing both sides of the equation by y and integrating over the time interval t = 0 to t yields:
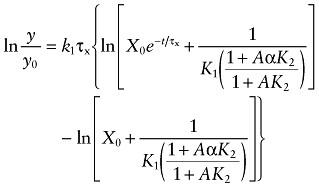 |
(4) |
In this equation, y0 denotes the total receptor density at the start of the reaction, and y denotes the residual, unalkylated receptors at time t. This equation reduces to:
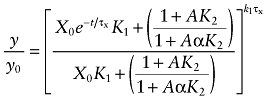 |
(5) |
We found that a small proportion of the M1 receptor population labelled by [3H]NMS was not alkylated by the aziridinium ion of BR384. Equation (5) was modified; therefore, by adding a constant (b) denoting the proportion of non-alkylated sites:
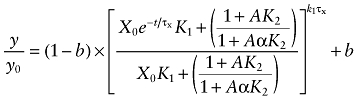 |
(6) |
Equation (6) describes the kinetics of the loss of [3H]NMS binding sites in the presence of BR384 and an allosteric modulator. When the negative cooperativity is very great (i.e. log α ≪ 0), then A behaves like a competitive inhibitor, and Equation (6) reduces to the following:
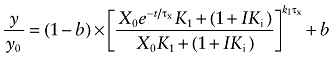 |
(7) |
in which A and K2 have been replaced with I and Ki to denote the concentration of the competitive inhibitor and its affinity constant respectively. In the absence of an allosteric modulator, Equation (6) reduces to:
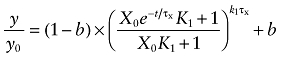 |
(8) |
It is well known that highly efficacious agonists, like ACh, exhibit competitive [3H]NMS binding curves with Hill slopes of less than one (Birdsall et al., 1978). Consequently, for the analysis of the effects of ACh on receptor alkylation by BR384, we modified Equations (6) and (7) by adding a Hill slope (n) to account for the complex binding properties of ACh:
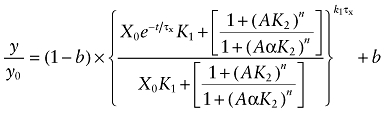 |
(9) |
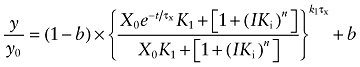 |
(10) |
Because the aziridinium ion of AChM is relatively stable at 37°C, we used simpler equations to describe the kinetics of receptor alkylation by AChM in the absence [Equation (11)] and presence of allosteric [Equation (12)] and competitive [Equation (13)] inhibitors:
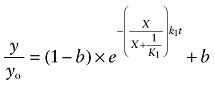 |
(11) |
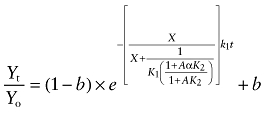 |
(12) |
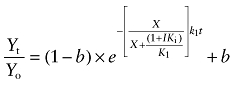 |
(13) |
These equations have been described previously (Suga et al., 2008) and are based on the assumption that the aziridinium ion of AChM remains constant during the incubation.
We also used a more empirical approach to analyse the effects of non-labelled inhibitors on the kinetics of receptor alkylation by AChM and BR384. The data for the loss of [3H]NMS binding as a function of time were fitted to a single exponential decay equation using Prism to estimate the observed rate constant for decay in the absence (kobs) and presence (kobs′) of inhibitors. With BR384, only the data obtained over the first 8 min were used to avoid estimation error caused by the decay in the aziridinium ion. The ratio (R) of time constants for receptor alkylation in the presence of the inhibitor (τ′) divided by that measured in its absence (τ) is defined as:
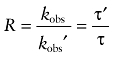 |
(14) |
The effect of allosteric [Equation (15)] and competitive [Equation (16)] inhibitors on the relative ratio of time constants (R) for receptor alkylation is described by:
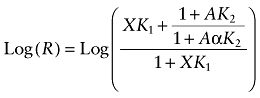 |
(15) |
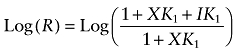 |
(16) |
The theoretical basis of Equations (15) and (16) has been described previously (Suga et al., 2008).
When fitting Equations (6)–(13) and (15) and (16) to the binding data by nonlinear regression analysis, the parameter K1 was constrained as a constant. The log molar values of this constant for the aziridinium ions of AChM and BR384 were 4.62 and 5.1, respectively, as determined from the data shown in Figure 4.
Figure 4.
The kinetics of alkylation of M1 receptors by AChM (A, B) and BR384 (C, D). Intact CHO hM1 cells were incubated with various concentrations of cyclized AChM (A) or BR384 (C) at 37°C, washed and then assayed for [3H]NMS binding. The theoretical curves represent the global least square fit of Equations (12) and (9) to the data for AChM and BR384 respectively. The observed rate constant for alkylation of M1 receptors was estimated for each concentration of AChM and BR384 in (A) and (C), and plotted against the mustard concentration in (B) and (D) respectively. A one-site model was fitted to each plot of the observed rate constants. The data represent the mean values ± SEM of two experiments, each in triplicate. The mean ± SEM denotes the range of the two measurements, because N = 2. The concentration of [3H]NMS was 1.0 nM. The concentration of cyclized nitrogen mustard is expressed as its initial concentration before transformation into the aziridinium ion.
To test whether the various allosteric models [Equations (6), (9), (12) and (15)] provided a better fit to the data than the corresponding competitive models [Equations (7), (10), (13) and (16)], we compared the residual estimate of the variance for both models using an F distribution as described previously (Suga et al., 2008).
Formation of the aziridinium ions of BR384 and AChM
BR384 was incubated at 37°C for 5 min in a mixture of 80% 50 mM phosphate buffer (pH 7.4) and 20% acetone to form its aziridinium ion (see Figure 1 for the transformation products of BR384 in aqueous solution at neutral pH). The small flask (10 mL) containing cyclized BR384 was attached to a rotary evaporator suspended in air at room temperature, and the acetone was quickly removed in vacuo (5 min). The solution quickly cooled to about 10°C because of the loss of the heat of vaporization. After removal of the acetone, the solution was placed on ice and used as soon as possible. At 37°C, the aziridinium ion of BR384 decays from its peak concentration of 54% (relative to the parent mustard) achieved after 5 min of incubation to 5.3% 30 min later (Ringdahl et al., 1990). AChM was cyclized to its aziridinium ion by incubation at 37°C for 15 min as described previously (Suga et al., 2008). The solution was placed on ice and used as soon as possible. The aziridinium ion of AChM is relatively stable at 37°C and only declines to 86% of its peak concentration in 60 min. All concentrations of the transformation products are given in the text as the starting concentration of the parent mustard.
Materials
Drugs and chemicals were obtained from the following sources: [3H]NMS (PerkinElmer Life and Analytical Sciences, Boston, MA, USA); F-12 media Kaighn's modification, fetal calf serum, trypsin–EDTA, G418 (Invitrogen, San Diego, CA, USA); ACh, NMS, gallamine, scopolamine, Na2S2O3, WIN 51708 (Sigma-Aldrich, St Louis, MO, USA). AChM and McN-A-343 were synthesized as described previously (Suga et al., 2008) using modifications of procedures described by Jackson and Hirst (1972) and Mellin et al. (1989) respectively. N-methylamitriptyline was synthesized as described by Suga et al. (2008), and BR384 was synthesized as described by Ringdahl et al. (1990).
Results
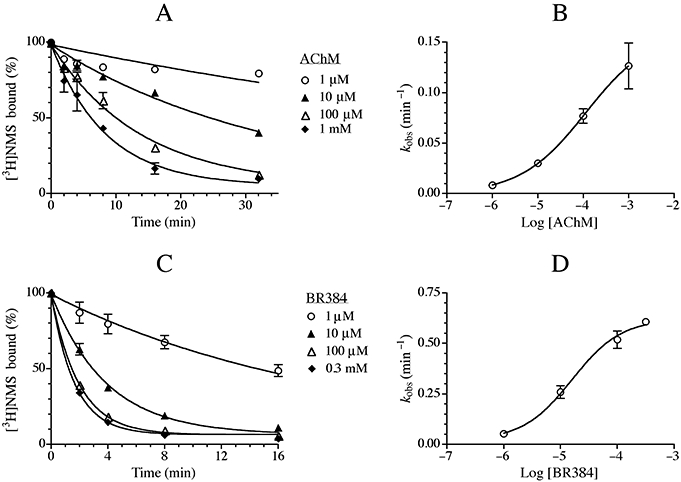
Effects of AChM and BR384 on the specific binding of [3H]NMS
To examine the influence of AChM and BR384 on the binding of [3H]NMS, CHO hM1 cells were incubated with the corresponding aziridinium ions at 37°C and then assayed for [3H]NMS binding at room temperature. In control CHO hM1 cells, [3H]NMS bound in a saturable fashion characterized by an observed log affinity constant (KA) of 9.41 ± 0.05; mean ± SEM (Figure 2). Treatment of CHO hM1 cells with the aziridinium ions of AChM (10 µM) and BR384 (1 µM) for 8 min at 37°C followed by washing caused a subsequent reduction in the binding capacity of [3H]NMS to 64 ± 1.8 and 44 ± 1.8%, respectively, without having a significant effect on KA (9.44 ± 0.04 and 9.52 ± 0.06 respectively). The data show that the aziridinium ions of AChM and BR384 bind irreversibly to the M1 muscarinic receptor.
