

Abstract
During gene expression, RNA polymerase (RNAP) encounters a major barrier at a nucleosome and yet must access the nucleosomal DNA. In vivo evidence suggests that multiple RNAPs might increase transcription efficiency through nucleosomes. Here we have quantitatively investigated this hypothesis using E. coli RNAP as a model system by directly monitoring its location on the DNA via a single molecule DNA unzipping technique. When an RNAP encountered a nucleosome, it paused with a distinctive 10-bp periodicity and was backtracked by ~10–15 bp. When two RNAPs elongated in close proximity, the trailing RNAP assisted the leading RNAP elongation, reducing its backtracking and enhancing its transcription through a nucleosome by a factor of 5. Taken together, our data indicate that histone-DNA interactions dictate RNAP pausing behavior, and alleviation of nucleosome-induced backtracking by multiple polymerases may serve as a mechanism for overcoming the nucleosomal barrier in vivo.
Nucleosomes are known to play an important role in the regulation of gene expression. During transcription, RNA polymerase (RNAP) must access DNA associated with nucleosomes, the fundamental packing units of chromatin. In vitro studies have shown that even a mononucleosome imposes a substantial barrier to transcription elongation by a single RNAP1–10. The presence of a nucleosome induces RNAP to pause/arrest due to backtracking, during which RNAP disengages its active site from the 3′ end of RNA and slides backwards non-catalytically along the DNA, resulting in an extrusion of 3′ RNA through its secondary channel6.
In contrast, in vivo data have shown that RNAP is able to elongate rapidly in the presence of nucleosomes11–13. If so, how does RNAP overcome the nucleosome barrier during elongation? To date, several mechanisms have been recognized, including direct elongation rate enhancement by transcription factors6, 14–16, and increasing DNA accessibility via histone modifications17–19 and/or nucleosome remodeling20, 21.
Additionally, in vivo evidence shows that multiple RNAPs often occur on active genes. A large number of human genes are found to have two or more active promoters which greatly increase the chance of recruitment of multiple RNAPs22. On a fully induced Drosophila hsp70 gene, ~30 transcribing polymerase molecules have been detected11. Live-cell imaging of transcription indicates that mammalian RNAP often enters a pause state for unexpectedly long times, which may allow trailing RNAPs to catch up to it13. More importantly, it has been shown that the density of RNAP is a major factor for defining the regions of nucleosome removal in transcribed genes23, 24. Therefore, it’s appealing to hypothesize that cooperation by multiple RNAPs may also contribute to efficient RNAP progression through a nucleosomal barrier. Several observations suggest that it may be plausible. Biochemical studies of E. coli RNAP show that when multiple initiation happens from the same promoter, the leading RNAP is able to more efficiently forward translocate through a bound protein such as EcoRQ111 or lac repressor, with a concomitant reduction in the RNAP arrest probability25, 26. In addition, single molecule studies show that both E. coli RNAP and Pol II are powerful molecular motors, capable of exerting forces and generating displacements16, 27. Thus an assisting force may be exerted by a trailing RNAP on a leading RNAP as the leading RNAP encounters a nucleosome barrier. Indeed an assisting external force has been shown to reduce RNAP backtracking while facilitating its forward translocation16, 28.
Here we have tested this hypothesis using E. coli RNAP as a model system because E. coli RNAP and Pol II are evolutionarily conserved in sequence, structure, and function29, 30, yet E.coli RNAP is structurally simpler and requires only the holoenzyme for initiation. Furthermore, E. coli RNAP has been shown to resemble yeast Pol II in all tested properties of transcription through a nucleosome in vitro5. In this work, we have ascertained how two RNAPs may work together to transcribe through a nucleosome.
