

Abstract
Approximately one half of patients who undergo antiviral therapy for chronic hepatitis C virus (HCV) genotype 1 infection will not respond to treatment. African Americans (AAs) are less responsive to treatment than Caucasian Americans (CAs) and the reasons for this disparity are largely unknown. Recent studies suggest that serum lipids may be associated with treatment response. The aims of this study were to evaluate baseline and changes in serum lipids during therapy, determine if serum lipids are associated with virological response, and assess if these measures explain the racial difference in efficacy. Participants were from Virahep-C, a prospective study of treatment naïve participants with type 1 HCV infection who received peginterferon alfa-2a (PEG-IFN) plus ribavirin therapy for up to 48 weeks. Fasting serum lipids were analyzed at baseline, during, and after therapy in 160 AAs and 170 CAs. A relative risk (RR) model was employed to evaluate characteristics associated with sustained virological response (SVR). Antiviral therapy was associated with changes in serum lipids during and after antiviral therapy, with the changes differing by race and the amount of PEG-IFN taken. Baseline lipid measures independently associated with a higher rate of SVR were lower TG and higher LDLc, with an interaction between high density lipoprotein cholesterol (HDLc) and gender. Lipid measures did not contribute significantly to explaining the racial difference in SVR.
Conclusion
Lipid levels are associated with SVR, although lipid parameters did not explain the racial difference in treatment response. Results are compatible with proposed biological mechanisms of HCV entry, replication, and secretion, and may underscore new potential therapeutic targets for HCV eradication.
Supplementary keywords: Serum lipids, treatment efficacy, cholesterol, lipoprotein, hepatitis C, hepatitis C virus, racial disparity, sustained virological response
INTRODUCTION
In the United States (US), chronic hepatitis C virus (HCV) infection is a major public health problem afflicting 3.6 million persons with direct health care costs, including liver transplantation, exceeding $1 billion dollars annually (1, 2). The current standard of treatment of combination pegylated interferon (PEG-IFN) alfa and ribavirin is not completely effective in patients with hepatitis C genotype 1, the predominant viral type in the US with approximately 46% people achieving sustained virological response (SVR) (3). Moreover, there is a racial difference in response with African Americans (AAs) having a significantly lower response to combination treatment compared to Caucasian Americans (CAs) (4–6). Factors that explain the racial disparity in efficacy are largely unknown (4).
Changes in serum lipid levels during interferon therapy have been reported, although results are inconsistent and differ by HCV genotype. Interferon therapy has been associated with increases in total cholesterol (TC) and triglyceride (TG) levels, with TC levels remaining significantly higher and TG levels returning to pretreatment levels after stopping therapy (7). Other work has found significant increases in TG levels, and no significant change in TC levels (8). Compared to pretreatment, significant increases in TC have been reported in a subgroup with HCV genotype 3, but not genotype 1 during therapy (9), whereas another study reported higher TG levels during therapy in a group with genotype 1, but not in non-genotype 1 (8).
Recent studies further suggest that pretreatment serum lipid measures may be important predictors of treatment response. Several studies indicate that high pretreatment low density lipoprotein cholesterol (LDLc) and TC levels are associated with higher rates of SVR in multivariable analyses (10–14). In addition, higher pretreatment TG levels have also been reported among virological responders compared to non-responders (7). These studies further suggest that associations between lipid measures and virological response may be specific to HCV genotype 1 and possibly genotype 2. Little is known about the association between changes in lipid measures while on therapy and treatment response.
Observations from in vitro studies suggest relationships between lipoproteins and HCV that are important for mechanisms of viral entry into hepatocytes, viral replication, and secretion. Several studies suggest that HCV may combine with lipoproteins in the serum, possibly obscuring the virus from the host immune response, which may in turn help in viral entry into the hepatocytes (15–18). Various receptors involved in lipoprotein-viral particle entry into hepatocytes are posited, including the scavenger receptor B1 (SR-B1) and LDL receptor (19–22). Direct entry of free HCV (i.e., not associated with lipoproteins) is also proposed to occur through binding of the HCV envelope glycoprotein E2 with SR-B1 or its human analogue CD81 (23–25). Within the hepatocyte endoplasmic reticulum, studies indicate that HCV replication may be reliant on cholesterol metabolism and a secretion process consisting of HCV and very low density lipoprotein (VLDL) conglomerate particles (26–30). Recent work suggests that interferon therapy leads to down-regulation of SR-B1 expression (31). This supports the notion that decreased lipoprotein expression may in turn impact serum lipoproteins and lipids profile measures. Therefore, associations between the serum lipids and treatment response are supported by biologically plausible mechanisms.
This study assesses changes in serum lipids among patients undergoing combination therapy for chronic hepatitis C, relationships between serum lipids (pretreatment levels and changes during treatment) and virological response, and whether serum lipids explain the racial disparity in treatment efficacy.
METHODS
Study population
Participants for this study were from the Virahep-C study, an investigation of resistance to antiviral therapy for genotype 1 chronic HCV infection, which has been described previously (4). In brief, Virahep-C sought to evaluate clinical, immunological, virological, and host genetic factors that contribute to the lack of virological response to antiviral treatment, and in particular, the racial difference in efficacy. The study enrolled approximately equal numbers of Caucasian Americans (CA) (n=205) and African Americans (AA) (n=196), all of whom underwent a combination PEG-IFN alfa-2a and ribavirin regimen for up to 48 weeks. At 24 weeks of therapy, participants were evaluated for the presence of HCV RNA and those with detectable levels were labeled as non-responders and discontinued therapy while the others continued therapy for an additional 24 weeks. All patients were followed for an additional 24 weeks after completion of therapy. The study protocol conformed to the ethical guidelines of the 1975 Declaration of Helsinki as reflected in a priori approval by the local institutional review board.
Of the 401 participants enrolled in Virahep-C, lipid profile analyses were performed among participants who granted genetic consent (n=374) approved by the local institutional review board and had stored fasting serum samples at baseline (n=335). Five participants who reported use of lipid lowering medications were excluded from this evaluation resulting in a final analysis sample of 330 participants (170 CA and 160 AA). During treatment (24 weeks after starting therapy) and post-treatment (24 weeks after stopping therapy) lipid profile data were additionally available for 253 and 245 of the participants, respectively.
Study measures
The primary outcome for Virahep-C was sustained virological response (SVR), defined as undetectable serum HCV RNA 24 weeks after the end of therapy. Serum lipid measures, TG, LDLc, high density lipoprotein cholesterol (HDLc), and TC, were obtained through analysis of stored fasting serum samples at the Heinz Nutrition Laboratory in the Department of Epidemiology, University of Pittsburgh. For serum samples with TG levels less than 400 mg/dL, the Friedewald formula was used to calculate LDLc indirectly (LDLc = TC – HDLc – 0.20 X TG) (32). For samples with TG levels of at least 400 mg/dL, LDLc was assessed directly. Dyslipidemia was defined using the cutoffs from the National Cholesterol Education Program (NCEP) Adult Treatment Panel (ATP) III recommendations as any of the following: LDLc greater than or equal to 130 mg/dL, HDLc less than 40 mg/dL, TC greater than or equal to 200 mg/dL, or TG greater than or equal to 150 mg/dL (33). A homeostasis model assessment (HOMA) variable, HOMA2, was calculated using fasting insulin and glucose measures with a Microsoft Excel HOMA 2 calculator and insulin resistance was defined as a score greater than or equal to 2 (34). Hepatic inflammation and fibrosis were assessed using the criteria of the Histological Activity Index (HAI) by a single hepatopathologist (35, 36). The amount of PEG-IFN and ribavirin taken by participants was estimated using data from the Medication Event Management System (MEMS) (Aardex, Zug, Switzerland) (37).