Figure 2.
Effects of AChM and BR384 on the binding of [3H]NMS to homogenates of CHO cells expressing the M1 receptor. Cellular homogenate was incubated with the aziridinium ion of BR384 (1 µM) or AChM (10 µM) for 8 min at 37°C. These concentrations refer to the starting concentration of the nitrogen mustard before transformation to the aziridinium ion. Sodium thiosulphate (1 mM) was added subsequently, and the homogenates were washed by centrifugation and suspension in fresh buffer. Control homogenate was treated in the same manner except for exposure to the mustards. The data represent the mean specific binding values ± SEM of two experiments, each in triplicate. The mean ± SEM denotes the range of the two measurements, because N = 2.
Competitive binding experiments with AChM, BR384 and their transformation products
We determined the extent to which the aziridinium ion of AChM alkylates the M1 muscarinic receptor at 0°C. CHO hM1 cells were incubated with four concentrations of the aziridinium ion of AChM for 60 min, and then washed prior to measuring specific [3H]NMS binding. The results in Figure 3A show negligible alkylation of the M1 receptor by the aziridinium ion of AChM at 0°C, indicating that it should be possible to estimate its dissociation constant for the M1 receptor in a competitive binding assay at 0°C. Figure 3B shows the competitive effects of ACh, AChM and its transformation products on the binding of [3H]NMS to intact CHO hM1 cells at 0°C. Both the parent mustard and the alcoholic hydrolysis product of AChM were without effect on the binding of [3H]NMS, whereas ACh and the aziridinium ion of AChM behaved as competitive inhibitors. Their IC50 values were corrected for the competitive effect of [3H]NMS to yield log affinity constants of 4.77 ± 0.12 and 4.24 ± 0.13 respectively.
Figure 3.
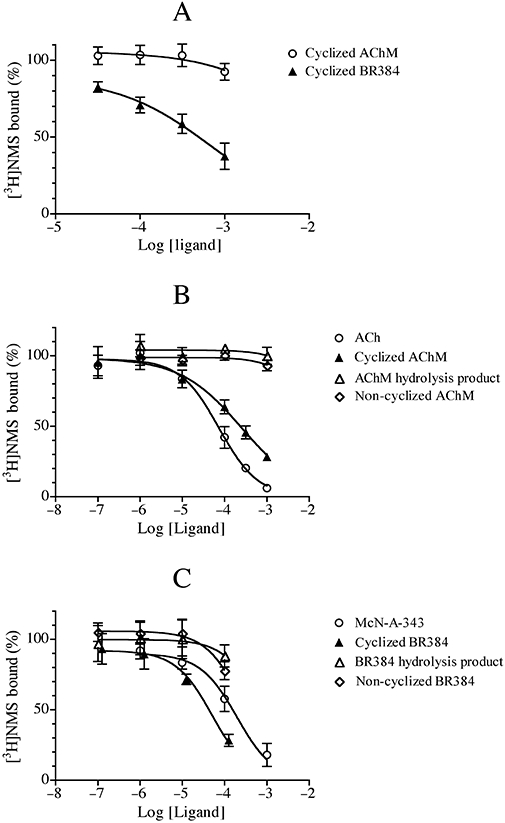
Effect of AChM, BR384, their transformation products, ACh and McN-A-343 on the binding of [3H]NMS to intact CHO hM1 cells at 0°C. (A) CHO hM1 cells were incubated with various concentrations of the aziridinium ions of AChM and BR384 for 1 h at 0°C, washed and then assayed for [3H]NMS binding at 0°C. (B) The competitive inhibition of the binding of [3H]NMS by various concentrations of ACh, AChM and its transformation products was measured at 0°C. (C) The binding of [3H]NMS in the presence of various concentrations of McN-A343, BR384 and its transformation products was measured at 0°C. The data represent the mean binding values ± SEM of three to four experiments, each in triplicate. The concentration of [3H]NMS was 1.0 nM, and its log affinity constant was estimated to be −9.53 in separate experiments at 0°C. The concentration of aziridinium ion is expressed as the total concentration of the parent mustard from which it was derived.
In contrast, the aziridinium ion of BR384 caused a moderate alkylation of the hM1 receptor after a 60 min incubation at 0°C as shown by the reduction in [3H]NMS binding measured after washing the reactive ligand from CHO hM1 cells (Figure 3A). We also examined McN-A-343, BR384 and its transformation products in competitive binding assays with [3H]NMS at 0°C (Figure 3C). Both the parent mustard and the alcoholic hydrolysis product had little effect on [3H]NMS binding, whereas McN-A-343 inhibited binding with a log affinity constant of 4.35 ± 0.14. The aziridinium ion of BR384 also inhibited binding, although the data are insufficient to resolve its reversible binding properties from its covalent interaction with the receptor. The data were analysed nonetheless using a competitive model to yield an ‘apparent’ log affinity constant of 4.93 ± 0.14.
Kinetics of hM1 receptor alkylation
We incubated M1 receptors with different concentrations of the aziridinium ions of the mustard analogs for various times, and then used [3H]NMS to measure the residual muscarinic receptor population after stopping the reaction and washing the cells (Figure 4A,C). The aziridinium ions of both AChM and BR384 caused a time- and concentration-dependent decrease in specific [3H]NMS binding. The kinetic data for AChM were fitted simultaneously to Equation (11) by nonlinear regression analysis to estimate the affinity constant of the aziridinium ion (K1) and the rate constant for alkylation (k1). A similar analysis was done for BR384 using Equation (8), which accounts for the decay in the aziridinium ion. These fits yielded K1 and k1 parameter estimates for AChM of 4.62 ± 0.07 and 0.10 ± 0.01 min−1, and for BR384 of 5.14 ± 0.03 and 0.55 ± 0.03 min−1 respectively. The estimates of the percentage of receptors not alkylated by AChM and BR384 were 8.1 and 4.5% respectively.
We also estimated the observed rate constant for alkylation (kobs) at each concentration of the mustards using an exponential decay equation. Only the data obtained during the first 8 min of incubation with BR384 were used to avoid error in the estimate of kobs due to decay of the aziridinium ion. A one-site equation was fitted to these estimates of kobs to yield the log affinity constant (K1) and the maximal rate constant for alkylation (k1) for the aziridinium ions of AChM (−4.08 ± 0.24 and 0.14 ± 0.015 min−1) and BR384 (−4.82 ± 0.09 and 0.61 ± 0.017 min−1) (Figure 4B,D). The latter values are similar to those estimated by global nonlinear regression analysis of the data according to Equations (8) and (11) as described above. The log affinity constant of the aziridinium ion of AChM, determined by the kinetic analysis at 37°C (4.62 ± 0.01), is also approximately similar to that estimated from the competitive binding assay at 0°C (4.24 ± 0.10; see Figure 3).
Effects of competitive inhibitors and allosteric modulators on receptor alkylation by AChM and BR384
To determine the site of action of the mustard analogs on the M1 receptor, we investigated how some known competitive inhibitors and allosteric modulators affect the kinetics of receptor alkylation by the mustard analogs. The rate of receptor alkylation by the aziridinium ion of AChM at concentrations of 0.1 mM (Figure 5A) and 1 mM (Figure 5B) was investigated in intact CHO hM1 cells in the presence of different concentrations of NMS. NMS caused a concentration-dependent slowing and ultimately stopped receptor alkylation by AChM at both the low and high concentrations of the mustard. Similar behaviour was observed in experiments with the aziridinium ion of BR384 at concentrations of 0.01 mM (Figure 5C) and 0.1 mM (Figure 5D). The data with both mustards are generally consistent with a competitive mechanism. To test this postulate, the competitive model [Equation (13)] was fitted to the data obtained at both concentrations of AChM by global nonlinear regression analysis sharing the estimate of the affinity constant of NMS between the data and setting the affinity constant of the aziridinium ion of AChM at a constant value (log K1 = 4.62) equivalent to that estimated from the data in Figure 3. A similar approach was used in the analysis of the data with BR384, except that Equation (10) was used to allow for the decay in the aziridinium ion of BR384. The estimates of the affinity constants of NMS in experiments with AChM and BR384 were 10.04 ± 0.07 and 9.97 ± 0.07 respectively. There was no significant improvement in residual error when the allosteric models [Equation (12) for AChM and Equation (9) for BR384] were fitted to the data.
