

Abstract
Background and purpose:
Current therapies for muscular dystrophy are based on corticosteroids. Significant side effects associated with these therapies have prompted several studies aimed at identifying possible alternative strategies. As inflammation and defects of nitric oxide (NO) generation are key pathogenic events in muscular dystrophies, we have studied the effects of combining the NO donor isosorbide dinitrate (ISDN) and the non-steroidal anti-inflammatory drug ibuprofen.
Experimental approach:
α-Sarcoglycan-null mice were treated for up to 8 months with ISDN (30 mg·kg−1) plus ibuprofen (50 mg·kg−1) administered daily in the diet. Effects of ISDN and ibuprofen alone were assessed in parallel. Drug effects on animal motility and muscle function, muscle damage, inflammatory infiltrates and cytokine levels, as well as muscle regeneration including assessment of endogenous stem cell pool, were measured at selected time points.
Key results:
Combination of ibuprofen and ISDN stimulated regeneration capacity, of myogenic precursor cells, reduced muscle necrotic damage and inflammation. Muscle function in terms of free voluntary movement and resistance to exercise was maintained throughout the time window analysed. The effects of ISDN and ibuprofen administered separately were transient and significantly lower than those induced by their combination.
Conclusions and implications:
Co-administration of NO and ibuprofen provided synergistic beneficial effects in a mouse model of muscular dystrophy, leading to an effective therapy. Our results open the possibility of immediate clinical testing of a combination of ISDN and ibuprofen in dystrophic patients, as both components are approved for use in humans, with a good safety profile.
Keywords: nitric oxide, inflammation, muscular dystrophy, skeletal muscle regeneration
Introduction
Muscular dystrophies are a group of genetic, hereditary muscle diseases characterized by defects in muscle proteins that induce progressive skeletal muscle damage accompanied by fibre necrosis and chronic local inflammation, leading to substitution of fibres by connective and adipose tissue (Emery, 2002). In the most severe forms, such as Duchenne muscular dystrophy, continuous and progressive skeletal muscle damage leads the patient to complete paralysis and death, usually by respiratory and/or cardiac failure (Emery, 2002). The pharmacological therapies currently in use are based on corticosteroids and provide only a temporary delay of the disease progression and is associated with severe side effects (Manzur et al., 2004). Alternative pharmacological strategies are thus continually proposed and investigated, including protease inhibitors, calcium antagonists and antioxidants, compounds that induce correct dystrophin gene expression, modulate muscle growth or stabilize the link between cytoskeleton and extracellular matrix. So far none of these therapies has yielded favourable outcomes in clinical trials (Wagner, 2008).
Molecular dystrophies have a complex pathogenesis as the original genetic defect leads to a host of concurrent pathogenic events. A possible successful pharmacological strategy is to use either individual drugs or drug combinations that address comprehensively the complexity of the dystrophic phenotype. There are two key pathological events that appear particularly susceptible to pharmacological intervention in muscular dystrophy. One is local inflammation, which is now recognized to play a significant role in fibre destruction and in progression of muscular dystrophies (Spencer and Tidball, 2001). Indeed, DNA microarray or biochemical data show that inflammatory mediators/effectors dominate the expression profile of muscles from the mdx mouse model of dystrophy (Porter et al., 2002).
A second pathogenic effect is reduced generation of nitric oxide (NO). Delocalization of nNOS and loss of NO generation that occur in muscular dystrophies (Bredt, 1998; Rando, 2001) have essential roles in muscle inactivity and fatigue (Kobayashi et al., 2008; Lai et al., 2009) and in the neurogenic defects associated with the disease (Deng et al., 2009) so much so that restoration of NO generation by transgenic expression of nNOS ameliorates the dystrophic phenotype (Wehling et al., 2001; Wehling-Henricks et al., 2009). NO protects muscle from damage during contractile activity by enhancing vasodilation and bioavailability of nutrients to muscle, as well as energy generation through enhanced glucose uptake, glycolysis and mitochondrial biogenesis (Stamler and Meissner, 2001; Nisoli et al., 2003; Wehling-Henricks et al., 2009). In addition, NO enhances repair of the damaged muscle via specific actions on survival, activation and differentiation of satellite cells, the mononuclear progenitors of myocytes, able to form new fibres (Anderson, 2000; Pisconti et al., 2006; Colussi et al., 2008; Percival et al., 2008; Filippin et al., 2009). Pharmacological strategies based on either anti-inflammatory drugs apart from corticosteroids or NO generation, tested in the mdx mouse model of the disease, have, however, shown a limited efficacy (Grounds and Torrisi, 2004; Voisin et al., 2005; Reutenauer et al., 2008; Huang et al., 2009; Wang et al., 2009).
We have previously reported that HCT 1026, belonging to a new class of NO-releasing drugs, in which the NO moiety is linked with a non-steroidal anti-inflammatory drug (NSAID), has significant and persistent therapeutic effects in the mdx mouse, with an efficacy superior to that of the corticosteroid prednisolone (Brunelli et al., 2007b). In the same study we found that HCT 1026 was effective also on the α-sarcoglycan (α-SG)-null mouse, a model of dystrophy that has a severe phenotype in the mouse with features resembling those of human Duchenne muscular dystrophy (Duclos et al., 1998). These results suggested that NO release and inhibition of inflammation synergized to yield a novel effective therapy in muscular dystrophy.
In this study we have tested this hypothesis studying the efficacy of the combined administration of the NO donor isosorbide dinitrate (ISDN) and the NSAID ibuprofen in the α-SG-null mouse. Our results show that the combination of these drugs has significant and persistent therapeutic effects due to both the anti-inflammatory and NO properties, through actions on both muscle preservation and regeneration. By contrast, ISDN and ibuprofen when administered as single drugs did not show significant therapeutic effects. ISDN and ibuprofen are approved for use in humans, including in paediatric patients; the results we report therefore open the possibility of their immediate clinical testing as a treatment in dystrophic patients. Of importance, a therapeutic regimen in which the NO and NSAID therapeutic activity are associated with different molecules allows their doses to be adjusted separately from each other to meet a more refined and personalized therapy.
Methods
Animals and treatment
All animal care and experimental procedures were in accordance with the European Community guidelines and were approved by the Institutional Ethical Committee. α-SG-null mice were a kind gift of Dr K Campbell (Iowa University, Iowa City, IA, USA). Animals were housed in the pathogen-free facility at our Institution. α-SG-null mice are characterized by the development of histopathological features of muscle dystrophy with ongoing fibre degeneration, fibrous tissue deposition and infiltration of inflammatory cells (Duclos et al., 1998). Animal groups (15 animals per group) were treated as follows. Animal were given in their diet (Mucedola, Milano, Italy) starting at 1 month of age for up to 8 months: (i) 30 mg·kg−1·day−1 of ISDN (Alexis, Lausen, Switzerland) and 50 mg·kg−1·day−1 of ibuprofen (Sigma-Aldrich, MO, USA) combination, (ii) 30 mg·kg−1·day−1 of ISDN; (iii) 50 mg·kg−1·day−1 of ibuprofen; and (iv) standard diet without drug additions. Diets were prepared based on the daily food intake measured for these animals (Brunelli et al., 2007b). Mice and diets were weighed and food intake was calculated weekly during the treatment. No significant differences in food intake and weight gain were observed among the experimental groups. Creatine kinase (CK) serum levels were assessed in each animal monthly from the first month of treatment (2 months of age) and functional tests (free wheel running and treadmill) were performed sequentially at the fourth and eighth month of treatment on seven animals per group. One week after completing the two functional tests, the seven mice for each treatment group were killed by neck dislocation for histological analyses, cytokine measurements and preparation of primary myoblasts.