RESULTS
Locating RNAP by unzipping DNA
To monitor how RNAP progresses through a nucleosome, we needed to be able to detect its physical location along DNA. This can not be readily achieved by conventional bulk transcription gel assays which measure the length of the RNA transcript, i.e., the 3′ RNA location along the DNA. Instead we used a single molecule assay to locate RNAP by mechanically unzipping dsDNA through a bound RNAP. Previously we had developed the DNA unzipping technique and demonstrated that it is a versatile and powerful tool for measurements of protein-DNA interactions with near basepair precision and accuracy31–33.
We first constructed a DNA template containing a single T7A1 promoter and then allowed a paused transcription complex (PTC) to form at the +20 nt position via depletion of UTP at room temperature (Fig. 1a and Methods). The PTC formation reaction was quenched by EDTA after 2 min. To unzip DNA through a PTC, an optical trap was used to sequentially convert dsDNA into ssDNA by mechanical separation of base pairs (Fig. 1a and Supplementary Fig. 1b and Methods). An RNAP-DNA interaction was detected whenever the unzipping force substantially deviated from the corresponding naked DNA unzipping force, a sequence-dependent baseline around 15 pN.
Figure 1.
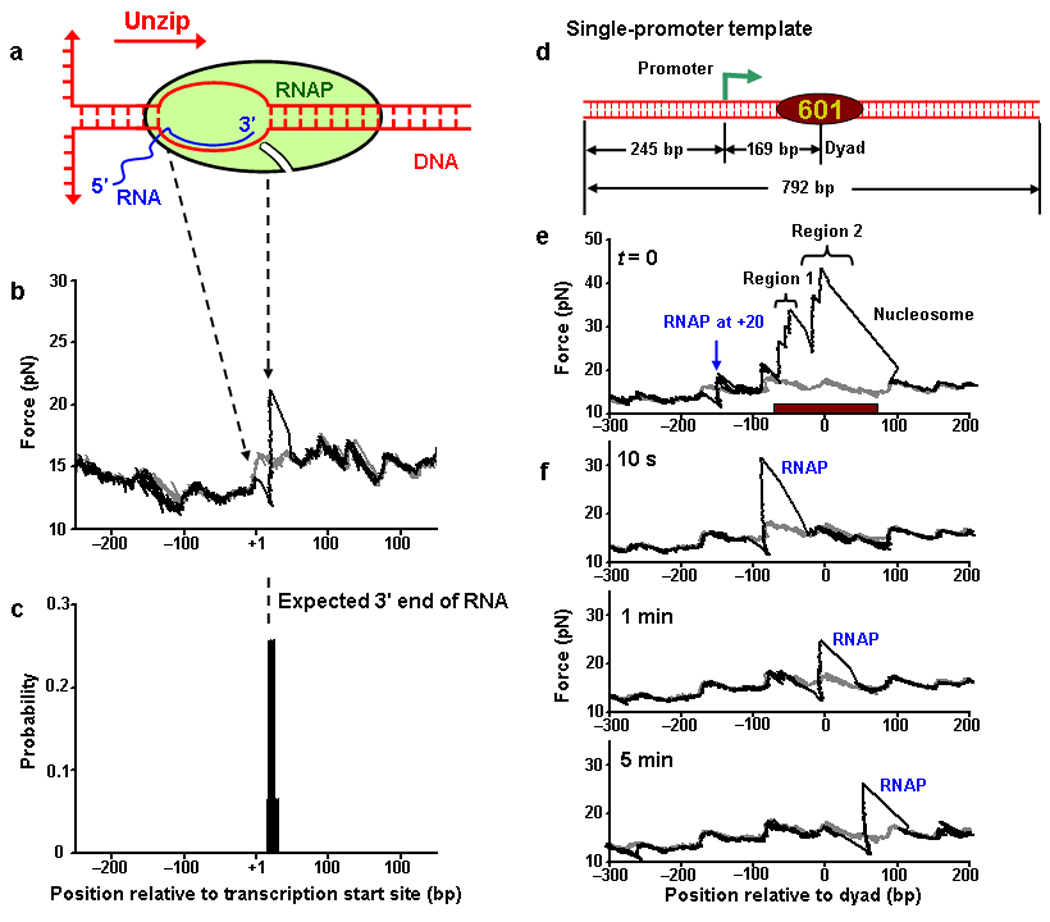
Locating an RNAP during elongation on nucleosomal DNA. (a) A cartoon of the transcription elongation complex. Unzipping direction is indicated by a red arrow. (b) An example trace of unzipping DNA through a PTC. RNAP was stalled at the +20 nt position relative to transcription start site. The RNAP unzipping force signature (black) shows a distinctive force drop immediately followed by a sharp force rise. The unzipping force of the corresponding naked DNA is shown for comparison (grey). (c) Location distribution of the unzipping force rise obtained by pooling a number of measurements such as that shown in (b). The dashed line indicates the expected location of the 3′ end of the transcribed RNA. (d) The single-promoter transcription template construct containing both a single T7A1 promoter and a 601 nucleosome positioning element (NPE). (e) An example unzipping trace of a template containing both a PTC stalled at +20 nt and a positioned nucleosome. Unzipping confirmed that the RNAP and the nucleosome were at their expected locations. Two regions of strong histone-DNA interactions in a nucleosome are indicated: Region 1 (off-dyad interactions) and Region 2 (dyad interactions). The brown bar indicates the 147-bp 601 NPE. (f) Representative traces of unzipping through an elongation complex. After transcription was resumed for an indicated duration, it was quenched and histones were dissociated. Unzipping revealed the location of the remaining RNAP. Each trace is from a different DNA molecule. The unzipping force of the corresponding naked DNA is shown for comparison (grey).