Figure 5.
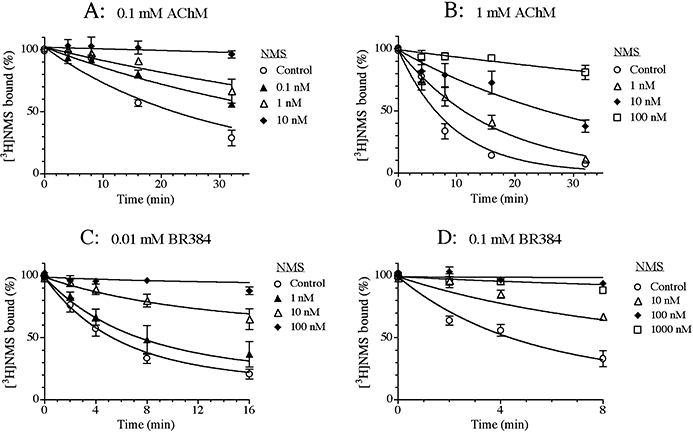
Effects of NMS on the kinetics of receptor alkylation by AChM (A, B) and BR384 (C, D). Intact CHO hM1 cells were incubated at 37°C with cyclized AChM at concentrations of 0.1 mM (A) or 1.0 mM (B), and in the presence of the indicated concentrations of NMS. Similar experiments were run with cyclized BR384 at concentrations of 0.01 mM (C) and 0.1 mM (D). Following incubation with the mustards, the cells were washed and assayed of [3H]NMS binding at a concentration of 1.0 nM. The data represent the mean binding values ± SEM of two (A), four (B), five (C) and five (D) experiments, each in triplicate. The concentration of cyclized nitrogen mustard is expressed as its initial concentration before transformation into the aziridinium ion.
Analogous experiments were carried out with the allosteric modulator gallamine in intact CHO hM1 cells (Figure 6). When the concentration of the aziridinium ion of AChM was low (0.1 mM), gallamine caused a concentration-dependent slowing in the rate of receptor alkylation (Figure 6A). When the concentration of the aziridinium ion of AChM was increased to 1 mM, however, the inhibitory effect of gallamine reached a plateau at high concentrations, consistent with an allosteric mechanism (Figure 6B). There was a substantial improvement in residual error when the data were fitted to the allosteric model [Equation (12)] as compared to the competitive model [Equation (13)] (F1,204 = 107.5; P = 1.7 × 10−20). The estimate of the log affinity constant of gallamine (K2) and its log cooperativity factor (α) for its interaction with the aziridinium ion of AChM were 5.06 ± 0.09 and −2.04 ± 0.07 respectively. We also observed similar behaviour in experiments with gallamine and BR384 at low (0.01 mM; Figure 6C) and high (0.1 mM; Figure 6D) concentrations. There was a highly significant improvement in residual error when the data were fitted to the allosteric model with [Equation (6)] as compared to the analogous competitive model [Equation (7)] (F1,110 = 135.4; P = 7.9 × 10−21). The estimates of the log affinity constant of gallamine for the unoccupied M1 receptor (K2) and the log of the cooperativity constant (α) for its interaction with the aziridinium ion of BR384 were 5.02 ± 0.08 and –1.73 ± 0.07 respectively.
Figure 6.
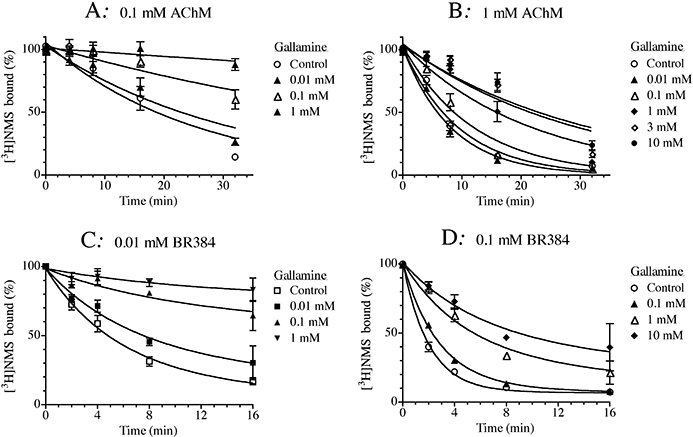
Effects of gallamine on the kinetics of receptor alkylation by AChM (A, B) and BR384 (C, D). Intact CHO hM1 cells were incubated at 37°C with cyclized AChM at concentrations of 0.1 mM (A) or 1.0 mM (B), and in the presence of the indicated concentrations of gallamine. Similar experiments were run with cyclized BR384 at concentrations of 0.01 mM (C) and 0.1 mM (D). Following incubation with the mustards, the cells were washed and assayed of [3H]NMS binding at a concentration of 1.0 nM. The data represent the mean binding values ± SEM of three (A), four (B), four (C) and two (D) experiments, each in triplicate. The concentration of cyclized nitrogen mustard is expressed as its initial concentration before transformation into the aziridinium ion.
The compound WIN 51708 is thought to bind to an allosteric site on the M1–M5 muscarinic receptors distinct from that of strychnine and gallamine (Lazareno et al., 2002), so we investigated its potential effects on M1 receptor alkylation by the aziridinium ion of BR384 in intact CHO hM1 cells (Figure 7). BR384 was used at a low concentration of 3 µM to optimize conditions for detecting a small allosteric effect. No influence on receptor alkylation was observed with WIN 51708 when used at concentrations of 3 and 10 µM. At a concentration (30 µM) approaching its limiting solubility, WIN 51708 reduced the observed rate constant for alkylation to 57% of the control value.
Figure 7.
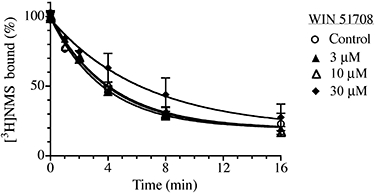
Effect of WIN 51708 on kinetics of receptor alkylation by BR384. Intact CHO hM1 cells were incubated at 37°C with cyclized BR384 at a concentration of 3.0 µM (initial concentration before cyclization) and in the presence of the indicated concentrations of WIN 51708. Following incubation with BR384, the cells were washed and assayed of [3H]NMS binding at a concentration of 1.0 nM. The data represent the mean binding values ± SEM of two experiments, each in triplicate. The mean ± SEM denotes the range of the two measurements, because N = 2.
The effects of McN-A-343 on receptor alkylation by the aziridinium ion of AChM at low (0.1 mM) and high (1.0 mM) concentrations are shown in Figure 8A and B respectively. These experiments used homogenates of CHO hM1 cells because the rate of alkylation was faster in homogenates compared to intact cells. This provided a greater range over which a slowing in the kinetics could be measured, which is useful for discriminating a large negatively cooperative effect from competitive inhibition. In homogenates, the average values of the observed rate constants of AChM (1.0 mM) and BR384 (0.1 mM) were 0.75 and 1.02 min−1, respectively, whereas the estimates of k1 in intact cells were 0.11 and 0.45 min−1 respectively. We have previously suggested that this difference may be related to recycling of receptors after the alkylation step (Suga et al., 2008). McN-A-343 caused a concentration-dependent slowing in the rate of receptor alkylation by AChM at both the high and low concentrations. Similar behaviour was noted in experiments investigating the influence of McN-A-343 on the alkylation of the M1 receptor by the aziridinium ion of BR384 (Figure 8C,D). Using the competitive model [Equation (13)], we estimated the affinity constant of McN-A-343 to be 5.23 ± 0.05 in experiments with AChM. The value estimated in experiments with BR384 was 5.21 ± 0.05 [using Equation (10)]. There was no significant improvement in residual error when the data were fitted to the allosteric models [Equation (12) for AChM and Equation (9) for BR384] as compared to the competitive models.
Figure 8.