To assess the steady state concentrations of ibuprofen, nitrites and nitrates reached during the treatment, four additional groups of mice (seven mice per group) were treated as above for 1 week after which drug concentrations were tested.
Ibuprofen plasma level determination
Plasma was obtained by cardiac puncture and centrifuged at 13 000×g at 4°C for 5 min to remove cells. Plasma ibuprofen concentrations were assessed using previously published methods (Farrar et al., 2002; Zhao et al., 2005) with minor modifications. Briefly, 25 mL of internal standard solution (indomethacin 100 mg·mL−1) and 500 mL of methanol were added to 100 mL of plasma in 1.5 mL polypropylene centrifuge tubes. The tubes were mixed for 30 s and centrifuged for 10 min at 10 000×g. The supernatant was transferred to a clean polypropylene tube and evaporated to dryness. The residue was reconstituted with 150 µL of a solution containing acetonitrile and 50 mM of acetate buffer, pH 4.5 (70/30), and 20 µL of this solution was injected for HPLC analysis using X Bridge C18 150 × 4.6, 3.5 mm (Waters Corporation, MA, USA) for the chromatographic separation. The detector was set at 220 nm, the flow was 1 mL·min−1 and the mobile phase consisted of acetonitrile and 50 mM of acetate buffer pH 4.5 (50/50). The retention times of ibuprofen and internal standard were 5.4 and 7.5 min respectively. The assay was linear from 0.5 to 100 mg·mL−1, imprecision and inaccuracy was below 10%.
Nitrite and nitrate plasma level determination
For quantitative measurements, prior to the derivatization procedure, plasma samples (100 µL) were spiked with the 15N-labelled nitrite and nitrate as internal standards to achieve final concentrations of 0.2 mM for [15N]nitrite and 2 mM for [15N]nitrate. Derivatization of nitrite and nitrate was performed as described (Tsikas, 2000). Briefly, acetone (400 µL) and pentafluorobenzyl bromide (PFB-Br; 10 µL) were added to the samples and they were incubated at 50°C for 60 min. After derivatization acetone was evaporated under a nitrogen stream, reaction products were extracted from the remaining aqueous phase by vortex-mixing with toluene (1 mL) for 1 min and a 700 µL aliquot of the organic phase was transferred into 1.5-mL glass vials for GC–MS analysis.
GC–MS was carried out on a Finnigan Trace (DSQ) 7000 apparatus (San Jose, CA, USA) connected directly to a Finnigan Trace gas chromatograph Ultra. The fused silica capillary column used was an Rtx-5MS (15 m × 0.25 mm I.D., 0.25 µm film thickness) from Restek (Bellefonte, USA). The following temperature programme was used in GC–MS: the column was held at 90°C for 5 min then increased to 200°C at a rate of 50°C/min. Helium (1.0 mL·min−1) and methane (2.2 mL·min−1) were used as the carrier and the reagent gases, respectively, for negative-ion chemical ionization. Injector, interface and ion-source were held constant at 200°C, 250°C and 200°C respectively. Aliquots (1 µL) were injected in the splitless mode. Calculation of concentrations of nitrites and nitrates in biological fluids was performed as described previously (Tsikas, 2000).
Analysis of biochemical and functional parameters
Serum CK levels were measured in blood samples obtained from the tail vein. The blood was centrifuged at 13 000×g at 4°C and the supernatant used to measure CK activity in an indirect colorimetric assay (Randox, UK) (Brunelli et al., 2007a).
Functional muscle activity was measured using both the running wheel, to assess free locomotor activity, and the exhaustion treadmill, to assess resistance to fatigue. The protocols used in these studies have been previously described (Brunelli et al., 2007b). In brief, for the running wheel test, mice were housed individually in cages (9 × 22 × 9 cm) equipped with a running wheel (20 cm in diameter, Trixie, Germany). Each wheel revolution was registered by a magnetic switch, which was connected to a speedometer. The distance was recorded continuously for 24 h. Three tests were performed on the same animal allowing 2 days in between each test. Results shown are average of the three tests.
For the exhaustion treadmill test, analyses were carried out using a six-lane motorized treadmill (Exeter 3/6 Treadmill; Columbus Instruments, Columbus, OH, USA) supplied with shocker plates. The initial trials were performed at low intensity and for short duration to get the mice accustomed to the exercise; the treadmill was then run at an inclination of 0° at 5 m·min−1 for 5 min, after which the speed was increased 1 m·min−1 every 3 min until a speed of 10 m·min−1 was reached. The test was stopped when the mouse remained on the shocker plate for 20 s without attempting to reengage the treadmill. The time to exhaustion was determined from the beginning of the test to the time of exhaustion. The treadmill inclination was maintained at 0° throughout the test. Three tests were performed on the same animal allowing 2 days in between each test. Results shown are the averages of the three tests.
Histological analysis
The animals were killed by cervical dislocation and tibialis anterior or diaphragm muscles were dissected and immediately frozen in liquid N2-cooled isopentane. Analyses were carried out as previously described (Brunelli et al., 2007b). In brief, serial muscle sections were obtained and stained with haematoxylin and eosin following standard procedures. Ten random images for each muscle were taken at 10× magnification with a S100 TV microscope (Carl Zeiss MicroImaging Inc, Iena, Germany). The number of necrotic and centronucleated fibres and the number of infiltrating inflammatory cells was counted and analysed using a digitized imaging systems (ImageJ 1.38× National Institute of Health). Necrotic cells were identified by hyper-eosinophilia, thinning and waviness and, eventually, presence of many nuclei (Ontell, 1981). To analyse the inflammatory infiltrates further, we performed an immunofluorescence staining of diaphragm sections obtained at 4 months from untreated and treated animals employing an anti-laminin antibody (Sigma-Aldrich, MO, USA) to identify skeletal muscle fibres and an anti-CD11b antibody for inflammatory infiltrate staining (BD PharMingen, CA, USA) followed by appropriated secondary antibodies (Alexa Fluor, Invitrogen,OR, USA). Ten random images for each muscle were taken at 40 × magnification using a Leica TCS SP2 Laser Scanning Confocal microcope (Leica, Mannheim, Germany) and analysed using a digitized imaging systems (ImageJ 1.38× National Institute of Health).
Cytokine determination
Tibialis muscles were isolated after death and rapidly homogenized in 20 mM Tris-HCl, containing 137 mM NaCl, 5 mM EDTA and a protease inhibitor cocktail (Roche, Basel, Switzerland), pH 8.0, and centrifuged at 3.000 rpm for 5 min at 4°C. Protein content in the samples was determined by the bicinchoninic acid assay (BCA, Pierce, Bezons, France). Concentrations of transforming growth factor β (TGF-β), tumour necrosis factor α (TNF-α) and CCL3/macrophage inflammatory protein 1α (MIP-1α) were determined on equal amounts of protein lysates using the Quantikine elisa kits (R&D System, Minneapolis, MN, USA) according to manufacturer's instructions. CCL2/monocyte chemoattractant protein-1 (MCP-1) was determined with an elisa kit from Biosource (CA, USA) Chemokine and other nomenclature follows Alexander et al. (2009).