Statistical analysis
Categorical measures were summarized as frequencies and percents with differences across nominally classified groups (i.e., race, gender, health insurance status, employment status, smoking status, alcohol consumption of at least 2 drinks per week, history of diabetes and hypertension, HCV genotype, and severe fibrosis) assessed using a Pearson’s chi-square test or the exact equivalent. Differences in categorical measures across ordinal groups (i.e., educational attainment and iron scores) were assessed using a Jonckeere-Terpstra test, or the exact equivalent. Continuous measures were summarized as medians and interquartile ranges with differences in group distributions assessed using a Wilcoxon rank sum test (for comparison of two groups) or a Kruskal-Wallis test (for comparison of more than two groups). To assess if changes in lipid profile measures significantly differed from baseline, a Wilcoxon sign rank test was used. Associations between the proportion of PEG-IFN and ribavirin taken with changes in serum lipids were assessed using Spearman’s correlation analyses. For all statistical tests, a p-value of <0.05 was considered statistically significant.
To evaluate factors associated with SVR, a relative risk model was employed with a robust variance estimator (38). In regression models, TG, HDLc, and TC were transformed to the natural logarithm (ln) scale to achieve normality. All continuous predictors were centered. The relationships between baseline and 24 week changes during treatment in lipid profile measures and the probability of SVR were graphically assessed using smoothing spline plots. Due to different patterns observed by gender, smoothing spline plots for HDLc were examined separately for males and females. Two types of multivariable models of sustained virological response were constructed using a stepwise approach. One type of multivariable model (models 1 and 2) allowed pretreatment characteristics and the amount of PEG-IFN alfa-2a taken during the first 24 weeks as eligible predictors. Model 2 allowed as additional eligible predictors the baseline lipid profile measures. A second type of multivariable model (model 3) also adjusted for body weight changes and allowed for the inclusion of variables representing baseline and changes in lipid profile measures during the first 24 weeks of therapy as eligible predictors. To compare the prediction of multivariable models, differences in the area under the receiver operating curves (AUROCs) were assessed using a non-parametric method (39).
RESULTS
Baseline characteristics of the 330 participants are shown in Table 1. AAs did not significantly differ from CAs by age, gender, employment status, health risk behaviors (smoking status and weekly alcohol consumption), viral level, AST, INR, WBC count, platelet count, percent Iron/TIBC, Ishak fibrosis, total HAI score, steatosis, TG, HDLc, or TC. Compared to CAs, a larger percentage of AAs had health insurance coverage (87% vs. 78%, p=0.04), public health insurance (31% vs. 18%, p=0.006), less education (p=0.008), a history of diabetes (15% vs. 4%, p<0.001), insulin resistance (46% vs. 33%, p=0.02), a history of hypertension (40% vs. 19%, p<0.001), and higher prevalence of HCV sub-genotype 1b (44% vs. 28%, p=0.002). As a group, AAs had higher body mass index (BMI) (median 29.3 vs. 27.4 kg/m2, p<0.001), higher HOMA2 (median 1.9 vs. 1.5 scores, p<0.001), higher alkaline phosphatase (median 83 vs. 78 U/L, p=0.043), and ferritin levels (median 246 vs. 149 ng/mL, p<0.001), and lower ALT (median 60 vs. 74.5 U/L, p<0.001), total bilirubin (median 0.06 vs. 0.07 mg/dL, p=0.004), albumin (median 4.2 vs. 4.2 g/dL, p=0.004), and LDLc levels (median 106.4 vs. 118.7 mg/dL, p=0.009) than CAs. The prevalence of dyslipidemia was 70% overall and did not significantly differ by race.
Table 1.
Cohort characteristics
| Feature* | Overall (n=330) | AA (n=160) | CA (n=170) | p |
|---|---|---|---|---|
| Demographics | ||||
| Age (years) | 48 (43,52) | 48 (45,52) | 47 (42,52) | 0.07 |
| Male | 216 (65.5) | 106 (66.3) | 110 (64.7) | 0.77 |
| Health insurance (m=6)a | 0.008 | |||
| Uninsured | 58 (17.9) | 21 (13.4) | 37 (22.2) | 0.04 |
| Public | 79 (24.4) | 49 (31.2) | 30 (18.0) | 0.006 |
| Private | 187 (57.7) | 87 (55.4) | 100 (59.9) | 0.42 |
| Education (m=8)b | 0.008 | |||
| Less than high school | 61 (18.9) | 36 (23.1) | 25 (15.1) | 0.07 |
| High school degree | 78 (24.2) | 41 (26.3) | 37 (22.3) | 0.16 |
| Some college | 105 (32.6) | 50 (32.1) | 55 (33.1) | 0.16 |
| College degree or more | 78 (24.2) | 29 (18.6) | 49 (29.5) | -- |
| Employed (m=4) | 204 (62.6) | 101 (63.9) | 103 (61.3) | 0.63 |
| Health risk behaviors | ||||
| Current smoker (m=6) | 128 (39.5) | 65 (41.7) | 63 (37.5) | 0.44 |
| Consumes ≥ 2 alcoholic drinks/week (m=7) | 66 (20.4) | 38 (24.2) | 28 (16.9) | 0.10 |
| General clinical features | ||||
| BMI (m=5) | 28.4 (25.2,32.4) | 29.3 (26.6,33.9) | 27.4 (24.4,31.4) | 0.0002 |
| Waist to hip ratio (m=23) | 0.9 (0.9,1.0) | 0.9 (0.8,1.0) | 0.9 (0.8,1.0) | 0.44 |
| Diabetic | 31 (9.4) | 24 (15.0) | 7 (4.1) | 0.0007 |
| HOMA2 (m=22) | 1.7 (1.0,2.7) | 1.9 (1.3,3.2) | 1.5 (0.9,2.3) | 0.0003 |
| Insulin resistant (m=22) | 120 (39.0) | 68 (45.6) | 52 (32.7) | 0.02 |
| Hypertensive | 97 (29.4) | 64 (40.0) | 33 (19.4) | <0.0001 |
| Viral characteristics | ||||
| Log10 HCV level (m=1) | 6.5 (5.6,6.7) | 6.4 (5.6,6.7) | 6.5 (5.6,6.8) | 0.25 |
| HCV genotypec | 0.002 | |||
| 1, NOS | 25 (7.6) | 10 (6.3) | 15 (8.8) | 0.38 |
| 1a | 177 (53.6) | 79 (49.4) | 98 (57.7) | 0.13 |
| 1a/b | 11 (3.3) | 1 (0.6) | 10 (5.9) | 0.01 |
| 1b | 117 (35.5) | 70 (43.8) | 47 (27.7) | 0.002 |
| Liver disease indicators | ||||
| ALT (IU/L) | 69 (45,108) | 60 (40,88) | 74.5 (52,139) | <0.0001 |
| AST (IU/L) | 52 (37,79) | 51.5 (35.5,71.5) | 53 (38,87) | 0.08 |
| Alk phosphatase (IU/L) | 79 (62,103) | 83 (62,108) | 78 (63,96) | 0.043 |
| Total bilirubin (mg/dL) | 0.6 (0.5,0.8) | 0.6 (0.4,0.8) | 0.7 (0.5,0.9) | 0.004 |
| INR (m=2) | 1.0 (0.9,1.1) | 1.0 (0.9,1.1) | 1.0 (0.9,1.1) | 0.95 |
| WBC count (103/mL) (m=3) | 6.0 (4.7,7.3) | 5.7 (4.6,7.3) | 6.25 (4.9,7.4) | 0.055 |