When a single DNA molecule was unzipped starting from upstream of the RNAP (Fig. 1b), the unzipping force initially followed that of the corresponding naked DNA. However, as the unzipping fork encountered the transcription bubble formed by the RNAP, the force dropped below the naked DNA baseline. Subsequently the force rose sharply above the baseline as the unzipping fork encountered the beginning of the dsDNA that was clamped downstream by the RNAP. The force then continued to follow that of the corresponding naked DNA. As expected for a thermally activated off-equilibrium process, the magnitude of the force drop and the rise varied from trace to trace.
For a PTC at +20 nt, the active site of the RNAP should be at +20 bp from the transcription start site and the downstream dsDNA should begin at around +23±1 bp34–36. The location of the force rise, indicative of the beginning of the downstream dsDNA, was detected at +22 bp, in excellent agreement with the expected location (Fig. 1c). Additional experiments also showed that depletion of Mg2+ by EDTA quenching minimized RNAP diffusive motion along the DNA in an elongation complex (Supplementary Fig. 2). Thus the unzipping force signature of an RNAP serves as a convenient and distinctive indicator of the RNAP location. The active site location was then taken to be 2 bp upstream from the measured force rise location for all subsequent experiments.
Locating RNAP during elongation on nucleosomal DNA
We next demonstrated that the DNA unzipping assay could also be used to locate an RNAP during elongation on nucleosomal DNA. For these experiments, we constructed a single-promoter DNA template containing a single T7A1 promoter followed by a 601 nucleosome positioning element (NPE) that is known to uniquely position a nucleosome37 (Fig. 1d). In this design, the 601 NPE was flanked by long stretches of DNA, in contrast to short DNA templates typically used in conventional biochemistry experiments. We then assembled a single nucleosome onto the 601 NPE using a salt dialysis method and subsequently formed a PTC at the +20 nt position (Methods). When this DNA template was unzipped, the characteristic force signatures for both the RNAP and the nucleosome were observed at their expected locations (Fig. 1e).
We found that the nucleosome was uniquely positioned within the 601 NPE and its unzipping force signatures were consistent with those of our previous work32: For a given nucleosome, there were three broad regions of strong interactions, with one around the dyad and the other two around ~ ±40 bp from the dyad. Unzipping from one direction typically only revealed the first two regions encountered but not the last one, due to histone dissociation from the 601 NPE upon disruption of the dyad region of interactions.