Primary myoblast cell isolation and analysis
Primary myoblasts were isolated from quadriceps and gastrocnemius muscles as described (Sciorati et al., 2006). Briefly, muscles were digested with a mixture of collagenase and dispase and the fibroblasts removed. Myoblasts were counted and stained with fluorescein isothiocyanate-conjugated CD34 antibody (AbD Serotec, Oxford, UK) and phycoerythrine-conjugated α7-integrin antibody (MBL, Woburn, MA, USA) according to manufacturer's protocol. The number of positive cells was measured by flow cytometry analysing 10.000 events for each sample (FACScalibur, Becton Dickinson, San Josè, CA, USA). Aliquots (1 × 105) of isolated cells were cultured using Matrigel-coated dishes in DMEM supplemented with 10% foetal bovine serum, 3% chick embryo extract, 100 U·mL−1 penicillin, 100 µg·mL−1 streptomycin and 50 µg·mL−1 gentamycin for 1 week. To induce myotube formation cells were than cultured for 24 h in differentiation medium (DMEM supplemented with 2% horse serum, 100 U·mL−1 penicillin and 100 µg·mL−1 streptomycin). Cells were lysed and analysed for differentiation proteins expression by SDS electrophoresis as described (Sciorati et al., 2006).
Statistical analysis
Results were expressed as the means ± SEM or median (plus 90% interval of confidence) according to their distribution based on results of the Kolmogorov–Smirnov normality test. For the parameters with a normal distribution, the anova test was used, whereas the Mann and Whitney test was used for parameters with non-normal distribution. A P-value of less than 0.05 was considered as statistically significant.
Results
Evaluation of the plasma concentrations of NO and ibuprofen in α-SG-null mice treated with ibuprofen and/or ISDN
We measured the plasma levels reached by the drugs when incorporated into the diet, assessing the steady state plasma concentrations of nitrites and nitrates (as a measure of NO), and ibuprofen after 1 week of treatment in α-SG-null mice receiving 50 mg·kg−1·day−1 of ibuprofen, 30 mg·kg−1·day−1 ISDN or ibuprofen plus ISDN. Table 1 shows the mean values of the plasma concentration of nitrites, nitrates and ibuprofen achieved with the different treatments. The values obtained for ibuprofen are in the range of those observed in patients treated with a paediatric dose of 150–300 mg·day−1 of the drug (Steen et al., 2000; Lotsch et al., 2001). The plasma concentrations of ibuprofen did not change when it was given alone or in combination with ISDN, showing no major pharmacokinetic interactions between the drugs.
Table 1.
Plasma ibuprofen, nitrite and nitrate levels
| Treatment | Ibuprofen (mg·mL−1 ± SEM) | Nitrites (µM ± SEM) | Nitrates (µM ± SEM) |
|---|---|---|---|
| Ibuprofen | 8.17 ± 0.73 | 3.13 ± 0.13 | 29.91 ± 2.27 |
| Ibuprofen plus ISDN | 7.35 ± 0.85 | 4.16 ± 0.11* | 46.16 ± 3.61* |
| ISDN | 4.32 ± 0.10* | 49.16 ± 2.37* | |
| STD | 3.27 ± 0.11 | 30.52 ± 3.89 |
P < 0.01 versus ibuprofen.
Plasma level of ibuprofen, nitrites and nitrates in mice treated for 1 week with standard diet (STD) or a diet containing ibuprofen, alone or together ISDN (n = 7).
ISDN, isosorbide dinitrate; STD, standard diet.
α-SG-null animal groups were then fed with standard diet or the various drug combinations in the diet for up to 8 months during which parameters assessing function, architecture, inflammatory status and ability to repair of the dystrophic muscle were evaluated. Weight and food intake were routinely assessed and no significant differences were observed between the animal groups.
Ibuprofen plus ISDN ameliorate muscle function persistently in α-SG-null mice
To assess muscle function we relied on in vivo analyses that allowed longitudinal functional evaluations in the same animal during a long period of treatment. We employed two independent tests: the free wheel test that measured the voluntary running capacity (Figure 1A) and the treadmill, that measured resistance to fatigue during a forced exercise (Figure 1B). Tests were applied after 4 and 8 months of treatment. The treatment with ibuprofen plus ISDN yielded significant amelioration of the functional parameters both at the free wheel and treadmill tests. By contrast, treatment with only ibuprofen or ISDN failed to ameliorate functional parameters at the treadmill test, and yielded only a slight, although significant, improvement in the free wheel test.
Figure 1.
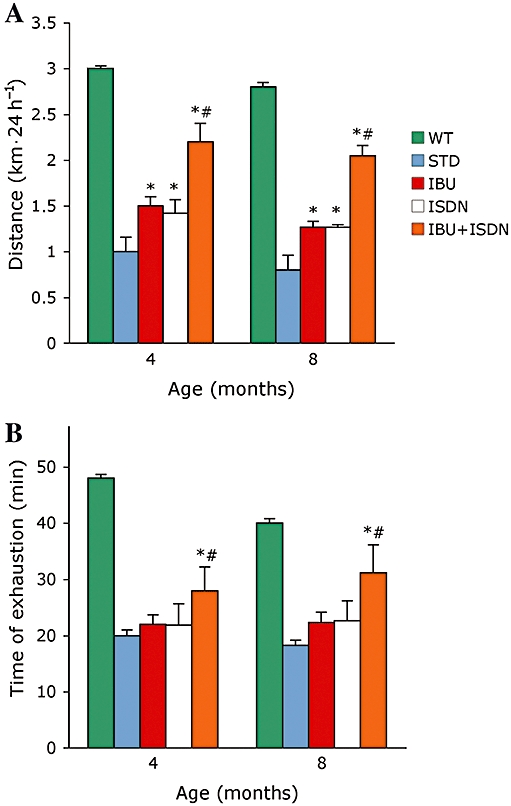
Ibuprofen plus ISDN ameliorate muscle function over the long term, in α-SG-null mice. Free wheel running to test spontaneous movement (A) or treadmill test to measure resistance to fatigue (B) was carried out with mice treated with standard diet (STD) or a diet containing ibuprofen (IBU), ISDN or the two drugs combined. Parameters measured in matched wild-type animals (WT) are shown for comparison. *P < 0.01 for the effect of each treatment versus STD, #P < 0.01 for the effect of IBU plus ISDN versus that of each single drug; n = 7. α-SG, α-sarcoglycan; ISDN, isosorbide dinitrate.
Ibuprofen plus ISDN affect CK release and reduce skeletal muscle necrosis in α-SG-null mice
As an in vivo indicator of skeletal muscle damage we analysed the serum levels of CK, a skeletal muscle enzyme released during fibre degeneration (Duclos et al., 1998). In untreated α-SG-null mice, the serum CK activity increased progressively up to the fourth month of age. In this time-window, treatment with ibuprofen plus ISDN reduced CK levels in the serum with respect to those observed in untreated animals (Figure 2A), suggesting that disease progression had been slowed by the treatment. In agreement, CK values in treated animals decreased progressively throughout the time window analysed. The observation that from the fifth month onwards, CK values in untreated animals decreased significantly, is consistent with the above observation as CK values depend not only on the severity of muscle damage (see Figures 2B and 3) but also on the extent of muscle mass that is damaged, and in these animals there is significant progressive substitution of muscular with fibrotic tissue (Duclos et al., 1998; Brunelli et al., 2007b). This may also explain why CK values in ibuprofen plus ISDN from fifth month onwards appear to be higher than those observed in the untreated animals at the same time-window. The fact that necrosis is still reduced by the treatment from the fifth month onwards sustains this hypothesis, although other hypothesis such as progressive damage to muscle at later stages of the disease cannot be excluded. The treatment with only ibuprofen or ISDN yielded reductions of CK serum levels which were less than those induced by the combined drug treatment and transient, as after the fourth month of treatment, values were similar to those observed in the untreated control group.
Figure 2.