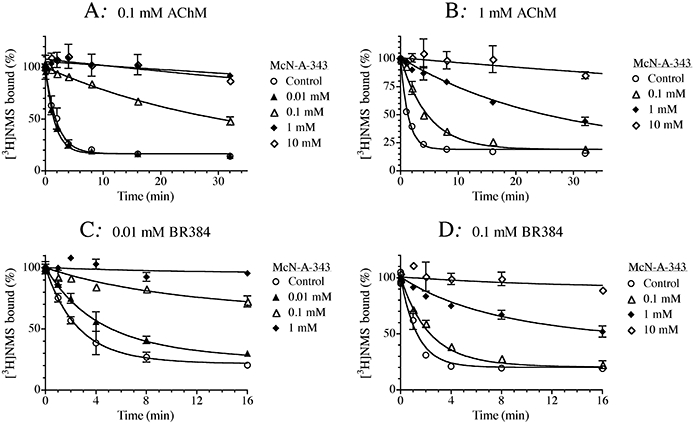
Effects of McN-A-343 on the kinetics of receptor alkylation by AChM (A, B) and BR384 (C, D). Homogenates of CHO hM1 cells were incubated at 37°C with cyclized AChM at concentrations of 0.1 mM (A) or 1.0 mM (B), and in the presence of the indicated concentrations of McN-A-343. Similar experiments were run with cyclized BR384 at concentrations of 0.01 mM (C) and 0.1 mM (D). Following incubation with the mustards, the cellular homogenates were washed and assayed of [3H]NMS binding at a concentration of 1.0 nM. The data represent the mean binding values ± SEM of two experiments, each in triplicate. The mean ± SEM denotes the range of the two measurements, because N = 2. The concentration of cyclized nitrogen mustard is expressed as its initial concentration before transformation into the aziridinium ion.
The influence of ACh on the rate of M1 receptor alkylation by the aziridinium ion of BR384 is shown in Figure 9. These experiments used homogenates of CHO hM1 cells using BR384 at concentrations of 0.01 mM (Figure 9A) and 0.1 mM (Figure 9B). The effect of ACh on receptor alkylation resembled that of a competitive inhibitor, with no evidence of a plateau in its inhibitory effect at high concentrations. The competitive model [Equation (10)] was fitted to the data to yield an estimate of the affinity constant of ACh (4.77 ± 0.08). No significant improvement in residual error occurred when the allosteric model [Equation (9)] was fitted to the data.
Figure 9.
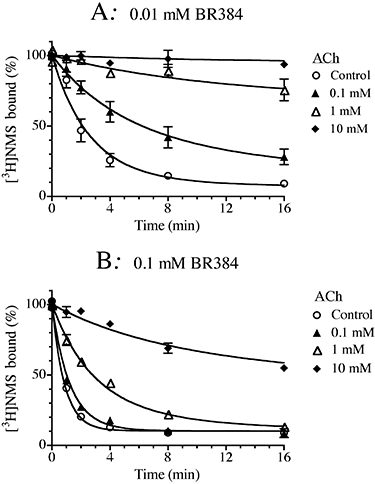
Effects of ACh on the kinetics of receptor alkylation by BR384. Homogenates of CHO hM1 cells were incubated at 37°C with cyclized BR384 at concentrations of 0.01 mM (A) or 0.1 mM (B), and in the presence of the indicated concentrations of ACh. Following incubation, cellular homogenate was washed and assayed of [3H]NMS binding at a concentration of 1.0 nM. The data represent the mean binding values ± SEM of two experiments, each in triplicate. The mean ± SEM denotes the range of the two measurements, because N = 2. The concentration of cyclized nitrogen mustard is expressed as its initial concentration before transformation into the aziridinium ion.
We also investigated the influence of NMS (Figure 10A) and gallamine (Figure 10B) on the rate of inactivation of muscarinic receptors in homogenates of rat cerebral cortex by the aziridinium ion of BR384 (0.1 mM). This brain region expresses mainly M1 and M4 muscarinic receptors, but also M2, M3 and M5 (Yasuda et al., 1993). NMS slowed the rate of receptor alkylation in a manner generally consistent with competition, whereas the inhibitory effect of gallamine reached a clear plateau at 170 µM and did not increase with a further increase in concentration. We did not analyse the data in detail because of the heterogeneity of the receptor population.
Figure 10.
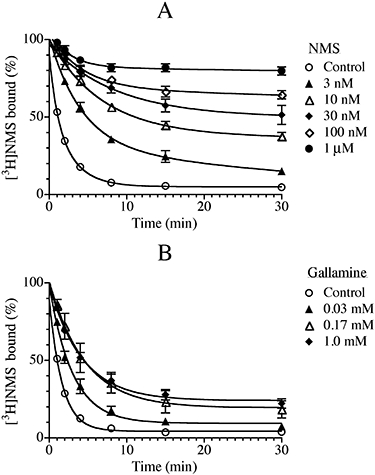
Effects of NMS (A) and gallamine (B) on the kinetics of receptor alkylation by BR384 in rat cortex. Tissue homogenates were incubated at 37°C with cyclized BR384 (0.1 mM, initial concentration before cyclization to the aziridinium ion) and in the presence of the indicated concentrations of NMS or gallamine. Following incubation with BR384, brain homogenate was washed and assayed of [3H]NMS binding at a concentration of 1.0 nM. The data represent the mean binding values ± SEM of two experiments, each in triplicate. The theoretical curves represent a best fit of an exponential decay equation to the data.
Effects of ACh, McN-A-343, NMS and gallamine on the time constant for receptor alkylation by AChM and BR384
A more intuitive way to analyse the kinetic data in Figures 5, 6, 8 and 9 is to plot the ratio of the time constant for alkylation in the presence of the inhibitor divided by that measured in its absence against the inhibitor concentration (Figure 11). Figure 11A shows the data obtained for the aziridinium ion of AChM at low (0.1 mM) and high (1.0 mM) concentrations. Both NMS and McN-A-343 increased the time constant for receptor alkylation in a manner that was proportional to their concentration. This effect occurred with both concentrations of AchM, and is indicative of a competitive mechanism. Although gallamine caused a concentration-dependent increase in the time constant at the low concentration of AChM, its inhibitory effect clearly reached a limit at the higher concentration of AChM. Similar behaviour is seen in the corresponding plot of the data for the aziridinium ion of BR384. The competitive model was fitted to the data for NMS, ACh and McN-A-343 (Figure 11B,C), whereas the allosteric model was fitted to the data for gallamine (Figure 11B). The results of this analysis were similar to those described above in connection with Figures 5, 6, 8 and 9, and are described in the legend to Figure 11. There was a tendency for the potency of NMS to be greater when antagonizing receptor alkylation by the higher concentration of BR384 (0.1 mM). As discussed later, this phenomenon may be attributed to the slow dissociation of NMS from the M1 receptor.
Figure 11.
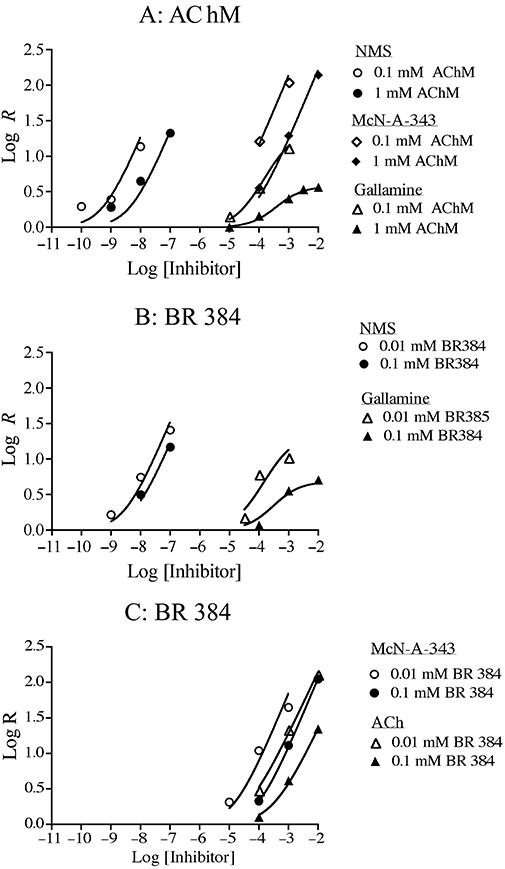
The effects of NMS, gallamine, McN-A-343 and ACh on the observed time constant for alkylation (τobs) of the M1 muscarinic receptor by AChM (A) and BR384 (B and C). The log ratio (R) of time constant for alkylation in the presence of the inhibitor divided by that measured in its absence is plotted against the log concentration of the inhibitor. The theoretical curves represent the best fit of Equations (16) (gallamine) and 17 (NMS, ACh and McN-A-343) to the data. The data obtained for a given test ligand at the two concentrations of AChM (0.1 and 1.0 mM) or BR384 (0.01 and 0.1 mM) were fitted simultaneously. Open symbols represent data obtained with 0.1 mM AChM or 0.01 mM BR384, whereas closed symbols denote data obtained with 1.0 mM AChM or 0.1 mM BR384. The concentration of cyclized nitrogen mustard is expressed as its initial concentration before transformation into the aziridinium ion. The R values were calculated from the data shown in Figures 5, 6, 8 and 9. The estimates of the parameters ± SEM in experiments with AChM are: NMS, pKi = 9.96 ± 0.12; gallamine, pK2 = 5.20 ± 0.11, log α = −2.08 ± 0.10; McN-A-343, pKi = 5.85 ± 0.05. The corresponding estimates for the experiments with BR384 are: gallamine, pK2 = 5.10 ± 0.17, log α = −1.77 ± 0.14; McN-A-343, pKi = 5.21 ± 0.06; ACh, pKi = 4.58 ± 0.04. With regard to NMS, the pK1 values at 0.01 and 0.1 mM BR384 were 9.47 ± 0.08 and 9.86 ± 0.13, respectively.