| Platelet count (103/mL) (m=4) | 207.5 (161,257) | 212 (159,268) | 207 (161,242) | 0.33 |
| Ferritin (ng/mL) (m=2) | 204 (96.8,366) | 246 (122,422) | 149 (78,287) | 0.0001 |
| Albumin (g/dL) (m=2) | 4.2 (4.0,4.4) | 4.2 (3.9,4.4) | 4.2 (4.0,4.5) | 0.004 |
| Iron/TIBC (m=8) | 34.1 (26.2,44.0) | 33.9 (25.7,41.9) | 34.3 (26.4,47.7) | 0.18 |
| Ishak fibrosis score (m=1) | 2 (1,3) | 2 (1,3) | 2 (1,3) | 0.85 |
| Ishak fibrosis score ≥ 3 (m=1) | 123 (37.4) | 58 (36.5) | 65 (38.2) | 0.74 |
| Fat score (m=1) | 0 (0,1) | 0 (0,1) | 0 (0,1) | 0.19 |
| Steatosis (>5 present) (m=1) | 209 (63.5) | 97 (61.0) | 112 (65.9) | 0.36 |
| Total HAI inflammation (m=1) | 8 (6,10) | 8 (7,10) | 9 (6,11) | 0.58 |
| Iron score (m=35) | 0.09 | |||
| 0 | 157 (53.2) | 66 (47.5) | 91 (58.3) | |
| 1 | 115 (39.0) | 62 (44.6) | 53 (34.0) | |
| 2 | 23 (7.8) | 11 (7.9) | 12 (7.7) | |
| Serum lipid measures | ||||
| TG (mg/dL) | 102.5 (75,146) | 105.5 (74.5,151) | 98.5 (76,137) | 0.21 |
| LDLc (mg/dL) | 115.1 (88.1,137.3) | 106.4 (83.4,133.4) | 118.7 (95.8,141.5) | 0.009 |
| HDLc (mg/dL) | 41.8 (33.7,53.8) | 42.3 (32.9,54.6) | 41.3 (33.8,52.0) | 0.66 |
| TC (mg/dL) | 185 (157,207) | 179 (153.5,204.5) | 187 (161,209) | 0.10 |
| Dyslipidemia | 232 (70.3) | 110 (68.8) | 122 (71.8) | 0.55 |
m=number with missing data
Each categorical variable is summarized as n (%) with p-values corresponding to a Pearson’s chi-square test (nominal variables) or the Jonckheere-Terpstra test (ordinal variables) or exact equivalents, where appropriate; each continuous variable is summarized as a median (interquartile range) with a p-value corresponding to a Wilcoxon rank sum test.
For features with two or more categories, the global p-value is listed in the first row of the feature.
Where the global p-value is <0.05, p-values correspond to Pearson’s chi-square with comparisons as follows:
Each health insurance status category compared to other categories combined;
Less than high school vs. high school degree or more; high school degree vs. more than high school degree; some college vs. more than some college;
Each genotype compared to other categories combined.
Compared to pretreatment, there were significant changes in serum lipids during therapy and after completion of therapy (Figure 1). During the initial 24 weeks of therapy, TG levels increased significantly (median +30 mg/dL), in contrast to significant declines in LDLc (−14.8 mg/dL), HDLc (median −8 mg/dL), and TC (median −17 mg/dL), (p<0.0001 for all). Post-therapy, statistically significant changes in lipid measures were limited only to the 24-week virological responders. Among 177 participants who underwent a 48 week course of therapy (24-week virological responders), post-treatment TG levels remained significantly higher than pretreatment (median +8 mg/dL, p=0.03), as did post-treatment LDLc (median +7.2 mg/dL, p<0.0001) and TC levels (median +9 mg/dL, p<0.0001), whereas HDLc levels did not significantly change (median +0.8 mg/dL, p=0.47). Among 62 participants who underwent a 24 week course of therapy (24-week virological non-responders) before stopping therapy, there were no significant changes in post-treatment serum lipids compared to pretreatment levels (TG: median +9 mg/dL, p=0.41; LDLc: median −3.2 mg/dL, p=0.36; TC: median −3 mg/dL, p=0.50; HDLc: +0.3 mg/dL, p=0.99).
Figure 1. Serum lipid profile changes during and after antiviral therapy.

* p<0.05;* * p<0.0001 (Wilcoxon signed rank test for differences from zero)
Tx=assessment at treatment week 24
F-up=assessment 24 weeks after stopping therapy
R=Among treatment week 24 virological responders (underwent a 48 week course of therapy) at week 72
NR=Among treatment week 24 virological nonresponders (underwent a 24 week course of therapy) at week 48
NOTE: Boxplots exclude extreme outliers for changes in TG levels: −768, −422, 477, 477, 562, 573, 629, and 1678 mg/dL on treatment; 434 mg/dL during follow-up among 6 month virological responders. Baseline median levels as follows: 100 mg/dL (TG); 115.5 mg/dL (LDLc); 41.1 (HLDc); and 183 (TC).
The proportion of PEG-IFN taken was significantly and directly associated with declines in LDLc (r = −0.22, p=0.005) and TC levels (r = −0.17, p=0.008) during the initial 24 weeks of therapy. The proportion of ribavirin taken was not significantly associated with any changes in serum lipid levels (p>0.05 for all). Race was significantly associated with changes in serum lipids during the first 24 weeks of therapy. Compared to CAs, AAs had significantly greater increases in TG and declines in LDLc levels (p=0.003 and p<0.0001, respectively). (Figure 2) The patterns of decreases in TC levels by race were similar to LDLc changes, although the differences were not statistically significant (p=0.054).
Figure 2. Race and serum lipid profile changes during therapy.

p-values correspond to Wilcoxon rank sum tests for racial differences
NOTE: Difference in lipid profile measures at treatment week 24 minus baseline
Baseline characteristics associated with SVR are summarized in Table 2. Features significantly associated with higher SVR in unadjusted analyses included CA race (RR=1.92, p<0.001, AA reference), education beyond high school (RR=0.64, p=0.002, less than high school degree reference), and insulin resistance (RR=0.63, p=0.003, not insulin resistant reference). Features inversely related to SVR included body weight (RR=0.95 per 5 kg increase, p=0.01), the natural log of HOMA2 (RR=0.77, p<0.001), baseline log10 HCV level (RR=0.77, p<0.001), and more disease severity as measured by fibrosis (Ishak) score (RR=0.90, p=0.02). Additionally, platelet count (RR=1.25 per 103 cells/mm3 increase, p=0.01) and the amounts of PEG-IFN (RR=1.41 per 10% increase, p<0.001) and ribavirin (RR=1.25 per 10% increase, p<0.001) taken during the first 24 weeks of therapy were directly related to the rate of SVR. With regard to lipid levels, baseline TG (natural log scale) was inversely related (RR=0.65, p=0.002) and LDLc was directly related (RR=1.05 per 10 mg/dL increase, p=0.002) to the rate of SVR. Baseline HDLc and TC levels were not significantly associated with SVR.