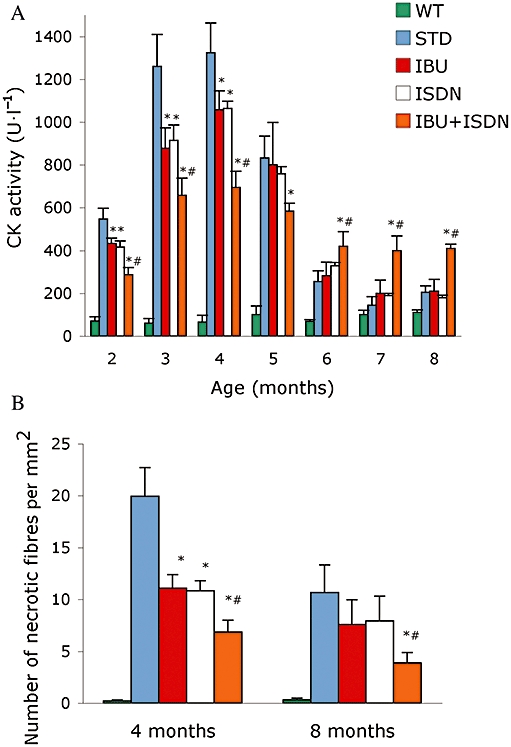
Ibuprofen plus ISDN reduce skeletal muscle damage long term in α-SG-null mice. CK activity serum levels (A) and number of necrotic fibres quantified in sections of diaphragm muscles (B) obtained from mice treated with standard diet (STD) or a diet containing ibuprofen (IBU), ISDN or the two drugs combined. Parameters measured in matched wild-type animals (WT) are shown for comparison *P < 0.01 for the effect of each treatment versus STD, #P < 0.01 for the effect of IBU plus ISDN versus that of each single drug; n = 7. α-SG, α-sarcoglycan; CK, creatine kinase; ISDN, isosorbide dinitrate.
Figure 3.
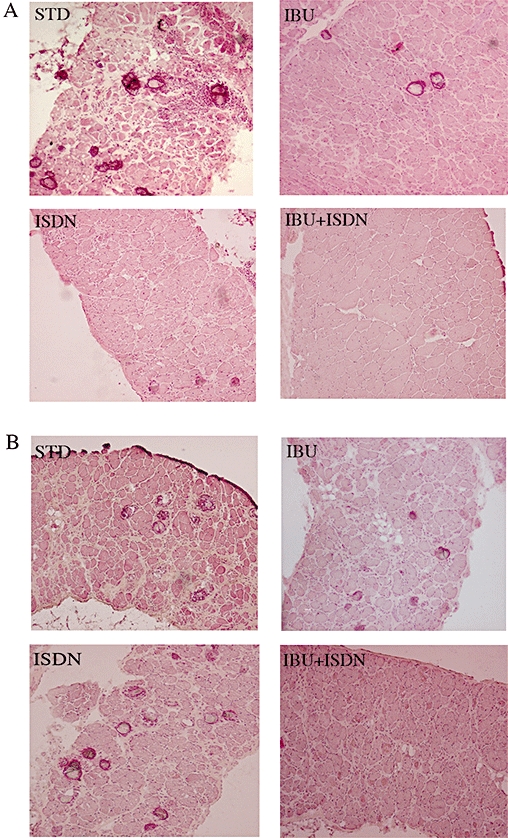
Histological analysis of dystrophic muscle in animals treated with standard diet, ibuprofen and/or ISDN. Representative haematoxylin and eosin-stained sections of diaphragms of mice treated with standard diet (STD) or a diet containing ibuprofen (IBU), ISDN or the two drugs combined, obtained after 4 (A) and 8 (B) months of treatment. ISDN, isosorbide dinitrate.
The efficacy of the various treatments in maintaining muscle integrity was assessed also by histological analyses. Evaluation of the number of necrotic fibres in sections of diaphragm muscles isolated from treated and untreated animals is shown in Figure 2B and representative images in Figure 3. All treatments yielded significant reduction of the number of necrotic fibres after 4 months of treatment, whereas after 8 months reduction was significant only in the groups receiving the combination of ibuprofen and ISDN. Similar results were obtained for tibialis anterior muscles (data not shown).
Ibuprofen and ibuprofen plus ISDN reduce skeletal muscle inflammation in α-SG-null mice
An abundant inflammatory infiltrate dispersed mostly in the fibrous/connective tissue that progressively replaces myofibres is a typical feature of dystrophic muscles (Tanabe et al., 1986; Spencer and Tidball, 2001). We evaluated inflammatory infiltrates in diaphragm and the expression of the pro-inflammatory cytokines TNF-α, TGF-β, CCL3/MIP-1α and CCL2/MCP-1 in homogenates of the tibialis anterior muscle. Ibuprofen, alone or combined with ISDN, reduced the number of infiltrating inflammatory cells as shown by the decreased cellular positivity to CD11b in muscle sections (Figure 4A and Table 2), and inhibited generation of MIP-1α, TGF-β and TNF-α (Figure 4B). By contrast, ISDN per se was without significant effects on the inflammatory parameters investigated. (Figure 4A and B).
Figure 4.
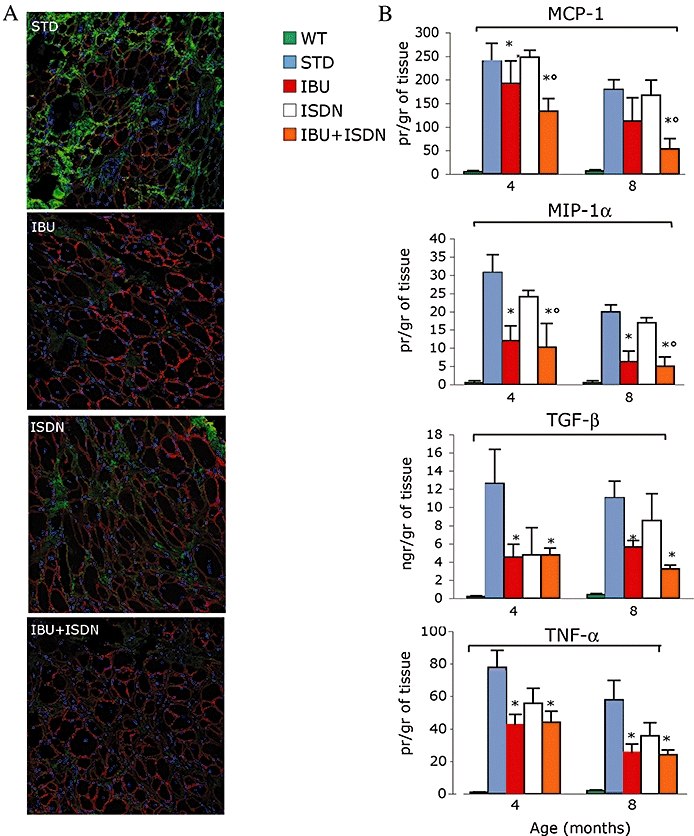
Ibuprofen and ibuprofen plus ISDN reduce skeletal muscle inflammation in α-SG-null mice. Inflammatory infiltrates were assessed by immunodetection with an anti CD11b antibody (green) (A). An anti laminin antibody was used to reveal the myofibres (red). The levels of CCL2/MCP-1, CCL3/MIP-1α, TGF-β and TNF-α were measured in homogenates of tibialis anterior muscles (B) obtained from mice treated with standard diet (STD) or a diet containing ibuprofen (IBU), ISDN or the two drugs combined. Parameters measured in matched wild-type animals (wt) are shown for comparison *P < 0.01 for the effect of each treatment versus STD, #P < 0.01 for the effect of IBU plus ISDN versus that of ISDN; n = 7. α-SG, α-sarcoglycan; ISDN, isosorbide dinitrate; MCP-1, monocyte chemoattractant protein-1; CCL2; MIP-1α, macrophage inflammatory protein 1α; CCL3; TGF-β, transforming growth factor β; TNF-α, tumour necrosis factor α.