When plotting the relative time constant for alkylation against the inhibitor concentration as shown in Figure 11, it is difficult to discriminate between R values when the rate of receptor alkylation is very slow. Consequently, in Figure 11, we have omitted the R value for AChM (0.1 mM) in the presence of 10 mM McN-A343 as calculated from the experiments shown in Figure 8A. The rate of alkylation in the presence of 1 and 10 mM McN-A-343 appears similar, which might suggest allosteric behaviour. Nonetheless, it is possible to resolve this issue by repeating these measurements using a higher concentration of AChM (1 mM) as shown in Figure 8B. Under this conditions, a clear distinction between the rate of alkylation in the presence of 1 and 10 mM McN-A-343 is possible, which illustrates a consistency with a competitive interaction as shown in Figure 11. In Figure 11, we also omitted the R value measured at 1 µM NMS in the presence of 0.1 mM BR384 as calculated from the experiments shown in Figure 5D. In this case, the rates of receptor alkylation by BR384 in the presence of 0.1 and 1.0 µM NMS are both approximately zero. This situation represents a limitation in our ability to resolve competitive inhibition from high negative cooperativity because it is difficult to use much higher concentrations of the aziridinium ion of BR384.
Discussion
Both AChM and BR384 bind irreversibly to the M1 muscarinic receptor because prior treatment with the aziridinium ions of the mustards followed by washing caused a decrease in the binding capacity of [3H]NMS with no change in affinity (Figure 2). The mechanism presumably involves the direct covalent binding of the aziridinium ion to the orthosteric binding site. Spalding et al. (1994) have shown that [3H]AChM labels aspartic acid 105 in the M1 muscarinic receptor, which presumably explains the actions of AChM described here.
Because ACh and McN-A-343 equilibrate rapidly with muscarinic receptors, it is likely that the aziridinium ions of AChM and BR384 do as well, and that their rate of alkylation of muscarinic receptors should be proportional to receptor occupancy. This appears to be the case because the affinity constant of the aziridinium ion of AChM estimated in a competitive binding assay with [3H]NMS (Figure 3) generally agreed with that measured from the kinetics of receptor alkylation (Figure 4), although the two assays were at different temperatures (0 and 37°C respectively). In addition, there was general agreement between the affinities of various ligands measured in equilibrium binding assays with [3H]NMS and those estimated from their inhibition of the kinetics of the alkylation by AChM or BR384.
Because M1 receptor alkylation by the aziridinium ions of AChM or BR384 was prevented by NMS, ACh and McN-A-343 in a manner proportional to their concentration (Figures 5, 8 and 9), it seems likely that the nitrogen mustards interact competitively with the former ligands at the orthosteric binding site. In contrast, increasing concentrations of the allosteric ligand, gallamine, did not cause a proportional increase in the protection of the M1 receptor from alkylation by high concentrations of the nitrogen mustards (Figure 6). This behaviour is consistent with an allosteric interaction with the M1 receptor. Our estimate of the log affinity constant of gallamine for the allosteric site on the M1 receptor, based on its ability to interfere with the kinetics of alkylation (5.02), agreed with that measured in equilibrium binding experiments with [3H]NMS (5.21) (Matsui et al., 1995).
Our estimate of the negative cooperativity between the binding of gallamine and the aziridinium ions of the mustard analogs is based on the assumption that the microscopic rate constants for alkylation of the free and the gallamine-occupied receptors are the same (see Scheme 1). The estimate of the cooperativity between AChM and gallamine (log α = −2.01) is approximately the same as that measured between gallamine and ACh in binding experiments on rat cerebral cortex (−1.82) (Stockton et al., 1983), which is rich in M1 muscarinic receptors (Yasuda et al., 1993). This agreement suggests that gallamine has little or no effect on the rate constant for alkylation of M1 receptors by AChM. It seems likely that the same is true for BR384 because there is no evidence that gallamine alters the equilibrium between ground and active states of the receptor. It often affects ligand affinity only, perhaps through allosteric modulation of a peripheral docking site (Ehlert and Griffin, 2008).
Although our experiments showed that the interaction between BR384 and ACh was consistent with competitive inhibition, we cannot rule out the possibility that there is very high negative cooperativity between the two. The minimum (log) estimate of this negative cooperativity was −3.33 (α = 0.00046), which is defined as that required to cause a significant increase in residual error, assuming an allosteric mechanism. For the interaction between McN-A-343 and AChM, the minimum logarithm of the estimate was approximately −3.01 (α = 0.001). Similarly, we cannot rule out the possibility that there is very high negative cooperativity between the binding of NMS and BR384.
The dissociation constant of WIN 51708 for its allosteric site on the M1 receptor has been estimated to be 1.6 µM (Lazareno et al., 2002). If BR384 were binding to the allosteric site for WIN 51708, then a significant inhibition in the rate of receptor alkylation should have been detected with WIN 51708 at concentrations of 3 and 10 µM. In addition, if WIN 51708 exhibits a significant cooperative interaction, then this effect should have been observed at the low concentration of BR384 used (3 µM). Our data show that BR384 does not alkylate the allosteric site for WIN 51708 on the M1 receptor (Figure 7).
We cannot rule out the possibility that BR384 binds covalently to a third allosteric site, distinct from that for gallamine and WIN 51708, and exhibits very high negative cooperativity with ACh and [3H]NMS. Alternatively, it might be possible that BR384 alkylates the allosteric site for gallamine at low concentrations, but then at high concentrations, alkylates the orthosteric site. The converse situation is also possible. Investigating how the alkylation of the receptor is affected by mutagenesis of nucleophilic residues in the binding pocket (e.g. D105) should settle these questions. The simplest model to explain the data so far is a competitive interaction between BR384 and orthosteric ligands.
It is often assumed that, because of high reactivity, an electrophilic ligand is capable of alkylating a nucleophilic residue residing beyond its binding pocket. Thus, it might be argued that BR384 binds reversibly to an allosteric site, but reacts covalently with the nearby nucleophilic D105 at the orthosteric site of the M1 receptor. This hypothesis could explain why orthosteric ligands, like ACh and NMS, protect the M1 receptor from alkylation by BR384, even if the latter bound reversibly to an allosteric site. This mechanism is described in Scheme 2 where X denotes the aziridinium ion of BR384, RX the reversible complex of X with the allosteric site, XR the reversible complex of X with the orthosteric site, X-R the covalent complex of X with the orthosteric site, KA the affinity constant of X for the allosteric site and KB the unimolecular constant describing the equilibration of X between the two sites. Also shown is a competitive inhibitor (I) for the orthosteric site having an affinity constant denoted by KI. It can be shown that X binds to this circuit of two sites in a manner consistent with a simple one-site model having an observed dissociation constant (Kobs) equal to 1/(KX+KXKB) [see Appendix, Equation (A4)]. According to this model, the rate of alkylation of the orthosteric site would be proportional to receptor occupancy of the two equilibrating sites, and the half-maximal rate would occur when the concentration of BR384 was equivalent to Kobs. In addition, at high concentrations, an orthosteric ligand could completely inhibit the alkylation.
Scheme 2.
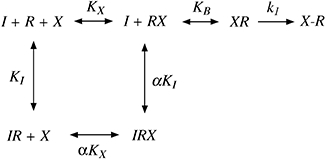
Model for the initial reversible interaction of the aziridinium ion (X) with an allosteric site on the M1 receptor followed by its subsequent irreversible binding to the orthosteric site in the presence of a competitive orthosteric ligand (I). The aziridinium ion first equilibrates with the receptor in the form of reversible complexes (XR and RX) as determined by the unimolecular constant KB. The RX complex converts to a covalent complex (R-X) as determined by the rate constant k1. The orthosteric ligand (I) only interacts reversibly with the orthosteric site. KX and KI denote the affinity constants of the reversible complexes of RX and IR, and α denotes the cooperativity between the binding of X and I.