Table 2.
Univariable models of SVR and selected predictors in serum lipids assessment subset
| Feature | RR (95%CI) | p |
|---|---|---|
| CA Race | 1.92 (1.45,2.56) | <0.001 |
| Male gender | 0.79 (0.61,1.02) | 0.07 |
| Age† | 0.96 (0.89,1.04) | 0.31 |
| Years infected | 1.005 (0.99,1.02) | 0.54 |
| ≤ High school education | 0.64 (0.48,0.85) | 0.002 |
| Weight (kg) † | 0.95 (0.91,0.99) | 0.01 |
| BMI (kg/m2) | 0.98 (0.96,1.01) | 0.23 |
| History of diabetes | 0.52 (0.27,1.01) | 0.054 |
| HOMA2** | 0.77 (0.65,0.90) | <0.001 |
| Insulin resistant | 0.63 (0.46,0.86) | 0.003 |
| History of hypertension | 0.76 (0.56,1.04) | 0.09 |
| Antidepressant drug use | 0.60 (0.18,2.00) | 0.40 |
| Current alcohol use | 0.92 (0.66,1.27) | 0.60 |
| Current smoker | 1.10 (0.85,1.43) | 0.48 |
| ALT (IU/L) # | 1.09 (0.94,1.25) | 0.26 |
| ALT (IU/L)** | 1.08 (0.90,1.31) | 0.40 |
| AST (IU/L) # | 0.83 (0.61,1.10) | 0.18 |
| AST (IU/L)** | 0.83 (0.66,1.04) | 0.11 |
| INR | 1.43 (0.48,4.26) | 0.52 |
| Hemoglobin (g/dL) | 0.95 (0.86,1.05) | 0.30 |
| White blood cells (per 103 cells/mm3) | 1.05 (0.99,1.11) | 0.12 |
| Platelet count (per 105 cells/mm3) | 1.25 (1.06,1.49) | 0.01 |
| Genotype 1a vs. non-1a | 0.88 (0.68,1.13) | 0.32 |
| Baseline viral level (IU)* | 0.77 (0.66,0.89) | <0.001 |
| Ishak fibrosis score | 0.90 (0.82,0.98) | 0.02 |
| Ishak fibrosis score ≥ 3 | 0.82 (0.62,1.08) | 0.16 |
| Cirrhosis (Fibrosis score 5–6) | 0.65 (0.34,1.24) | 0.20 |
| Steatosis score | 0.85 (0.67,1.07) | 0.17 |
| Steatosis (>5% present) | 0.86 (0.66,1.11) | 0.24 |
| HAI inflammation score | 0.995 (0.95,1.05) | 0.85 |
| Proportion of peg-IFN taken` | 1.41 (1.18,1.68) | <0.001 |
| Proportion of ribavirin taken` | 1.25 (1.15,1.35) | <0.001 |
| Lipid parameters:*** | ||
| TG** (mg/dL) | 0.65 (0.49,0.86) | 0.002 |
| LDLc^ (mg/dL) | 1.05 (1.02,1.09) | 0.002 |
| HDLc* (mg/dL) | 0.96 (0.64,1.44) | 0.84 |
| TC* (mg/dL) | 1.59 (0.84,3.01) | 0.15 |
Transfomations as noted:
Log10 transformed;
Natural log transformed
Relative risk:
per 5 unit increase;
per 10 unit increase;
per 100 unit increase;
per 10% increase in dose
Eligible for entry in multivariable model 2
The relationships between the probability of SVR and baseline and changes in TG, LDLc, HDLc, and TC, and during 24 weeks of therapy are displayed (smoothing spline plots) in Supplementary Figures 1a–e. Although the probability of SVR was negatively associated with baseline TG, it was positively related to increases in TG during therapy. On the other hand, the probability of SVR was positively associated with baseline LDLc, but negatively associated with increases in LDLc from baseline during 24 weeks of therapy. Among males, HDLc appeared to be inversely related to SVR rates (Supplemental Figure 1c), while in females the relationship was opposite (Supplemental Figure 1d). The probability of SVR based on baseline and on-treatment changes in TC levels revealed similar patterns as LDLc.
In crude and race-adjusted regression models, the relationships between variables representing the changes in lipid profile measures (both during and after therapy) and the rate of SVR are summarized in Table 3. Adjusting for race, SVR rates were directly and significantly associated with increases in TG (natural log scale; RR=1.29, p=0.02) and declines in LDLc (RR=0.97, p=0.02, per 5 mg/dL increase) during 24 weeks of therapy, compared to pretreatment. Post-treatment changes from pretreatment values in both LDLc (RR=1.04, p=0.001, per 5 mg/dL increase) and TC (natural log scale; RR=4.10, p<0.001) were directly and significantly related to the rate of SVR.
Table 3.
Evaluation of lipid profile measure changes during and after therapy as predictors of SVR
| Lipid profile measure | SVR | |||
|---|---|---|---|---|
| Unadjusted | Race-adjusted | |||
| RR (95%CI) | p | RR (95%CI) | p | |
| Δ: On Tx – Baseline | n=253 | n=253 | ||
| TG* (mg/dL) | 1.43 (1.15,1.78) | 0.001 | 1.29 (1.05,1.59) | 0.02 |
| LDLc† (mg/dL) | 0.96 (0.92,0.98) | <0.001 | 0.97 (0.95,0.995) | 0.02 |
| HDLc* (mg/dL) | 1.13 (0.63,2.02) | 0.68 | 1.13 (0.65,1.96) | 0.66 |
| TC* (mg/dL) | 0.49 (0.23,1.08) | 0.08 | 0.64 (0.30,1.35) | 0.24 |
| Δ: F-up – Baseline | n=245 | n=245 | ||
| TG* (mg/dL) | 1.29 (0.98,1.70) | 0.07 | 1.24 (0.97,1.61) | 0.09 |
| LDLc† (mg/dL) | 1.04 (1.02,1.06) | 0.001 | 1.04 (1.01,1.06) | 0.001 |
| HDLc* (mg/dL) | 0.94 (0.54,1.66) | 0.84 | 0.92 (0.54,1.58) | 0.78 |
| TC* (mg/dL) | 4.64 (2.46,8.76) | <0.001 | 4.10 (2.14,7.85) | <0.001 |
Natural log transformed
per 5 mg/dL change
The multivariable model reported by Conjeevaram et al.(4) based on 400 participants was fit for the 329 participants for whom serum lipid and covariate data were available. (Table 4) In model 1, factors significantly associated with SVR included male gender (RR=0.80, p=0.049), Ishak fibrosis score (RR=0.90, p=0.009), and the amount of PEG-IFN taken during the first 24 weeks (RR=1.38, p<0.001 per 10% dose increase). In addition, there was a significant interaction between race and baseline viral level (p=0.005), indicating that the magnitude of the inverse relationship between viral level and the rate of SVR differed by race, which was documented and described graphically in a previous report based on the Virahep-C cohort (4). Using the same eligible predictors as model 1 and also allowing the baseline lipid profile variables to be eligible for entry, model 2 was created. In model 2, a significant interaction between HDLc and gender (p=0.02) was found. Assessed graphically in Figure 3a for an AA male and female adjusting for other parameters in the model, whereas HDLc (natural log scale) was inversely related to the rate of SVR for males, the relationship was direct among females. Additional multivariable analyses on a sample of 307 patients with available insulin resistance data did not indicate a significant relationship between insulin resistance measures and SVR, accounting for other parameters in model 2 (data not shown). The prediction of SVR did not significantly differ between the models 1 and 2 (AUROCs=0.801 vs. 0.811, respectively, p=0.42) (Figure 4a).