To resume elongation, 1 mM of NTPs was supplemented, together with competitor DNA containing a T7A1 promoter to prevent re-initiation (Methods and Supplementary Fig. 3a). The reaction was then quenched by excess EDTA at specified time points. When the RNAP was not in the immediate vicinity of the nucleosome, the unzipping force signatures for both the RNAP and the nucleosome were readily discernable (as shown in Fig. 1e). However when the RNAP had encountered a nucleosome, we observed a much more complex and variable force signature that did not readily distinguish between the RNAP and the nucleosome. To examine only the RNAP location, heparin was used to dissociate the histones from the DNA immediately after the chase reaction was quenched (Methods and Supplementary Fig. 3b). Control experiments showed that neither the competitor DNA nor heparin dissociated RNAP or altered RNAP locations (Supplementary Fig. 3a and its Legend and Supplementary Fig. 3d). As shown from the representative traces at three transcription times (Fig. 1f), RNAP was clearly distinguishable on DNA molecules after histone dissociations by heparin. When RNAP moved through a nucleosome, it encountered strong interactions preceding the dyad region (10 s trace) followed by strong interactions at the dyad region (1 min trace), and then moved out of the nucleosome (5 min trace).
Transcription pausing pattern at a nucleosome
We carefully examined nucleosome-induced pause sites using bulk transcription assays on a single-promoter DNA template (Fig. 2a, top and Methods). The lengths of the RNA, indicative of the 3′ end location of the RNA transcript on DNA, were determined using denaturing PAGE. Consistent with previous observations1, 4–8, the presence of a nucleosome dramatically reduced the transcription rate. While essentially all RNAPs reached the runoff end of a naked DNA template within 1 min, only ~ 50% of RNAPs were able to reach the runoff end in the presence of a nucleosome, even after 30 min. In addition, as RNAP proceeded into the nucleosome, a distinct periodicity of ~ 10 bp highlighted the nucleosome-induced pause sites: −60 bp (strong), −50 bp (weak), −40 bp (strong) and −30 bp (strongest) from the dyad. Since the RNAP leading edge is located ~ 20 bp downstream of the active site38, these pause sites coincided with the two strong histone-DNA interaction regions that the RNAP encountered. As the leading edge of the RNAP passed the dyad region, pausing immediately disappeared, indicating the absence of major obstacles. It is noteworthy that the pausing pattern, including the 10 bp periodicity, remained unchanged when the DNA downstream of the nucleosome was truncated, indicating that this segment of the DNA was not essential for the pausing pattern (Supplementary Fig. 4).
Figure 2.
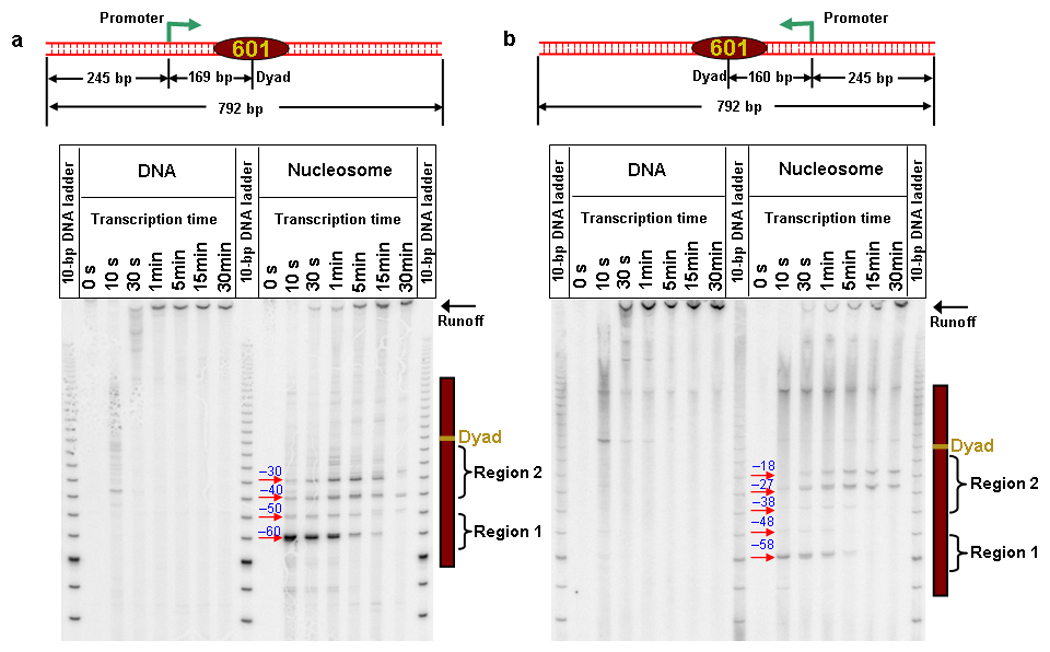