There are four reasons, however, why this model cannot explain our data. First, the maximal rate constant for alkylation of the orthosteric site would be less than if it alkylated the site directly. For example, if BR384 alkylated the orthosteric site with a rate constant of k1, its observed rate constant (k1-obs) would be equivalent to [1/(1 + 1/KB)]k1[see Appendix, Equation (A9)]. Thus, if BR384 spent 90% of its time on the allosteric site and only 10% of its time on the orthosteric site (i.e. KB = 0.1), then k1-obs≈ 0.1k1. The relatively high rate constant of BR384 compared to AChM (see below) argues against Scheme 2. A ligand with a higher KB value would alkylate the orthosteric site with a faster rate, yet, by definition, such a ligand would be more orthosteric.
The second argument against Scheme 2 is that, under conditions where the aziridinium ion of BR384 does not bind covalently (i.e. low temperature), its maximal inhibition of orthosteric ligand binding would be incomplete and only reach a non-zero plateau of α/(1 +KB) times control binding when the radioligand concentration is low [see Appendix, Equation (A18)]. Both McN-A-343 and BR384 are capable of fully displacing [3H]NMS binding from the M1 receptor, however, as shown in Figure 3. If the negative cooperativity is very great (i.e. α << 1), however, then near complete displacement of binding could occur. This seems unlikely because the requisite negative cooperativity would need to be very high (−log α > 3).
A third reason against Scheme 2 is that, according to this model, allosteric ligands should competitively inhibit the covalent binding of BR384 to R2, yet gallamine and WIN 51708 do not (Figures 6 and 7).
Finally, the consequences of Scheme 2 predict that a large increase in the concentration of BR384 should be unable to overcome the inhibitory effect of orthosteric ligands on receptor alkylation [see Appendix, Equation (A12)], yet our data with ACh show a reciprocal competitive interaction (i.e. an increase in the concentration of one ligand can overcome the effect of an increase in the concentration of the other). In the case of NMS, it exhibited moderately greater potency at inhibiting receptor alkylation at the higher concentration of BR384, suggesting some deviation from a reciprocal competitive interaction. In these experiments, NMS was allowed to incubate with the receptor to reach equilibrium first, and then the aziridinium ion of BR384 was added to start the reaction at time zero. After this starting condition, it seems unlikely that the reversible binding interaction between NMS and the aziridinium ion is at equilibrium because NMS dissociates slowly from the M1 receptor. As a result, the estimate of the affinity constant of NMS is overestimated, particularly when it is used at high concentrations (Suga et al., 2008). The data with NMS illustrate a limitation in our analysis, because the latter is based on the assumption that the reversible ligand interactions equilibrate quickly compared to the rate of receptor alkylation. We expect that high-affinity ligands (i.e. log K1 > 8.0) would not satisfy this criterion.
The rate constant for alkylation of the M1 receptor by the aziridinium ion of AChM in intact cells was only one-fifth that of BR384, but approximately equal to that of a nitrogen mustard analog of oxotremorine-M (BM123) for alkylating muscarinic receptors in rat cortex (Ehlert and Jenden, 1985). The aziridinium ion of the latter compound alkylates superhigh- and high-affinity agonist binding sites in rat cerebral cortical homogenate at much slower rates (0.014 and 0.022 min−1) compared to that of the more abundant low-affinity agonist site (0.26 min−1). Because a greater proportion of superhigh- and high-affinity agonist–receptor complexes is likely to be in the active state, it has been suggested that BM123 alkylates the ground state of the receptor at a faster rate than the active state (Ehlert and Jenden, 1985). Spalding et al. (1994) have also given a similar explanation for the slower rate of aklylation of the M1 receptor by AChM compared to benzilylcholine mustard. A readily alkylated ground state may account, at least in part, for the larger rate constant of BR384 compared to that of AchM, because McN-A-343 is less efficacious than ACh, and the same relation probably applies to the aziridinium ions of BR384 and AChM.
Structural features of the aziridinium ion unrelated to efficacy are also capable of influencing the alkylation rate. For example, the rate constant for alkylation of cortical muscarinic receptors by 4-DAMP mustard (Thomas et al., 1992) is approximately one-fourth that of benzilylcholine mustard. Presumably, the piperidine ring of 4-DAMP mustard hinders the alkylation reaction as compared to the more flexible aliphatic ethylamine moiety of benzilylcholine mustard. In addition, the enantiomers of a nitrogen mustard analog of N-methyloxotremorine (BM130, N-[4-(2-chloromethylpyrrolidine)-2-butynyl]-2-pyrrolidone) having an asymmetric carbon atom in the quaternary aziridinum ring exhibit similar agonist activity on the guinea pig ileum and similar affinity for rat cortical muscarinic receptors, but the (+) enantiomer alkylates muscarinic receptors at a fourfold faster rate (Ringdahl et al., 1989).
We found that the rate constant for alkylation of the M1 receptor by AChM was faster in cellular homogenates than in intact cells. We observed similar behaviour at the M2 receptor and have suggested that the mechanism might involve recycling of internalized receptors after treatment with AChM (Suga et al., 2008). While this complication causes an underestimation in the rate constant for alkylation, it does not appear to alter the pattern of receptor protection causes by competitive or allosteric ligands.
The structural similarity of McN-A-343 with the aziridinium ion of BR384 (Figure 1) strongly suggests that the two ligands interact at the same site on the M1 muscarinic receptor. Our estimates of the log affinity constants of the two ligands were approximately the same (about 5.0), but varied somewhat across different experimental paradigms.
Our experiments with BR384 on rat cerebral cortex (Figure 10) are consistent with the behaviour that we observed at the human M1 receptor. The rat cortex is known to express M1 and M4 receptors in substantial abundance (35 and 40%, respectively) in addition to a moderate amount of the M2 receptor (15%) and less of the M3 and M5 subtypes (Yasuda et al., 1993). Our results suggest an orthosteric interaction with at least a majority of the sites in cortex. Studying recombinant muscarinic receptors in isolation should yield a more unambiguous conclusion. It has previously been shown that prior treatment of the rat cerebral cortex with the aziridinium ion of BR384 causes an irreversible decrease in the binding capacity of [3H]NMS without affecting its binding affinity (Ringdahl et al., 1990).
Measurement of the ability of a test drug to affect the irreversible binding of a site-directed electrophile is a powerful means of determining whether the drug interacts at allosteric or orthosteric sites on the receptor (Ehlert and Jenden, 1985; Suga et al., 2008). In the conventional binding experiment, the interaction of a drug with a radioligand having a defined binding site is usually studied. But, in cases where there is high negative cooperativity, it may not be feasible to measure radioligand binding at a high-enough concentration to discriminate a competitive inhibitor from negative allosteric modulator. Our approach does not suffer from this constraint because the primary interaction is between the electrophile and the test drug, both of which can be used over a wide range of concentrations. Evidence of their interactive behaviour is preserved after washing the receptor preparation because the electrophile binds covalently. The amount of unalkylated receptors can be determined easily using a low, feasible concentration of radioligand. As described herein, allosteric and competitive inhibitors differ in how they protect radioligand-binding sites from alkylation by a site-directed electrophile.
Kinetics are often viewed as a solution to the problem of discriminating between competitive inhibition and high negative cooperativity because, in the simplest case, a competitive inhibitor should have no effect on kinetics, whereas a negative allosteric modulator should increase dissociation kinetics. But at muscarinic receptors, mere occupancy of the allosteric site can prevent ligand traffic to and from the orthosteric site. This means that a major component of the effect of an allosteric modulator on the kinetics of radioligand binding may be unrelated to the microscopic rate constants for states of the orthosteric binding pocket, but rather, to a simple blockade of ligand traffic (Proska and Tucek, 1994). There is also a true allosteric component related to the change in the conformation of the receptor. Nonetheless, the capping effect of allosteric modulators on kinetics can be the predominant effect, and ligands that occupy the allosteric site without causing a change in the conformation of the orthosteric site can have large effects on kinetics without allosterically modulating the orthosteric site. While knowledge of whether a drug can occupy the allosteric site is useful, one can never determine from this type of kinetic experiment to which site the putative modulator binds with higher affinity – orthosteric or allosteric (Birdsall and Lazareno, 2005).