Table 2.
IBU and IBU + ISDN reduce inflammatory CD11b+ infiltrates
| Treatment | % of CD11b positive area |
|---|---|
| STD | 36.4 ± 4.2 |
| IBU | 17.5 ± 3.3* |
| ISDN | 33.3 ± 5.2 |
| IBU + ISDN | 15.8 ± 1.0* |
P < 0.01 versus STD.
CD11b-positive areas in sections of the diaphragm of animals exposed to the various treatments or standard diet (STD) and killed at 4 months of age, were quantified and expressed as percentage of total analysed areas (n = 7).
IBU, ibuprofen; ISDN, isosorbide dinitrate; STD, standard diet.
Ibuprofen plus ISDN increase muscle regeneration, myogenic precursor cells number and regenerative potential
The α-SG-null mice skeletal muscles degeneration is accompanied in the early phases of the disease by appearance of centronucleated, regenerating fibres. Later on, a significant decrease of regenerative capacity is observed, most likely due to depletion of the pool of myogenic precursor cells, an event that contributes to disease progression and muscle wasting (Jejurikar and Kuzon, 2003).
We found that ibuprofen plus ISDN significantly increased the number of centronucleated fibres in both diaphragm and tibialis anterior muscles, whereas ibuprofen or ISDN were ineffective when given alone (Figure 5A).
Figure 5.
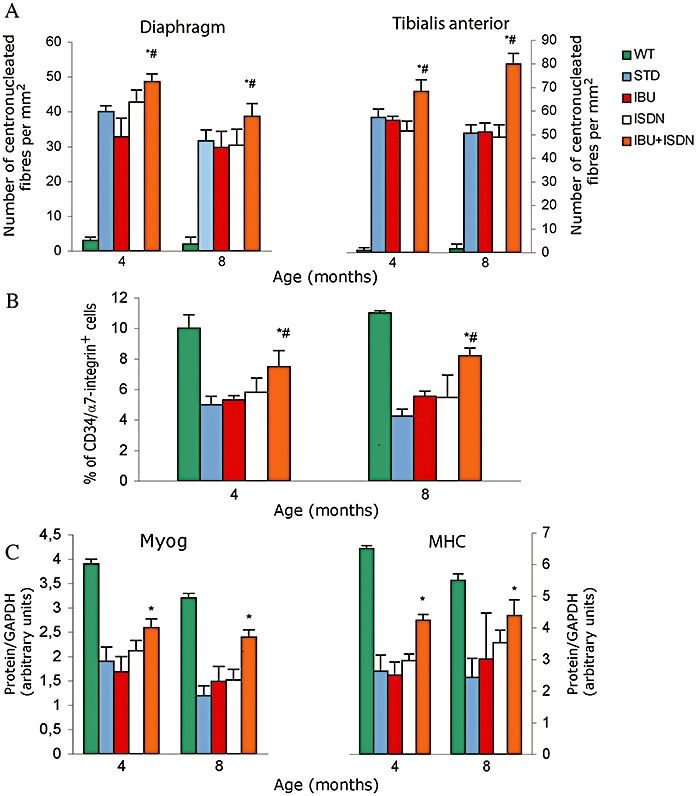
Ibuprofen plus ISDN increase muscle regeneration, myogenic precursor cells number and regenerative potential. Number of centronucleated regenerating fibres quantified in sections of diaphragm and tibialis anterior muscles (A), number of CD34/α7 integrin-positive cells isolated from tibialis anterior muscles quantified by flow cytometry (B) and expression of the differentiation markers, myogenin (Myog) and myosin heavy chain (MHC), in myogenic precursor cells isolated and then cultured for 1 week (C) from mice treated with standard diet (STD) or a diet containing ibuprofen (IBU), ISDN or the two drugs combined. Parameters measured in matched wild-type animals (WT) are shown in panel A for comparison. *P < 0.01 for the effect of each treatment versus STD, #P < 0.01 for the effect of IBU plus ISDN versus that of each single drug; n = 7. ISDN, isosorbide dinitrate.
We also isolated from the same animal groups the myogenic precursor cells and analysed their number, by assessing co-expression of the markers CD34 and α7-integrin, as well as their ability to differentiate in vitro, a good measure of their regenerative potential (Buckingham et al., 2003). Ibuprofen plus ISDN enhanced both the number and differentiation ability of myogenic precursor cells significantly. Compared with untreated control cells; by contrast, ibuprofen and ISDN when administered alone did not have significant effects on these parameters (Figure 5B and C).
Discussion
The main finding of our study is that a combination of ibuprofen and ISDN proved to be a long-term effective therapy for muscular dystrophy in the α-SG-null mice model. Furthermore, because this combination was of two well characterized drugs, our finding opens the door to immediate testing for efficacy in humans.
To assess the efficacy of the therapy in mice, we carried out a careful evaluation for 8 months of several unrelated markers of muscle function, damage and regeneration, and inflammatory responses, investigated using multiple different approaches in treated and untreated α-SG-null mice, as well as in matched healthy animals. Muscle function was evaluated studying both the spontaneous animal movement and the resistance to fatigue; the level of muscle damage was assessed by measuring CK release in plasma and by counting the number of necrotic fibres in sections of both a voluntary and an involuntary muscle (tibialis anterior and diaphragm). Inflammatory reactions were studied by histological and biochemical parameters, while the regenerative capacity of muscle was assessed by analysing the proliferative and differentiative capacities of myogenic precursor cells both in vivo and ex vivo. Of importance, the treatment allowed a significant, albeit not full, restoration of parameters towards those observed in WT animals, clearly indicating the efficacy of the therapy we propose. In addition, the data obtained from the various experimental approaches especially at the eighth month strongly suggest that the therapeutic action of ibuprofen plus ISDN results from converging beneficial effects that limit inflammation and damage to the muscle and concurrently enhance its regenerative potential, although progression of damage to muscles cannot be excluded in the treated animals, in view of the persisitence of high CK levels, at later stages of treatment. Likewise, it is important to mention that the experimental protocol was designed in such a way that the ex vivo analyses were carried out, 1 week later, on the same animals that performed the functional tests, i.e. conditions under which we cannot exclude the presence of muscle damage, inflammation and regeneration induced by the physical exercise during functional tests. Whereas this protocol allows data from all groups to be internally consistent and comparable, we cannot exclude that part of the effects of drug treatments were due to actions on exercise-induced inflammation, damage and regeneration and not solely due to the effect of treatments on dystrophy.
The observation that NO or NSAID therapies may have some degree of efficacy in muscular dystrophy is not new; however, this is the first report that demonstrate the synergism of a combined treatment and its long-term efficacy. Approaches with only NO-donors or L-arginine yielded some amelioration of the mdx dystrophic phenotype. However, these treatmens were not investigated over the long term and, moreover, in most studies only amelioration of muscle morphology, but not functional recovery was reported (Barton et al., 2005; Marques et al., 2005; Voisin et al., 2005; Hnia et al., 2008; Benabdellah et al., 2009; Wang et al., 2009). Similar considerations apply to therapies aimed at controlling inflammation. The pharmacological approaches proposed thus far in the mdx mouse model yielded amelioration of only single muscle force and/or the treatment was limited to a few weeks (Pierno et al., 2007; Radley et al., 2008; Reutenauer et al., 2008; Burdi et al., 2009; Dorchies et al., 2009; Huang et al., 2009). An open pilot study with the TNF-α inhibitor pentoxifylline suggested potential stabilization of strength in Duchenne patients but the treatment was poorly tolerated (Escolar et al., 2007).