Transcription through a nucleosome shows a distinctive 10 bp periodicity pausing pattern. (a) RNAP transcribed through a nucleosome in the forward direction of the 601 NPE as indicated by the template cartoon (identical to Fig. 1d). PAGE analysis of transcription through naked DNA and nucleosomal DNA shows that as RNAP proceeded into the nucleosome, a distinctive periodicity of ~ 10 bp highlighted all nucleosome-induced pause sites within Regions 1 and 2. Transcription pause sites are marked as distances from the dyad. (b) RNAP transcribed through a nucleosome from the reverse direction of 601 NPE as indicated by the template cartoon. Although RNAP effectively transcribed a different sequence, all nucleosome-induced pauses were again highlighted by a distinctive ~ 10 bp periodicity within Regions 1 and 2. The pause site at the end of the 601 NPE might be intrinsic pausing (compare transcription through naked DNA and nucleosomal DNA). Also note that at this pause site the leading edge of the RNAP was ~ 20 bp downstream of the 601 NPE.
To examine whether these observations were specific to the DNA sequence transcribed, we placed the promoter on the distal site of the 601NPE and allowed the RNAP to elongate into the nucleosome from the reverse direction (Fig. 2b). Since the 601NPE sequence is not palindromic, RNAP effectively transcribed a new sequence. We found that all nucleosome-induced pauses were still highlighted by a distinctive 10 bp periodicity. The pause patterns from the two sequences share substantial similarities, indicating that the nucleosome-induced pausing pattern described here is not specific to the sequences used here, although DNA sequence may influence the strengths of the pause sites.
To substantiate this conclusion, we compared the intrinsic pause sites obtained at low [NTP] on naked DNA with nucleosome-induced pause sites (Supplementary Fig. 5). As shown, the intrinsic pausing sites do not display a 10-bp periodicity and in general do not completely coincide with the pausing sites at a nucleosome. Therefore a nucleosome does not simply enhance intrinsic pausing.
Remarkably, these pausing features bear resemblance to the resistance encountered during mechanical unzipping through a nucleosome32: the unzipping fork paused at the first off-dyad and dyad regions of interactions. In addition, the unzipping fork paused with a 5-bp periodicity, likely resulting from alternating interactions of the histone core with the two strands of dsDNA at each minor groove32, 39. Since RNAP paused every 10 bp, it may cooperatively disrupt a pair of interactions at each DNA minor groove. Therefore we conclude that the transcription pausing pattern at a nucleosome is predominantly determined by the nucleosome structure. Other factors, such as the type of RNAP, DNA sequence, and the uniqueness of nucleosome positioning, may also contribute to the pausing pattern.