Although our results illustrate clear differences between allosteric and competitive inhibitors, the site-directed electrophiles that we used (AChM and BR384) are less than ideal because of their moderately low affinity (log K1 = 4.5–5.0). This places an upper limit of about 100-fold on the highest concentration (1 mM) of these agents that can be used relative to their dissociation constants. This working concentration range establishes a limit for the maximum amount of negative cooperativity that could be distinguished from competitive inhibition using our approach. Highly potent alkylating agents, like benzilylcholine mustard, are undesirable because of their bulky size and slow equilibration. For example, gallamine prevents the access and egress of [3H]NMS from the orthosteric-binding pocket of the M2 receptor, and greatly slows down its binding kinetics (Stockton et al., 1983). It is expected to do the same for benzilylcholine mustard. An ideal agent would be one of small size and moderate affinity (e.g. log K1 = 7.0). These properties would ensure rapid equilibration and reduce the possibility that bulky allosteric modulators would interfere with trafficking of the electrophile to and from the receptor. Various oxotremorine analogs display a broad range of potency and efficacy at muscarinic receptors, including antagonists with moderate affinity (log K1 = 7.0) that differ from oxotremorine only by the addition of a single methyl group (Ringdahl and Jenden, 1983; Ringdahl, 1988). Nitrogen mustard derivatives of these compounds may have utility in studying allosterism using the approach described here. The working concentration range of an alkylating agent having a dissociation constant of 0.1 µM could easily be 10 000-fold. This large range would greatly extend the maximum detectable amount of negative cooperativity between it and an interacting ligand. When used in combination with site-directed mutagenesis, our method has considerable power in discriminating competitive inhibition from negative allosteric modulation.
Acknowledgments
Initial feasibility studies were carried out with Don Jenden and the late Bjorn Ringdahl at UCLA under the support of the National Institutes of Health grant MH 17691. The work on rat cerebral cortex described here was supported by the National Institutes of Health grant NS 26511, and the remainder of the work was supported by the National Institutes of Health Grant GM 69829 and a Predoctoral Fellowship from the PhRMA Foundation (K.W.F.).
Glossary
Abbreviations:
- AChM
ACh mustard
- BR384
4-[(2-bromoethyl)methyl-amino]-2-butynyl N-(3-chlorophenyl)carbamate
- CHO hM1 cells
Chinese hamster ovary cells stably transfected with the human M1 muscarinic receptor
- KRB
Krebs–Ringer bicarbonate
- McN-A-343
[4-[[N-(3-chlorophenyl)carbamoyl]oxy]-2-butynyl] trimethylammonium chloride
- NMS
N-methylscopolamine
- WIN 51708
17-β-hydroxy-17-α-ethynyl-5-α-androstano[3,2-β]pyrimido[1,2-α]benzimidazole
Appendix
It is often suggested that because of its high reactivity, a nitrogen mustard can bind reversibly with its primary target, but then reacts covalently with a different nucleophilic target nearby. We have adapted this idea to consider how BR384 (X) would alkylate the M1 receptor if it bound reversibly with an allosteric site, but then alkylated the nearby orthosteric site coordinated by D105 in the M1 sequence (see Scheme 2). In this model, the affinity constant of BR384 for the allosteric site is denoted by KX. We assume that BR384 rapidly equilibrates with both sites in the form of reversible receptor complexes (i.e. XR and RX, orthosteric and allosteric, respectively) as described by the unimolecular constant, KB (KB = [RX]/[XR]). The rate constant for alkylation of the orthosteric site is denoted by k1, and the affinity of the competitive inhibitor (I) for the orthosteric site is denoted by KI. The cooperativity between the binding of I and X is denoted by α. The covalent complex of the aziridinium ion of BR384 with the orthosteric site is denoted by X-R. The affinity constants are defined in the conventional manner as the product of the products divided by that of the reactants [e.g. KX = [RX]/([X][R])]. Fractional receptor occupancy of this circuit of two sites by the aziridinium ion of BR384 in the form of reversible complexes (XR and RX) and in the presence of I is given by:
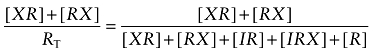 |
(A1) |
in which RT denotes the total amount of receptors. Each receptor complex in Equation (A1) can be replaced with an expression in terms of affinity constants and ligand concentrations. For example, RX = XKX[R]. Making analogous substitutions for the other receptor complexes and factoring out the term R yield:
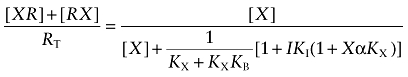 |
(A2) |
In the absence of I, Equation (A2) reduces to:
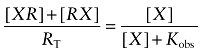 |
(A3) |
in which Kobs denotes the observed dissociation constant:
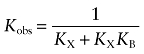 |
(A4) |
Fractional occupancy of the orthosteric site by the aziridinium ion of BR384 in the form of a reversible complex (XR) and in the presence of the orthosteric competitive inhibitor (I) is given by:
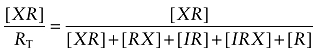 |
(A5) |
Replacing each receptor complex with an expression in terms of ligands and affinity constants yields:
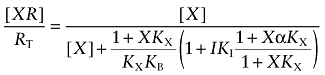 |
(A6) |
The observed rate constant for alkylation of the orthosteric site (kobs) in the presence of I is equivalent to the product of k1 and the fractional occupancy of the orthosteric site by the aziridinium ion of BR384, which is derived by multiplying Equation (A6) by k1:
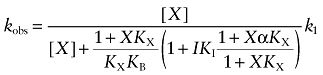 |
(A7) |
In the absence of I, Equation (A7) reduces to:
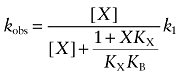 |
(A8) |
The maximal observed rate constant (kobs-max) for receptor alkylation in the absence of I can be determined by taking the limit of Equation (A8) as X approaches infinity:
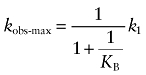 |
(A9) |
If KB is very small, and the aziridinium ion of BR384 spends most of its time on the allosteric site, then the maximal rate constant is approximately equal to:
| (A10) |
In contrast, if KB is large, then the observed rate constant for alkylation is approximately equal to:
| (A11) |
In this case, however, it is inappropriate to refer to BR384 as an allosteric ligand because it primarily binds to the orthosteric site and uses the allosteric site as a relay site for access to the orthosteric site.
In the presence of I, the maximal observed rate constant for alkylation (kobs-max′) can be derived by taking the limit of Equation (A7) as X approaches infinity:
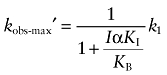 |
(A12) |
Equation (A12) shows that as the concentration of I increases to very high levels, the maximal observed rate constant for alkylation is approximately equal to 0. Thus, at high concentrations, an orthosteric ligand can completely prevent the alkylation of the orthosteric site by X in a manner that is insurmountable by an increasing concentration of X.
It is possible to use the model in Scheme 2 to predict how a reversible ligand, like McN-A-343 (D), inhibits the binding of a radioligand (L) to the orthosteric site. It is assumed that D binds reversibly in the same manner as the aziridinium ion of BR384, but does not alkylate the orthosteric site (k1 = 0). Fractional receptor occupancy (y) of orthosteric radioligand L in the presence of D is given by:
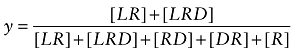 |
(A13) |
Replacing each receptor complex in Equation (A13) with an expression in terms of ligands and affinity constants, and then simplifying yields:
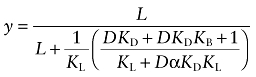 |
(A14) |
In this equation, KD and KL are used to denote the affinity constants of D and L in place of the corresponding values for X and I (KA and KI) used in Scheme 2. The fractional binding of the radioligand at a fixed concentration (F) in the presence of various concentrations of D can be described as the ratio of the right side of Equation (A14) divided by the same expression with the variable D = 0:
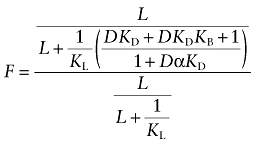 |
(A15) |
This equation reduces to:
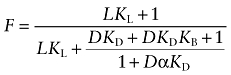 |
(A16) |
By taking the limit of Equation (A16) as L approaches 0, it is possible to predict how various concentrations of D inhibit the binding of radioligand L when the latter is at a very low fixed concentration:
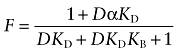 |
(A17) |
Under this condition (i.e. L approaches 0), the fractional binding of the radioligand in the presence of a maximally effective concentration of D can be predicted by taking the limit of Equation (A17) as D approaches infinity:
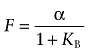 |
(A18) |
The maximal inhibition of radioligand binding by D (Imax) when L is very low can be estimated by subtracting Equation (A13) from 1:
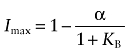 |
(A19) |
The concentration of D causing half of its maximal inhibition of the binding of L (IC50) can be estimated by subtracting one-half of Imax from 1, and setting the result equal to the right-hand side of Equation (A17), with D replaced with the variable IC50:
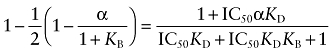 |
(A20) |
Solving for IC50 yields:
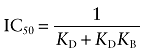 |
(A21) |
Because this equation was derived under the condition of a very low concentration of L, the IC50 in Equation (A21) represents the observed dissociation constant of D for the receptor system illustrated in Scheme 2. It is analogous to that described above in Equation (A4) for the reversible binding of the aziridinium ion.