A study reported functional recovery of treatment with the combination of L-arginine and the corticosteroid drug deflazacort on muscle voluntary exercise for 3 months (Archer et al., 2006). At variance with that study however, we used a strategy that avoided corticosteroid administration and was effective long-term.
The therapy we propose also solves some specific drawbacks reported with NSAIDs in muscle. NSAIDS have been described to negatively regulate the activation of myogenic precursor cells (Thorsson et al., 1998; Mendias et al., 2004; Mackey et al., 2007) and the concomitant administration of NO appears to prevent this inhibitory effect in a way similar to that already described for the combination of NO with prednisolone (Betters et al., 2008).
The results we describe in the mouse model are important in terms of possible treatments for the human muscular dystrophies and open the door to immediate testing in patients. Ibuprofen and ISDN are drugs long established in the clinic with a good profile of safety (Ferdinand, 2005; Leroy et al., 2007), and we did not detect overt signs of toxicity during the treatments, including the group of animals receiving both drugs. In addition, the doses chosen for this study for each drugs yield plasma level similar to those measured in humans and, in the case of ibuprofen, in the range of those obtained using paediatric doses of the drug (Steen et al., 2000; Lotsch et al., 2001). Finally, in terms of pharmacokinetics, no significant interactions affecting bioavailability were detected as demonstrated by the similar steady-state plasma concentrations reached by each drug administered alone or in combination. A phase IIb clinical trial using this combination of drugs is currently being planned in Duchenne muscular dystrophy patients.
An important aspect of the study was in the comparison between the pharmacodynamic effects of ibuprofen plus ISDN and those of ISDN or ibuprofen administered alone. This approach allowed us to conclude that the efficacy of the combination of ISDN and ibuprofen therapy is indeed attributable to synergistic effects of NO release and NSAID activity, as no long-term amelioration of muscle function or histological/biochemical parameters were observed when ISDN or ibuprofen were administered alone. It remains to be elucidated which mechanisms beyond the specific protective effects of NO on muscle and reduction of inflammation by ibuprofen contribute to the synergistic effect. One possibility is an enhanced cytoprotection resulting from the combination of the inhibitory effects of NO on several death-inducing pathways (Liu and Stamler, 1999; Clementi et al., 2003) with the protective effect resulting from inhibition of inducible cyclo-oxygenase (Mollace et al., 2005). In particular such synergism might be expressed as the preservation of mitochondrial integrity and function in analogy with other degenerative diseases (Deigner et al., 2000; Takuma et al., 2001).
The combination of ISDN plus ibuprofen had an impressive effect on the regenerative potential of myogenic precursor cells, an effect not observed when the drugs were administered separately. Although we still have to elucidate the mechanism(s) of this enhanced pro-regenerative potential, it is most likely due to the direct effects of NO as promoter of activation proliferation and fusogenicity of myogenic cells (Anderson, 2000; Pisconti et al., 2006; Colussi et al., 2008; Percival et al., 2008; Filippin et al., 2009) and it is of great significance from a perspective of potential therapies. A therapy that controls fibre necrosis and limits damage may not be effective if the residual regenerative capacity to bring back significant muscle mass and strength is exhausted. Maintenance, self-renewal and differentiation of the myogenic precursor cells pool are thus key aspects required for an effective pharmacological therapy for muscular dystrophy as they would allow the muscle to maintain its self-repairing ability, which is especially needed in dystrophic muscles, subjected to continuous damage (Cossu and Sampaolesi, 2004). The preservation of the regenerative capacity of muscle by the combination of ISDN plus ibuprofen may explain why this therapy was effective long-term.
The possibility of treating muscular dystrophy with an approach other than steroids is attractive for several reasons. In Duchenne muscular dystrophy, several studies suggest that steroids are effective when administered in daily dose regimens; however, they are often not tolerated in the long term; over the course of several years, severe adverse effects in approximately a third of treated patients are reported (Manzur et al., 2004). Alternative regimens have been proposed, with the hope of reducing the adverse effects but a final assessment of whether the beneficial effects of steroids justify the adverse effects is not defined (Manzur and Muntoni, 2009).
In conclusion, this study demonstrate that NO and NSAIDs exert synergistic and persistent beneficial effect in dystrophic mice. Inhibition of muscle damage and inflammation, as well as maintenance of muscle regenerative capacity appear to account for the therapeutic effect of this combination. The fact that the therapy is obtained by combining drugs already approved for paediatric use paves the way to immediate testing of its efficacy in human patients.
Acknowledgments
This work was supported by Telethon Italia (GP007006, to EC), European Community 7th framework programme (OPTISTEM and ENDOSTEM large cooperative programmes, to EC and SB), Cariplo (to EC and SB), Fondazione Romeo ed Enrica Invernizzi (to EC), Italian Ministry of Health RF2007 and RC 2010 (to EC), Parent Project Onlus Italia (To EC and GD'A) and Association Française des Myopathies (13478, to EC).
Glossary
Abbreviations
- α-SG
α-sarcoglycan
- CK
creatine kinase
- ISDN
isosorbide dinitrate
- MCP-1
monocyte chemoattractant protein-1; CCL2
- MIP-1α
macrophage inflammatory protein 1α; CCL3
- NOS
nitric oxide synthase
- TGF-β
transforming growth factor β
- TNF-α
tumour necrosis factor α
Conflict of interest
None.
References
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC) Br J Pharmacol. (4th edn.) 2009;158(Suppl 1):S1–S254. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson JE. A role for nitric oxide in muscle repair: nitric oxide-mediated activation of muscle satellite cells. Mol Biol Cell. 2000;11:1859–1874. doi: 10.1091/mbc.11.5.1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Archer JD, Vargas CC, Anderson JE. Persistent and improved functional gain in mdx dystrophic mice after treatment with L-arginine and deflazacort. FASEB J. 2006;20:738–740. doi: 10.1096/fj.05-4821fje. [DOI] [PubMed] [Google Scholar]
- Barton ER, Morris L, Kawana M, Bish LT, Toursel T. Systemic administration of L-arginine benefits mdx skeletal muscle function. Muscle Nerve. 2005;32:751–760. doi: 10.1002/mus.20425. [DOI] [PubMed] [Google Scholar]