RNAP backtracking at a nucleosome
We investigated the extent of backtracking during nucleosome-induced transcription pausing by comparing the location of the RNAP active site on DNA with the corresponding transcript length. This allowed a direct measurement of the backtracking distance, as compared with conventional methods which typically can only detect transcript length and therefore rely on sensitivity to cleavage factors (TFIIS or GreA/B) for evidence of backtracking.
A line scan (Fig. 3b) of the transcription gel of the single-promoter nucleosome template (Fig. 2a) shows that the distribution of the 3′ end of RNA peaked at the –60 bp position from the dyad (upon encountering the off-dyad region of interactions) after 10 s of transcription (Fig. 3b). The corresponding distribution of the location of the RNAP active site, as determined by DNA unzipping, resembles that of the 3′ end of RNA, but peaked at –75 bp from the dyad (Fig. 3c) with a broader distribution which lacks the 10 bp periodicity. This clearly shows that a substantial fraction of RNAP was backtracked to various distances at a given pause and on average the nucleosome-induced backtracking was ~15 bp (Fig. 3a). After 5 min of transcription, the RNAP progressed further into the nucleosome and encountered the dyad region of strong interactions as indicated by the strong pause sites at –40 bp and –30 bp before the dyad (Fig. 2a and Supplementary Fig. 6a). RNAP again backtracked with a mean backtracking distance of ~ 10 bp while a small fraction elongated through the nucleosome (Supplementary Fig. 6b). Compared with the 10 s data, a fraction of RNAP that initially paused continued to elongate, indicating that this fraction was either not backtracked or not backtracked extensively, as has been previously reported6, 8. However, a substantial fraction was not able to elongate through the nucleosome even after 30 min of transcription (Fig. 2), indicating that extensive backtracking occurred in this fraction.
Figure 3.
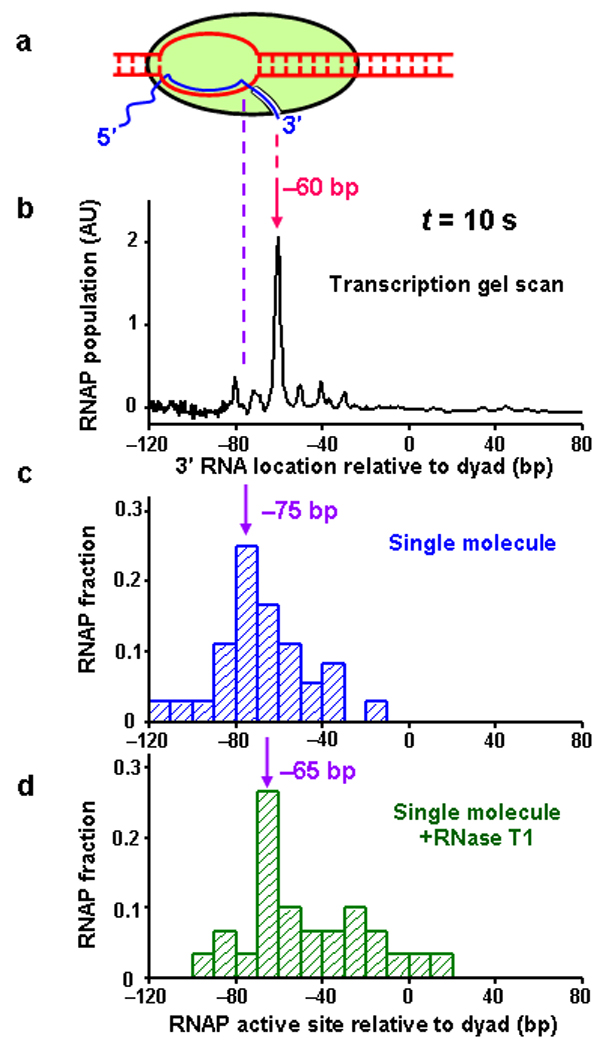
Histone-DNA interactions induce RNAP backtracking and prevention of backtracking facilitates transcription. All experiments were conducted using the single-promoter DNA template and for 10 s transcription time. The predominant peak position in each distribution is indicated by an arrow. (a) A cartoon of a backtracked transcription elongation complex. Pink dashed line indicates the location of the 3′ end of RNA, and the purple dashed line indicates the location of RNAP active site. (b) An intensity scan of the gel shown in Figure 2a. The 3’ RNA location is specified relative to the dyad. (c) Distribution of RNAP active site location as determined by the unzipping method. The active site location is specified relative to the dyad. The displacement between the peak location of the active site and that of the 3′ end of the RNA indicates the backtracking distance. (d) Distribution of RNAP active site location in the presence of RNase T1.