Equation (A16) can also be used to determine the effect of an increase in the radioligand concentration on the ability of D to inhibit radioligand binding. It can be shown that as the concentration of L increases, the maximal inhibition of radioligand binding decreases. At sufficiently high concentrations of L, D has no effect on radioligand binding. This behaviour is inconsistent with the influence of McN-A-343 on [3H]NMS binding (see Figure 4).
Conflicts of interest
F.J.E. and H.S. have no conflicts of interest to declare. After completing this work, one of us (K.W.F.) took up a position with Johnson & Johnson.
References
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC) Br J Pharmacol. (4th edn.) 2009;158(Suppl. 1):S1–S254. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birdsall NJ, Lazareno S. Allosterism at muscarinic receptors: ligands and mechanisms. Mini Rev Med Chem. 2005;5:523–543. doi: 10.2174/1389557054023251. [DOI] [PubMed] [Google Scholar]
- Birdsall NJ, Burgen AS, Hulme EC. The binding of agonists to brain muscarinic receptors. Mol Pharmacol. 1978;14:723–736. [PubMed] [Google Scholar]
- Birdsall NJ, Burgen AS, Hulme EC, Stockton JM, Zigmond MJ. The effect of McN-A-343 on muscarinic receptors in the cerebral cortex and heart. Br J Pharmacol. 1983;78:257–259. doi: 10.1111/j.1476-5381.1983.tb09388.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehlert FJ, Griffin MT. Two-state models and the analysis of the allosteric effect of gallamine at the M2 muscarinic receptor. J Pharmacol Exp Ther. 2008;325:1039–1060. doi: 10.1124/jpet.108.136960. [DOI] [PubMed] [Google Scholar]
- Ehlert FJ, Jenden DJ. The binding of a 2-chloroethylamine derivative of oxotremorine (BM 123) to muscarinic receptors in the rat cerebral cortex. Mol Pharmacol. 1985;28:107–119. [PubMed] [Google Scholar]
- Figueroa KW, Griffin MT, Ehlert FJ. Selectivity of agonists for the active state of M1–M4 muscarinic receptor subtypes. J Pharmacol Exp Ther. 2008;328:331–342. doi: 10.1124/jpet.108.145219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammer R, Giachetti A. Muscarinic receptor subtypes: M1 and M2 biochemical and functional characterization. Life Sci. 1982;31:2991–2998. doi: 10.1016/0024-3205(82)90066-2. [DOI] [PubMed] [Google Scholar]
- Jackson CH, Hirst M. Syntheses and pharmacological actions of 2-((2-chloroethyl)methylamino)ethyl acetate and some of its derivatives on the isolated guinea pig ileum. J Med Chem. 1972;15:1183–1184. doi: 10.1021/jm00281a026. [DOI] [PubMed] [Google Scholar]
- Lazareno S, Farries T, Birdsall NJ. Pharmacological characterization of guanine nucleotide exchange reactions in membranes from CHO cells stably transfected with human muscarinic receptors M1–M4. Life Sci. 1993;52:449–456. doi: 10.1016/0024-3205(93)90301-i. [DOI] [PubMed] [Google Scholar]
- Lazareno S, Popham A, Birdsall NJ. Analogs of WIN 62 577 define a second allosteric site on muscarinic receptors. Mol Pharmacol. 2002;62:1492–1505. doi: 10.1124/mol.62.6.1492. [DOI] [PubMed] [Google Scholar]
- McKinney M, Miller JH, Gibson VA, Nickelson L, Aksoy S. Interactions of agonists with M2 and M4 muscarinic receptor subtypes mediating cyclic AMP inhibition. Mol Pharmacol. 1991;40:1014–1022. [PubMed] [Google Scholar]
- Martin JR. Pressor effect of the putative M1 muscarinic receptor agonist MCN-A-343 in the conscious rat. Life Sci. 1996;59:1839–1852. doi: 10.1016/s0024-3205(96)00531-0. [DOI] [PubMed] [Google Scholar]
- Matsui H, Lazareno S, Birdsall NJ. Probing of the location of the allosteric site on M1 muscarinic receptors by site-directed mutagenesis. Mol Pharmacol. 1995;47:88–98. [PubMed] [Google Scholar]
- May LT, Avlani VA, Langmead CJ, Herdon HJ, Wood MD, Sexton PM, et al. Structure–function studies of allosteric agonism at M2 muscarinic acetylcholine receptors. Mol Pharmacol. 2007;72:463–476. doi: 10.1124/mol.107.037630. [DOI] [PubMed] [Google Scholar]
- Mellin C, Vargas HM, Ringdahl B. Dimethylsulfonium analogues of the muscarinic agent McN-A-343: [4-[[N-(3- or 4-halophenyl)carbamoyl]oxy]-2-butynyl] dimethylsulfonium perchlorates. J Med Chem. 1989;32:1590–1593. doi: 10.1021/jm00127a031. [DOI] [PubMed] [Google Scholar]
- Proska J, Tucek S. Mechanisms of steric and cooperative actions of alcuronium on cardiac muscarinic acetylcholine receptors. Mol Pharmacol. 1994;45:709–717. [PubMed] [Google Scholar]
- Ringdahl B. 5-Methyl-2-pyrrolidone analogues of oxotremorine as selective muscarinic agonists. J Med Chem. 1988;31:683–688. doi: 10.1021/jm00398a031. [DOI] [PubMed] [Google Scholar]
- Ringdahl B, Jenden DJ. Pharmacological properties of oxotremorine and its analogs. Life Sci. 1983;32:2401–2413. doi: 10.1016/0024-3205(83)90365-x. [DOI] [PubMed] [Google Scholar]
- Ringdahl B, Katz ED, Roch M, Jenden DJ. Muscarinic actions and receptor binding of the enantiomers of BM 130, an alkylating analog of oxotremorine. J Pharmacol Exp Ther. 1989;249:210–215. [PubMed] [Google Scholar]
- Ringdahl B, Mellin C, Ehlert FJ, Roch M, Rice KM, Jenden DJ. Tertiary 2-haloethylamine derivatives of the muscarinic agent McN-A-343, [4-[[N-(3-chlorophenyl)carbamoyl]oxy]-2-butynyl]trimethylammonium chloride. J Med Chem. 1990;33:281–286. doi: 10.1021/jm00163a046. [DOI] [PubMed] [Google Scholar]
- Roszkowski AP. An unusual type of sympathetic ganglionic stimulant. J Pharmacol Exp Ther. 1961;132:156–170. [PubMed] [Google Scholar]
- Spalding TA, Birdsall NJ, Curtis CA, Hulme EC. Acetylcholine mustard labels the binding site aspartate in muscarinic acetylcholine receptors. J Biol Chem. 1994;269:4092–4097. [PubMed] [Google Scholar]
- Stockton JM, Birdsall NJ, Burgen AS, Hulme EC. Modification of the binding properties of muscarinic receptors by gallamine. Mol Pharmacol. 1983;23:551–557. [PubMed] [Google Scholar]
- Suga H, Figueroa KW, Ehlert FJ. Use of acetylcholine mustard to study allosteric interactions at the M(2) muscarinic receptor. J Pharmacol Exp Ther. 2008;327:518–528. doi: 10.1124/jpet.108.141234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sur C, Mallorga PJ, Wittmann M, Jacobson MA, Pascarella D, Williams JB, et al. N-desmethylclozapine, an allosteric agonist at muscarinic 1 receptor, potentiates N-methyl-d-aspartate receptor activity. Proc Natl Acad Sci U S A. 2003;100:13674–13679. doi: 10.1073/pnas.1835612100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas EA, Hsu HH, Griffin MT, Hunter AL, Luong T, Ehlert FJ. Conversion of N-(2-chloroethyl)-4-piperidinyl diphenylacetate (4-DAMP mustard) to an aziridinium ion and its interaction with muscarinic receptors in various tissues. Mol Pharmacol. 1992;41:718–726. [PubMed] [Google Scholar]
- Valant C, Gregory KJ, Hall NE, Scammells PJ, Lew MJ, Sexton PM, et al. A novel mechanism of G protein-coupled receptor functional selectivity. Muscarinic partial agonist McN-A-343 as a bitopic orthosteric/allosteric ligand. J Biol Chem. 2008;283:29312–29321. doi: 10.1074/jbc.M803801200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waelbroeck M. Identification of drugs competing with d-tubocurarine for an allosteric site on cardiac muscarinic receptors. Mol Pharmacol. 1994;46:685–692. [PubMed] [Google Scholar]
- Yasuda RP, Ciesla W, Flores LR, Wall SJ, Li M, Satkus SA, et al. Development of antisera selective for M4 and M5 muscarinic cholinergic. Mol Pharmacol. 1993;43:149–157. [PubMed] [Google Scholar]