Table 4.
Multivariable models of SVR and selected predictors
| Feature | Model 1 | Model 2 | Model 3 | |||
|---|---|---|---|---|---|---|
| RR (95%CI) | p | RR (95%CI) | p | RR (95%CI) | p | |
| CA Race | 1.98 (1.47,2.67) | <0.001 | 1.81 (1.33,2.45) | <0.001 | 2.28 (1.58,3.30) | <0.001 |
| Male gender | 0.80 (0.64,0.999) | 0.049 | 0.81 (0.62,1.07) | 0.14 | 0.96 (0.72,1.27) | 0.76 |
| Baseline viral level(IU)* and CA race interaction | 1.51 (1.13,2.00) | 0.005 | 1.49 (1.12,1.97) | 0.006 | 1.72 (1.23,2.41) | 0.001 |
| Ishak fibrosis score | 0.90 (0.83,0.97) | 0.009 | 0.91 (0.83,0.98) | 0.02 | 0.90 (0.83,0.98) | 0.02 |
| Amount of peg-IFN taken` | 1.38 (1.18,1.62) | <0.001 | 1.37 (1.18,1.60) | <0.001 | 0.98 (0.85,1.12) | 0.77 |
| Lipid parameters:*** | ||||||
| TG** (mg/dL) | 0.69 (0.52,0.91) | 0.009 | ||||
| HDLc** (mg/dL) and male gender interaction | 0.40 (0.18,0.87) | 0.02 | 0.36 (0.16,0.77) | 0.009 | ||
| LDLc^ (mg/dL) | 1.03 (1.001,1.07) | 0.046 | ||||
| ΔLDLc^ (mg/dL) | 0.97 (0.95,0.996) | 0.02 | ||||
Model 1=Replication of Conjeevaram et al. model 9 on subset of participants with available baseline lipid profiledata (n=330)
Model 2=Baseline lipid profile variables as eligible for entry
Model 3=Multivariable model with baseline and on treatment changes from baseline in lipid profile variables as eligible for entry (n=253). Model also adjusts for changes in body weight (not shown).
Transformations as follows:
Log10 transformed;
Natural log transformed
RR estimate adjustment as follows:
per 5 unit increase;
per10% increase in dose
NA=Not applicable due to significant interaction with gender (refer to genderspecific coefficients)
Figure 3.
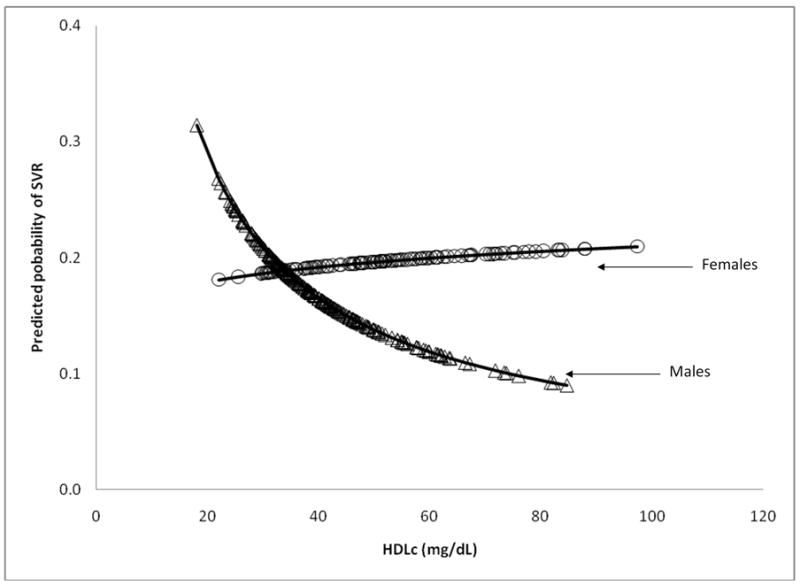
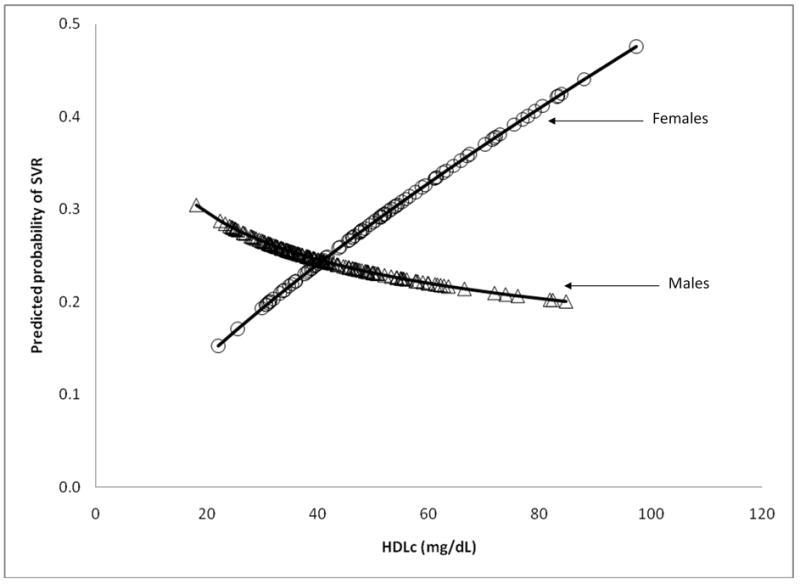
Figure 3a. The predicted probabilities of SVR and HDLc by gender from model 2
Predicted probabilities were calculated adjusting for other parameters in the model for an individual with the follow characteristics: an AA, Ishak fibrosis score of 2, took 80% of the prescribed PEG-IFN during 24 weeks of therapy, and a mean baseline viral level (6.3 log10 IU/mL), natural log of TG (4.7 mg/dL), and LDLc (116.2 mg/dL).
Figure 3b. Predicted probabilities of SVR and HDLc by gender from model 3
Predicted probabilities were calculated adjusting for other parameters in the model for an individual with the follow characteristics: an AA, Ishak fibrosis score of 2, took 80% of the prescribed PEG-IFN during 24 weeks of therapy, and a mean baseline viral level (6.3 log10 IU/mL) and on treatment change in LDLc from pretreatment (− 6.8 mg/dL).
Figure 4.