To substantiate this conclusion, we conducted an experiment in which RNase T1 was added during the transcription chase reaction (Methods) to remove most of the 5′ end of the exposed nascent RNA. This truncation is expected to reduce the extent of backtracking so as to facilitate transcription through a nucleosomal template6. Such an effect is difficult to observe using traditional methods that typically measure the length of intact RNA, but the unzipping assay allows direct detection of the RNAP position and thus circumvents this problem. As a control experiment, we verified that the presence of RNase T1 did not alter the unzipping force signature of the RNAP or the nucleosome (Supplementary Fig. 3c).
In the presence of RNase T1, after 10 s of transcription, the active site location distribution peaked at −65 bp from the dyad and the peak was better defined (Fig. 3d). This indicates that when the leading edge of the RNAP encountered the first off-dyad region of interactions in the nucleosome, it still paused but the backtracking distance was largely reduced (compare Fig. 3b, 3c, and 3d). The reduced backtracking is expected to be less inhibitory to elongation. Consistent with this, a greater fraction of RNAP elongated further along the template (compare Fig. 3c and 3d; also compare Supplementary Fig. 6b and Supplementary Fig. 6c for the 5 min transcription).
Taken together, these results suggest that backtracking is the major cause of nucleosome-induced RNAP pausing and any mechanism that reduces backtracking should facilitate transcription through nucleosomes.
Elongation by two RNAPs through a nucleosome