- Benabdellah F, Yu H, Brunelle A, Laprevote O, De La Porte S. MALDI reveals membrane lipid profile reversion in MDX mice. Neurobiol Dis. 2009;36:252–258. doi: 10.1016/j.nbd.2009.07.013. [DOI] [PubMed] [Google Scholar]
- Betters JL, Long JH, Howe KS, Braith RW, Soltow QA, Lira VA, et al. Nitric oxide reverses prednisolone-induced inactivation of muscle satellite cells. Muscle Nerve. 2008;37:203–209. doi: 10.1002/mus.20915. [DOI] [PubMed] [Google Scholar]
- Bredt DS. NO skeletal muscle derived relaxing factor in Duchenne muscular dystrophy. Proc Natl Acad Sci USA. 1998;95:14592–14593. doi: 10.1073/pnas.95.25.14592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunelli S, Rovere-Querini P, Sciorati C, Manfredi AA, Clementi E. Nitric oxide: emerging concepts about its use in cell-based therapies. Expert Opin Investig Drugs. 2007a;16:33–43. doi: 10.1517/13543784.16.1.33. [DOI] [PubMed] [Google Scholar]
- Brunelli S, Sciorati C, D'Antona G, Innocenzi A, Covarello D, Galvez BG, et al. Nitric oxide release combined with nonsteroidal antiinflammatory activity prevents muscular dystrophy pathology and enhances stem cell therapy. Proc Natl Acad Sci USA. 2007b;104:264–269. doi: 10.1073/pnas.0608277104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckingham M, Bajard L, Chang T, Daubas P, Hadchouel J, Meilhac S, et al. The formation of skeletal muscle: from somite to limb. J Anat. 2003;202:59–68. doi: 10.1046/j.1469-7580.2003.00139.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burdi R, Rolland JF, Fraysse B, Litvinova K, Cozzoli A, Giannuzzi V, et al. Multiple pathological events in exercised dystrophic mdx mice are targeted by pentoxifylline: outcome of a large array of in vivo and ex vivo tests. J Appl Physiol. 2009;106:1311–1324. doi: 10.1152/japplphysiol.90985.2008. [DOI] [PubMed] [Google Scholar]
- Clementi E, Borgese N, Meldolesi J. Interactions between nitric oxide and sphingolipids and the potential consequences in physiology and pathology. Trends Pharmacol Sci. 2003;24:518–523. doi: 10.1016/j.tips.2003.08.008. [DOI] [PubMed] [Google Scholar]
- Colussi C, Mozzetta C, Gurtner A, Illi B, Rosati J, Straino S, et al. HDAC2 blockade by nitric oxide and histone deacetylase inhibitors reveals a common target in Duchenne muscular dystrophy treatment. Proc Natl Acad Sci USA. 2008;105:19183–19187. doi: 10.1073/pnas.0805514105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cossu G, Sampaolesi M. New therapies for muscular dystrophy: cautious optimism. Trends Mol Med. 2004;10:516–520. doi: 10.1016/j.molmed.2004.08.007. [DOI] [PubMed] [Google Scholar]
- Deigner HP, Haberkorn U, Kinscherf R. Apoptosis modulators in the therapy of neurodegenerative diseases. Expert Opin Investig Drugs. 2000;9:747–764. doi: 10.1517/13543784.9.4.747. [DOI] [PubMed] [Google Scholar]
- Deng B, Glanzman D, Tidball JG. Nitric oxide generated by muscle corrects defects in hippocampal neurogenesis and neural differentiation caused by muscular dystrophy. J Physiol. 2009;587:1769–1778. doi: 10.1113/jphysiol.2008.166256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorchies OM, Wagner S, Buetler TM, Ruegg UT. Protection of dystrophic muscle cells with polyphenols from green tea correlates with improved glutathione balance and increased expression of 67LR, a receptor for (-)-epigallocatechin gallate. Biofactors. 2009;35:279–294. doi: 10.1002/biof.34. [DOI] [PubMed] [Google Scholar]
- Duclos F, Straub V, Moore SA, Venzke DP, Hrstka RF, Crosbie RH, et al. Progressive muscular dystrophy in alpha-sarcoglycan-deficient mice. J Cell Biol. 1998;142:1461–1471. doi: 10.1083/jcb.142.6.1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emery AE. The muscular dystrophies. Lancet. 2002;359:687–695. doi: 10.1016/S0140-6736(02)07815-7. [DOI] [PubMed] [Google Scholar]
- Escolar D, Tesi-Rocha C, Clemens P, Iannaccone S, Pestronk A, Kuntz N, et al. Immediate release oral pentoxifylline is poorly tolerated in Duchenne muscular dystrophy boys. Neuromuscul Disord. 2007;17:817. [Google Scholar]
- Farrar H, Letzig L, Gill M. Validation of a liquid chromatographic method for the determination of ibuprofen in human plasma. J Chromatogr B Analyt Technol Biomed Life Sci. 2002;780:341–348. doi: 10.1016/s1570-0232(02)00543-3. [DOI] [PubMed] [Google Scholar]
- Ferdinand KC. Isosorbide dinitrate and hydralazine hydrochloride: a review of efficacy and safety. Expert Rev Cardiovasc Ther. 2005;3:993–1001. doi: 10.1586/14779072.3.6.993. [DOI] [PubMed] [Google Scholar]
- Filippin LI, Moreira AJ, Marroni NP, Xavier RM. Nitric oxide and repair of skeletal muscle injury. Nitric Oxide. 2009;21:157–163. doi: 10.1016/j.niox.2009.08.002. [DOI] [PubMed] [Google Scholar]
- Grounds MD, Torrisi J. Anti-TNFalpha (Remicade) therapy protects dystrophic skeletal muscle from necrosis. FASEB J. 2004;18:676–682. doi: 10.1096/fj.03-1024com. [DOI] [PubMed] [Google Scholar]
- Hnia K, Gayraud J, Hugon G, Ramonatxo M, De La Porte S, Matecki S, et al. L-arginine decreases inflammation and modulates the nuclear factor-kappaB/matrix metalloproteinase cascade in mdx muscle fibers. Am J Pathol. 2008;172:1509–1519. doi: 10.2353/ajpath.2008.071009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang P, Zhao XS, Fields M, Ransohoff RM, Zhou L. Imatinib attenuates skeletal muscle dystrophy in mdx mice. FASEB J. 2009;23:2539–2548. doi: 10.1096/fj.09-129833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jejurikar SS, Kuzon WM., Jr Satellite cell depletion in degenerative skeletal muscle. Apoptosis. 2003;8:573–578. doi: 10.1023/A:1026127307457. [DOI] [PubMed] [Google Scholar]
- Kobayashi YM, Rader EP, Crawford RW, Iyengar NK, Thedens DR, Faulkner JA, et al. Sarcolemma-localized nNOS is required to maintain activity after mild exercise. Nature. 2008;456:511–515. doi: 10.1038/nature07414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai Y, Thomas GD, Yue Y, Yang HT, Li D, Long C, et al. Dystrophins carrying spectrin-like repeats 16 and 17 anchor nNOS to the sarcolemma and enhance exercise performance in a mouse model of muscular dystrophy. J Clin Invest. 2009;119:624–635. doi: 10.1172/JCI36612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leroy S, Mosca A, Landre-Peigne C, Cosson MA, Pons G. [Ibuprofen in childhood: evidence-based review of efficacy and safety] Arch Pediatr. 2007;14:477–484. doi: 10.1016/j.arcped.2007.01.012. [DOI] [PubMed] [Google Scholar]
- Liu L, Stamler JS. NO: an inhibitor of cell death. Cell Death Differ. 1999;6:937–942. doi: 10.1038/sj.cdd.4400578. [DOI] [PubMed] [Google Scholar]
- Lotsch J, Muth-Selbach U, Tegeder I, Brune K, Geisslinger G. Simultaneous fitting of R- and S-ibuprofen plasma concentrations after oral administration of the racemate. Br J Clin Pharmacol. 2001;52:387–398. doi: 10.1046/j.1365-2125.2001.01451.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackey AL, Kjaer M, Dandanell S, Mikkelsen KH, Holm L, Dossing S, et al. The influence of anti-inflammatory medication on exercise-induced myogenic precursor cell responses in humans. J Appl Physiol. 2007;103:425–431. doi: 10.1152/japplphysiol.00157.2007. [DOI] [PubMed] [Google Scholar]