Figure 4a. Receiver operator curves of multivariable models 1 and 2 (n=329) AUROC=area under the receiver operator curve
Model 1=Includes race, gender, baseline viral level, baseline viral level and race interaction, Ishak fibrosis score, and the amount of PEG-IFN taken
Model 2=Includes variables in model 1, plus baseline TG, baseline HDLc, baseline HDLc and gender interaction, and baseline LDLc
Figure 4b. Receiver operator curves of multivariable Models 1 and 3
AUROC=area under the receiver operator curve
Model 1=Includes race, gender, baseline viral level, baseline viral level and race interaction, Ishak fibrosis score, and the amount of PEG-IFN taken
Model 3=Includes variables in model 1, body weight change, baseline HDLc, baseline HDLc and gender interaction, and the change in LDLc during 24 weeks of therapy
In multivariable modeling, model 3 was constructed using 250 participants who had covariate, baseline lipid profile, and treatment week 24 lipid profile data (Table 3). Model 3 evaluated the relationships between SVR and lipid profile changes during therapy along with those variables eligible for entry into model 2. Variables in models 2 and 3 were similar, though the baseline TG (natural log scale) and LDLc levels were not significantly associated with SVR in model 3, whereas the change in LDLc during the first 24 weeks was retained. Similar to model 2, there was a significant interaction (p=0.009) in model 3 between HDLc (natural log scale) and gender, a relationship which was inverse among males and direct among females (Figure 3b). The AUROC for model 3 was not significantly different than that of model 1 fit to the same 250 participants (AUROC=0.799 vs. 0.779, p=0.19) (Figure 4b). In separate multivariable assessments based on a patient subsample with available insulin resistance data (n=231) and accounting for parameters in model 3, insulin resistant status as a dichotomous measure, but not HOMA2 as a continuous measure, was significantly associated with SVR (data not shown). In all three multivariable models, race remained a significant predictor of SVR and the magnitude of the association was little changed by the addition of serum lipid measures.
DISCUSSION
This evaluation of serum lipids and virological response showed a relationship between baseline lipid measure measurements and on-treatment changes in lipids with antiviral response to therapy. Pretreatment TG and LDLc levels were inversely and directly related to the rate of SVR, respectively. Also, baseline LDLc was significantly higher in CAs compared to AAs. Furthermore, changes in these two parameters during the first 24 weeks of therapy were associated with virologic response and differed significantly by race. In regression models, several lipid profile parameters at baseline (TG and LDLc, HDLc [with an interaction with gender]), and in addition on-treatment changes (TG and LDLc) were significant predictors of SVR. However, including serum lipid measures did not significantly improve the prediction of SVR compared to models without these measures, nor did serum lipid measures account for the racial difference in treatment efficacy between CAs and AAs.
Few studies have assessed in detail changes in serum lipids during and after therapy for chronic HCV infection. Compared to pretreatment, the significant increase in TG levels during therapy found here is consistent with findings in other studies that reported mean TG statistically significant increases of 45 mg/dL and 60 mg/dL (8, 9). We note that compared to pretreatment, the studies did not report significant changes in TC during or after therapy, in contrast to our study which found significant declines in TC during treatment (8, 9). However, the significant increase in TC post-therapy compared to pretreatment is consistent with one study (7) which reported an approximate 10.5 mg/dL significant mean increase, whereas another study did not report significant changes. The difference in findings across studies may be due to variable sample sizes, disparate treatment regimens, inclusion of patients with different HCV genotypes in these studies, and other participant characteristics. The direct relationship between pretreatment LDLc levels and the rate of SVR is consistent with findings from several other studies (10–14).
The study by Hamamoto et al. (7) reported an association between higher pretreatment TG levels and virologic response, which is opposite of the relationship in our study, possibly a reflection of HCV genotype or host lipid receptor genetic differences. Whereas only people infected with HCV genotype 1 were included in Virahep-C, genotype 2 was predominant genotype in the previously referenced study. Single nucleotide polymorphisms in the receptors involved in the serum lipoprotein particle uptake into hepatocytes (SR-B1 and LDL receptors) may also account for the different relationships observed in the two study populations. In multivariable analyses, significant interactions between HDLc levels and gender in relation to virologic response were found, which have not been previously reported. These relationships warrant further investigation and validation in other cohorts to clarify if lipid profile measures are important predictors of treatment response. Post-therapy, increases from baseline in LDLc and TC were found to be associated with SVR, which may correspond to HCV eradication and the subsequent resolution of HCV-induced liver damage.
With evidence from in vitro work supporting several possible mechanisms involving serum lipoproteins, cholesterol metabolism, lipoprotein receptors, and HCV entry, replication and secretion, the significant relationships between both baseline and changes from baseline LDLc and TG levels and rates of SVR are biologically feasible (15–22, 26–30). The direct relationship between LDLc and SVR may partially be explained by competition for LDL receptor sites preventing viral entry into hepatocytes, increasing exposure of HCV to the host immune response in the serum. These findings suggest that serum lipids may yield some prognostic value in determining the probability of treatment success and possibly highlight new therapeutic targets. Further prospective investigation of the impacts of dietary modification and lipid lowering agents on virological response may be warranted. Treatment trials investigating statins and fibrates to improve virological response have yielded mixed results (40–42). As previously documented in the Virahep-C cohort (43), insulin resistance may also contribute to the relationship between serum lipids and SVR.
In conclusion, this study suggests that serum lipid measures are predictors of SVR, but that their predictive ability is ameliorated by race such that these measures do not explain the racial disparity in treatment efficacy between CAs and AAs. However, this study underscores the potential relevance of serum lipids to virologic response. Future investigations may seek to assess relationships between SVR, other characterizations relevant to serum lipids, and genetic determinants of lipid metabolism.
Supplementary Material
*Number of participants within interval for changes in ln(TG) from baseline
**Number of participants within interval for baseline ln(TG)
*Number of participants within interval for changes in LDLc from baseline
**Number of participants within interval for baseline LDLc
*Number of males within interval for changes in ln(HDLc) from baseline
**Number of males within interval for baseline ln(HDLc)
*Number of females within interval for changes in ln(HDLc) from baseline
**Number of females within interval for baseline ln(HDLc)
*Number of participants within interval for changes in ln(TC) from baseline
**Number of participants within interval for baseline ln(TC)
Acknowledgments
Financial Support: The Virahep-C study was conducted as a cooperative agreement funded by the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) with co-funding from the National Center on Minority Health and Health Disparities (NCMHD) and a Cooperative Research and Development Agreement (CRADA) with Roche Laboratories. The specific grant numbers and membership of the Virahep-C Study Group are provided in the primary publication of the study (reference 4). Funding for additional lipid profile analyses utilizing stored serum samples was obtained for an ancillary study of the pathogenesis of steatosis and insulin resistance in chronic HCV infection (KL2 RR024154-02 to LJY).
Abbreviations used
- TC
Total cholesterol
- LDLc
low density lipoprotein cholesterol
- TG
triglyceride
- AA
African American
- CA
Caucasian American
- HDLc
high density lipoprotein cholesterol
- HCV
hepatitis C virus
- VLDL
very low density lipoprotein
- HAI
histological activity index
- ALT
alanine aminotransferase
- AST
aspartate aminotransferase
Footnotes
Financial Disclosures: The authors of this manuscript have no financial disclosures to report.