- Manzur AY, Kuntzer T, Pike M, Swan A. Glucocorticoid corticosteroids for Duchenne muscular dystrophy. Cochrane Database Syst Rev. 2004 doi: 10.1002/14651858.CD003725.pub2. CD003725. [DOI] [PubMed] [Google Scholar]
- Manzur AY, Muntoni F. Diagnosis and new treatments in muscular dystrophies. J Neurol Neurosurg Psychiatry. 2009;80:706–714. doi: 10.1136/jnnp.2008.158329. [DOI] [PubMed] [Google Scholar]
- Marques MJ, Luz MA, Minatel E, Neto HS. Muscle regeneration in dystrophic mdx mice is enhanced by isosorbide dinitrate. Neurosci Lett. 2005;382:342–345. doi: 10.1016/j.neulet.2005.03.023. [DOI] [PubMed] [Google Scholar]
- Mendias CL, Tatsumi R, Allen RE. Role of cyclooxygenase-1 and -2 in satellite cell proliferation, differentiation, and fusion. Muscle Nerve. 2004;30:497–500. doi: 10.1002/mus.20102. [DOI] [PubMed] [Google Scholar]
- Mollace V, Muscoli C, Masini E, Cuzzocrea S, Salvemini D. Modulation of prostaglandin biosynthesis by nitric oxide and nitric oxide donors. Pharmacol Rev. 2005;57:217–252. doi: 10.1124/pr.57.2.1. [DOI] [PubMed] [Google Scholar]
- Nisoli E, Clementi E, Paolucci C, Cozzi V, Tonello C, Sciorati C, et al. Mitochondrial biogenesis in mammals: the role of endogenous nitric oxide. Science. 2003;299:896–899. doi: 10.1126/science.1079368. [DOI] [PubMed] [Google Scholar]
- Ontell M. Muscle fiber necrosis in murine dystrophy. Muscle Nerve. 1981;4:204–213. doi: 10.1002/mus.880040306. [DOI] [PubMed] [Google Scholar]
- Percival JM, Anderson KN, Gregorevic P, Chamberlain JS, Froehner SC. Functional deficits in nNOSmu-deficient skeletal muscle: myopathy in nNOS knockout mice. PLoS One. 2008;3:e3387. doi: 10.1371/journal.pone.0003387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierno S, Nico B, Burdi R, Liantonio A, Didonna MP, Cippone V, et al. Role of tumour necrosis factor alpha, but not of cyclo-oxygenase-2-derived eicosanoids, on functional and morphological indices of dystrophic progression in mdx mice: a pharmacological approach. Neuropathol Appl Neurobiol. 2007;33:344–359. doi: 10.1111/j.1365-2990.2007.00798.x. [DOI] [PubMed] [Google Scholar]
- Pisconti A, Brunelli S, Di Padova M, De Palma C, Deponti D, Baesso S, et al. Follistatin induction by nitric oxide through cyclic GMP: a tightly regulated signaling pathway that controls myoblast fusion. J Cell Biol. 2006;172:233–244. doi: 10.1083/jcb.200507083. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Porter JD, Khanna S, Kaminski HJ, Rao JS, Merriam AP, Richmonds CR, et al. A chronic inflammatory response dominates the skeletal muscle molecular signature in dystrophin-deficient mdx mice. Hum Mol Genet. 2002;11:263–272. doi: 10.1093/hmg/11.3.263. [DOI] [PubMed] [Google Scholar]
- Radley HG, Davies MJ, Grounds MD. Reduced muscle necrosis and long-term benefits in dystrophic mdx mice after cV1q (blockade of TNF) treatment. Neuromuscul Disord. 2008;18:227–238. doi: 10.1016/j.nmd.2007.11.002. [DOI] [PubMed] [Google Scholar]
- Rando TA. Role of nitric oxide in the pathogenesis of muscular dystrophies: a ‘two hit’ hypothesis of the cause of muscle necrosis. Microsc Res Tech. 2001;55:223–235. doi: 10.1002/jemt.1172. [DOI] [PubMed] [Google Scholar]
- Reutenauer J, Dorchies OM, Patthey-Vuadens O, Vuagniaux G, Ruegg UT. Investigation of Debio 025, a cyclophilin inhibitor, in the dystrophic mdx mouse, a model for Duchenne muscular dystrophy. Br J Pharmacol. 2008;155:574–584. doi: 10.1038/bjp.2008.285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sciorati C, Galvez BG, Brunelli S, Tagliafico E, Ferrari S, Cossu G, et al. Ex vivo treatment with nitric oxide increases mesoangioblast therapeutic efficacy in muscular dystrophy. J Cell Sci. 2006;119:5114–5123. doi: 10.1242/jcs.03300. [DOI] [PubMed] [Google Scholar]
- Spencer MJ, Tidball JG. Do immune cells promote the pathology of dystrophin-deficient myopathies? Neuromuscul Disord. 2001;11:556–564. doi: 10.1016/s0960-8966(01)00198-5. [DOI] [PubMed] [Google Scholar]
- Stamler JS, Meissner G. Physiology of nitric oxide in skeletal muscle. Physiol Rev. 2001;81:209–237. doi: 10.1152/physrev.2001.81.1.209. [DOI] [PubMed] [Google Scholar]
- Steen AE, Reeh PW, Geisslinger G, Steen KH. Plasma levels after peroral and topical ibuprofen and effects upon low pH-induced cutaneous and muscle pain. Eur J Pain. 2000;4:195–209. doi: 10.1053/eujp.2000.0173. [DOI] [PubMed] [Google Scholar]
- Takuma K, Phuagphong P, Lee E, Mori K, Baba A, Matsuda T. Anti-apoptotic effect of cGMP in cultured astrocytes: inhibition by cGMP-dependent protein kinase of mitochondrial permeable transition pore. J Biol Chem. 2001;276:48093–48099. doi: 10.1074/jbc.M108622200. [DOI] [PubMed] [Google Scholar]
- Tanabe Y, Esaki K, Nomura T. Skeletal muscle pathology in X chromosome-linked muscular dystrophy (mdx) mouse. Acta Neuropathol. 1986;69:91–95. doi: 10.1007/BF00687043. [DOI] [PubMed] [Google Scholar]
- Thorsson O, Rantanen J, Hurme T, Kalimo H. Effects of nonsteroidal antiinflammatory medication on satellite cell proliferation during muscle regeneration. Am J Sports Med. 1998;26:172–176. doi: 10.1177/03635465980260020401. [DOI] [PubMed] [Google Scholar]
- Tsikas D. Simultaneous derivatization and quantification of the nitric oxide metabolites nitrite and nitrate in biological fluids by gas chromatography/mass spectrometry. Anal Chem. 2000;72:4064–4072. doi: 10.1021/ac9913255. [DOI] [PubMed] [Google Scholar]
- Voisin V, Sebrie C, Matecki S, Yu H, Gillet B, Ramonatxo M, et al. L-arginine improves dystrophic phenotype in mdx mice. Neurobiol Dis. 2005;20:123–130. doi: 10.1016/j.nbd.2005.02.010. [DOI] [PubMed] [Google Scholar]
- Wagner KR. Approaching a new age in Duchenne muscular dystrophy treatment. Neurother. 2008;5:583–591. doi: 10.1016/j.nurt.2008.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G, Burczynski FJ, Hasinoff BB, Zhang K, Lu Q, Anderson JE. Development of a nitric oxide-releasing analogue of the muscle relaxant guaifenesin for skeletal muscle satellite cell myogenesis. Mol Pharm. 2009;6:895–904. doi: 10.1021/mp800226z. [DOI] [PubMed] [Google Scholar]
- Wehling M, Spencer MJ, Tidball JG. A nitric oxide synthase transgene ameliorates muscular dystrophy in mdx mice. J Cell Biol. 2001;155:123–131. doi: 10.1083/jcb.200105110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wehling-Henricks M, Oltmann M, Rinaldi C, Myung KH, Tidball JG. Loss of positive allosteric interactions between neuronal nitric oxide synthase and phosphofructokinase contributes to defects in glycolysis and increased fatigability in muscular dystrophy. Hum Mol Genet. 2009;18:3439–3451. doi: 10.1093/hmg/ddp288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Chen D, Li K, Wang D. Sensitive liquid chromatographic assay for the simultaneous determination of ibuprofen and its prodrug, ibuprofen eugenol ester, in rat plasma. Yakugaku Zasshi. 2005;125:733–737. doi: 10.1248/yakushi.125.733. [DOI] [PubMed] [Google Scholar]