References
- 1.Armstrong GL, Wasley A, Simard EP, McQuillan GM, Kuhnert WL, Alter MJ. The prevalence of hepatitis C virus infection in the United States, 1999 through 2002. Ann Intern Med. 2006;144:705–714. doi: 10.7326/0003-4819-144-10-200605160-00004. [DOI] [PubMed] [Google Scholar]
- 2.Kim WR. The burden of hepatitis C in the United States. Hepatology. 2002;36:S30–34. doi: 10.1053/jhep.2002.36791. [DOI] [PubMed] [Google Scholar]
- 3.Fried MW, Shiffman ML, Reddy KR, Smith C, Marinos G, Goncales FL, Jr, Haussinger D, et al. Peginterferon alfa-2a plus ribavirin for chronic hepatitis C virus infection. N Engl J Med. 2002;347:975–982. doi: 10.1056/NEJMoa020047. [DOI] [PubMed] [Google Scholar]
- 4.Conjeevaram HS, Fried MW, Jeffers LJ, Terrault NA, Wiley-Lucas TE, Afdhal N, Brown RS, et al. Peginterferon and ribavirin treatment in African American and Caucasian American patients with hepatitis C genotype 1. Gastroenterology. 2006;131:470–477. doi: 10.1053/j.gastro.2006.06.008. [DOI] [PubMed] [Google Scholar]
- 5.Jeffers LJ, Cassidy W, Howell CD, Hu S, Reddy KR. Peginterferon alfa-2a (40 kd) and ribavirin for black American patients with chronic HCV genotype 1. Hepatology. 2004;39:1702–1708. doi: 10.1002/hep.20212. [DOI] [PubMed] [Google Scholar]
- 6.Muir AJ, Bornstein JD, Killenberg PG. Peginterferon alfa-2b and ribavirin for the treatment of chronic hepatitis C in blacks and non-Hispanic whites. N Engl J Med. 2004;350:2265–2271. doi: 10.1056/NEJMoa032502. [DOI] [PubMed] [Google Scholar]
- 7.Hamamoto S, Uchida Y, Wada T, Moritani M, Sato S, Hamamoto N, Ishihara S, et al. Changes in serum lipid concentrations in patients with chronic hepatitis C virus positive hepatitis responsive or non-responsive to interferon therapy. J Gastroenterol Hepatol. 2005;20:204–208. doi: 10.1111/j.1440-1746.2004.03526.x. [DOI] [PubMed] [Google Scholar]
- 8.Naeem M, Bacon BR, Mistry B, Britton RS, Di Bisceglie AM. Changes in serum lipoprotein profile during interferon therapy in chronic hepatitis C. Am J Gastroenterol. 2001;96:2468–2472. doi: 10.1111/j.1572-0241.2001.04055.x. [DOI] [PubMed] [Google Scholar]
- 9.Fernandez-Rodriguez CM, Lopez-Serrano P, Alonso S, Gutierrez ML, Lledo JL, Perez-Calle JL, Temino R, et al. Aliment Pharmacol Ther. 2006;24:507–512. doi: 10.1111/j.1365-2036.2006.03000.x. [DOI] [PubMed] [Google Scholar]
- 10.Akuta N, Suzuki F, Kawamura Y, Yatsuji H, Sezaki H, Suzuki Y, Hosaka T, et al. Predictive factors of early and sustained responses to peginterferon plus ribavirin combination therapy in Japanese patients infected with hepatitis C virus genotype 1b: Amino acid substitutions in the core region and low-density lipoprotein cholesterol levels. J Hepatol. 2007;46:403–410. doi: 10.1016/j.jhep.2006.09.019. [DOI] [PubMed] [Google Scholar]
- 11.Backus LI, Boothroyd DB, Phillips BR, Mole LA. Predictors of response of US veterans to treatment for the hepatitis C virus. Hepatology. 2007;46:37–47. doi: 10.1002/hep.21662. [DOI] [PubMed] [Google Scholar]
- 12.Economou M, Milionis H, Filis S, Baltayiannis G, Christou L, Elisaf M, Tsianos E. Baseline cholesterol is associated with the response to antiviral therapy in chronic hepatitis C. J Gastroenterol Hepatol. 2007 doi: 10.1111/j.1440-1746.2007.04911.x. [DOI] [PubMed] [Google Scholar]
- 13.Gopal K, Johnson TC, Gopal S, Walfish A, Bang CT, Suwandhi P, Pena-Sahdala HN, et al. Correlation between beta-lipoprotein levels and outcome of hepatitis C treatment. Hepatology. 2006;44:335–340. doi: 10.1002/hep.21261. [DOI] [PubMed] [Google Scholar]
- 14.Martinez-Bauer E, Crespo J, Romero-Gomez M, Moreno-Otero R, Sola R, Tesei N, Pons F, et al. Development and validation of two models for early prediction of response to therapy in genotype 1 chronic hepatitis C. Hepatology. 2006;43:72–80. doi: 10.1002/hep.21002. [DOI] [PubMed] [Google Scholar]
- 15.Andre P, Komurian-Pradel F, Deforges S, Perret M, Berland JL, Sodoyer M, Pol S, et al. Characterization of low- and very-low-density hepatitis C virus RNA-containing particles. J Virol. 2002;76:6919–6928. doi: 10.1128/JVI.76.14.6919-6928.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Andre P, Perlemuter G, Budkowska A, Brechot C, Lotteau V. Hepatitis C virus particles and lipoprotein metabolism. Semin Liver Dis. 2005;25:93–104. doi: 10.1055/s-2005-864785. [DOI] [PubMed] [Google Scholar]
- 17.Dreux M, Cosset FL. HCV and lipoproteins: is oxLDL an Achilles’ heel of the Trojan horse? Hepatology. 2006;43:903–905. doi: 10.1002/hep.21202. [DOI] [PubMed] [Google Scholar]
- 18.Monazahian M, Kippenberger S, Muller A, Seitz H, Bohme I, Grethe S, Thomssen R. Binding of human lipoproteins (low, very low, high density lipoproteins) to recombinant envelope proteins of hepatitis C virus. Med Microbiol Immunol. 2000;188:177–184. doi: 10.1007/s004300000032. [DOI] [PubMed] [Google Scholar]
- 19.Carriere M, Rosenberg AR, Conti F, Chouzenoux S, Terris B, Sogni P, Soubrane O, et al. Low density lipoprotein receptor transcripts correlates with liver hepatitis C virus RNA in patients with alcohol consumption. J Viral Hepat. 2006;13:633–642. doi: 10.1111/j.1365-2893.2006.00737.x. [DOI] [PubMed] [Google Scholar]
- 20.Molina S, Castet V, Fournier-Wirth C, Pichard-Garcia L, Avner R, Harats D, Roitelman J, et al. The low-density lipoprotein receptor plays a role in the infection of primary human hepatocytes by hepatitis C virus. J Hepatol. 2007;46:411–419. doi: 10.1016/j.jhep.2006.09.024. [DOI] [PubMed] [Google Scholar]
- 21.Monazahian M, Bohme I, Bonk S, Koch A, Scholz C, Grethe S, Thomssen R. Low density lipoprotein receptor as a candidate receptor for hepatitis C virus. J Med Virol. 1999;57:223–229. doi: 10.1002/(sici)1096-9071(199903)57:3<223::aid-jmv2>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 22.Petit JM, Minello A, Duvillard L, Jooste V, Monier S, Texier V, Bour JB, et al. Cell surface expression of LDL receptor in chronic hepatitis C: correlation with viral load. Am J Physiol Endocrinol Metab. 2007;293:E416–420. doi: 10.1152/ajpendo.00091.2007. [DOI] [PubMed] [Google Scholar]
- 23.Kapadia SB, Barth H, Baumert T, McKeating JA, Chisari FV. Initiation of hepatitis C virus infection is dependent on cholesterol and cooperativity between CD81 and scavenger receptor B type I. J Virol. 2007;81:374–383. doi: 10.1128/JVI.01134-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Molina S, Castet V, Pichard-Garcia L, Wychowski C, Meurs E, Pascussi JM, Sureau C, et al. Serum-derived hepatitis C virus infection of primary human hepatocytes is tetraspanin CD81 dependent. J Virol. 2008;82:569–574. doi: 10.1128/JVI.01443-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.von Hahn T, Lindenbach BD, Boullier A, Quehenberger O, Paulson M, Rice CM, McKeating JA. Oxidized low-density lipoprotein inhibits hepatitis C virus cell entry in human hepatoma cells. Hepatology. 2006;43:932–942. doi: 10.1002/hep.21139. [DOI] [PubMed] [Google Scholar]
- 26.Gastaminza P, Cheng G, Wieland S, Zhong J, Liao W, Chisari FV. Cellular determinants of hepatitis C virus assembly, maturation, degradation and secretion. J Virol. 2007 doi: 10.1128/JVI.02053-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huang H, Sun F, Owen DM, Li W, Chen Y, Gale M, Jr, Ye J. Hepatitis C virus production by human hepatocytes dependent on assembly and secretion of very low-density lipoproteins. Proc Natl Acad Sci U S A. 2007;104:5848–5853. doi: 10.1073/pnas.0700760104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nielsen SU, Bassendine MF, Burt AD, Martin C, Pumeechockchai W, Toms GL. Association between hepatitis C virus and very-low-density lipoprotein (VLDL)/LDL analyzed in iodixanol density gradients. J Virol. 2006;80:2418–2428. doi: 10.1128/JVI.80.5.2418-2428.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Perlemuter G, Sabile A, Letteron P, Vona G, Topilco A, Chretien Y, Koike K, et al. Hepatitis C virus core protein inhibits microsomal triglyceride transfer protein activity and very low density lipoprotein secretion: a model of viral-related steatosis. Faseb J. 2002;16:185–194. doi: 10.1096/fj.01-0396com. [DOI] [PubMed] [Google Scholar]
- 30.Ye J. Reliance of host cholesterol metabolic pathways for the life cycle of hepatitis C virus. PLoS Pathog. 2007;3:e108. doi: 10.1371/journal.ppat.0030108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Murao K, Imachi H, Yu X, Cao WM, Nishiuchi T, Chen K, Li J, et al. Interferon alpha decreases expression of human scavenger receptor class BI, a possible HCV receptor in hepatocytes. Gut. 2008;57:664–671. doi: 10.1136/gut.2006.111443. [DOI] [PubMed] [Google Scholar]
- 32.Friedewald WT, Levy RI, Fredrickson DS. Estimation of the concentration of low-density lipoprotein cholesterol in plasma, without use of the preparative ultracentrifuge. Clin Chem. 1972;18:499–502. [PubMed] [Google Scholar]
- 33.Executive Summary of The Third Report of The National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation And Treatment of High Blood Cholesterol In Adults (Adult Treatment Panel III) Jama. 2001;285:2486–2497. doi: 10.1001/jama.285.19.2486. [DOI] [PubMed] [Google Scholar]
- 34.A software implimentation of the HOMA2 model. 2.2. Oxford: Diabetes Trials Unit, University of Oxford; 2004. [Google Scholar]
- 35.Knodell RG, Ishak KG, Black WC, Chen TS, Craig R, Kaplowitz N, Kiernan TW, et al. Formulation and application of a numerical scoring system for assessing histological activity in asymptomatic chronic active hepatitis. Hepatology. 1981;1:431–435. doi: 10.1002/hep.1840010511. [DOI] [PubMed] [Google Scholar]
- 36.Ishak K, Baptista A, Bianchi L, Callea F, De Groote J, Gudat F, Denk H, et al. Histological grading and staging of chronic hepatitis. J Hepatol. 1995;22:696–699. doi: 10.1016/0168-8278(95)80226-6. [DOI] [PubMed] [Google Scholar]
- 37.Kruse W, Rampmaier J, Ullrich G, Weber E. Patterns of drug compliance with medications to be taken once and twice daily assessed by continuous electronic monitoring in primary care. Int J Clin Pharmacol Ther. 1994;32:452–457. [PubMed] [Google Scholar]
- 38.Zou G. A modified poisson regression approach to prospective studies with binary data. Am J Epidemiol. 2004;159:702–706. doi: 10.1093/aje/kwh090. [DOI] [PubMed] [Google Scholar]
- 39.DeLong ER, DeLong DM, Clarke-Pearson DL. Comparing the areas under two or more correlated receiver operating characteristic curves: a nonparametric approach. Biometrics. 1988;44:837–845. [PubMed] [Google Scholar]
- 40.Bader T, Fazili J, Madhoun M, Aston C, Hughes D, Rizvi S, Seres K, et al. Fluvastatin Inhibits Hepatitis C Replication in Humans. Am J Gastroenterol. 2008 doi: 10.1111/j.1572-0241.2008.01876.x. [DOI] [PubMed] [Google Scholar]
- 41.Fujita N, Kaito M, Kai M, Sugimoto R, Tanaka H, Horiike S, Konishi M, et al. Effects of bezafibrate in patients with chronic hepatitis C virus infection: combination with interferon and ribavirin. J Viral Hepat. 2006;13:441–448. doi: 10.1111/j.1365-2893.2005.00718.x. [DOI] [PubMed] [Google Scholar]
- 42.O’Leary JG, Chan JL, McMahon CM, Chung RT. Atorvastatin does not exhibit antiviral activity against HCV at conventional doses: a pilot clinical trial. Hepatology. 2007;45:895–898. doi: 10.1002/hep.21554. [DOI] [PubMed] [Google Scholar]
- 43.Conjeevaram HS, Kleiner DE, Everhart JE, Hoofnagle JH, Zacks S, Afdhal NH, Wahed AS. Race, insulin resistance and hepatic steatosis in chronic hepatitis C. Hepatology. 2007;45:80–87. doi: 10.1002/hep.21455. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
*Number of participants within interval for changes in ln(TG) from baseline
**Number of participants within interval for baseline ln(TG)
*Number of participants within interval for changes in LDLc from baseline
**Number of participants within interval for baseline LDLc
*Number of males within interval for changes in ln(HDLc) from baseline
**Number of males within interval for baseline ln(HDLc)
*Number of females within interval for changes in ln(HDLc) from baseline
**Number of females within interval for baseline ln(HDLc)
*Number of participants within interval for changes in ln(TC) from baseline
**Number of participants within interval for baseline ln(TC)