In vivo, the concerted action of multiple RNAPs which elongate in the same direction may facilitate transcription through nucleosomal DNA. In order to test this hypothesis, we constructed a two-promoter DNA template containing two T7A1 promoters, each followed by identical sequences of 36 bp, and both oriented towards a downstream 601 NPE (Fig. 4a and Fig. 5a). The locations of the two RNAPs were then monitored by the unzipping method which, unlike a bulk transcription assay, does not suffer from complications caused by overlapping in pause sites from the two RNAPs.
Figure 4.
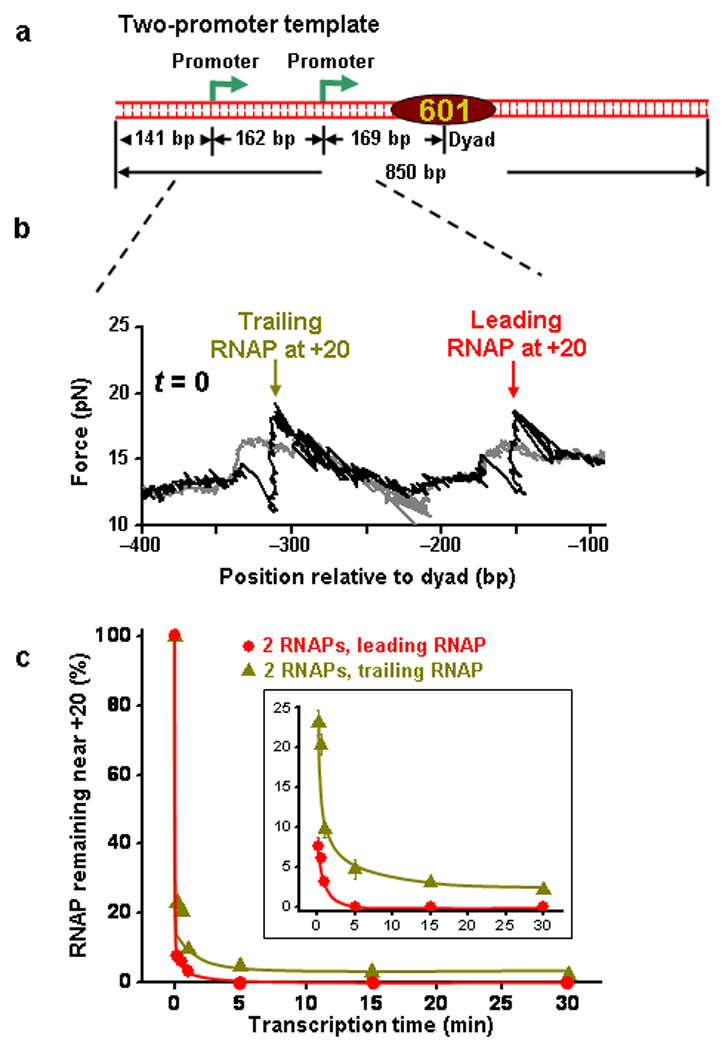
Trailing RNAP assists leading RNAP to exit an arrested state. (a) The two-promoter transcription template construct contains two T7A1 promoters followed by a single 601 NPE. (b) Example unzipping trace from the template shown in (a) containing two PTCs at their respective +20 nt positions. The two RNAPs were detected at their expected locations. (c) Percentage of RNAP that remained near the +20 nt position versus transcription time for leading and trailing RNAPs. The inset more clearly shows the percentage of the RNAP remaining near the +20 nt.
Figure 5.
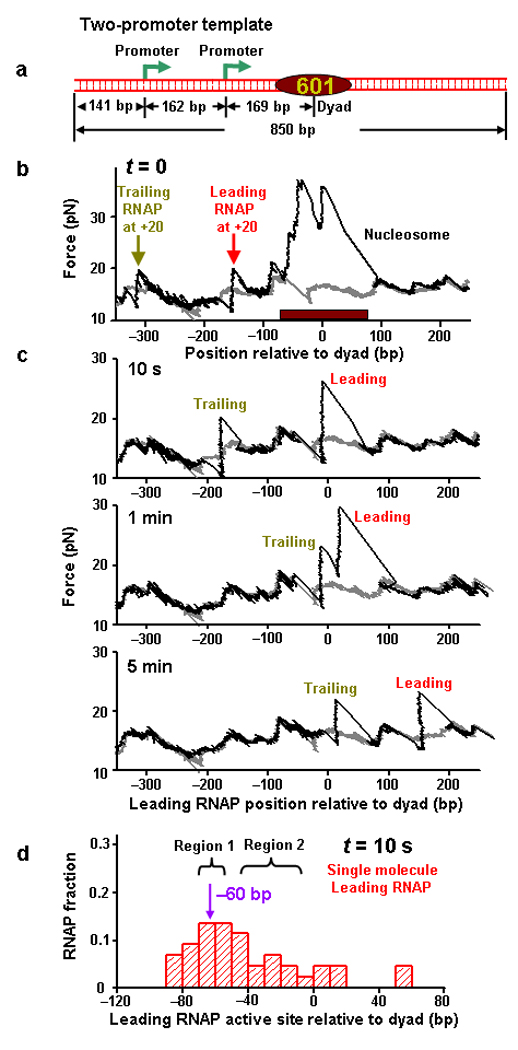
Two RNAPs work synergistically to overcome a nucleosomal barrier. (a) The two-promoter transcription template construct contains two T7A1 promoters followed by a single 601 NPE (same as Fig. 4a). (b) Example unzipping trace from the template shown in (a) containing two PTCs at their respective +20 bp positions and a positioned nucleosome before transcription resumption. The two RNAPs and the nucleosome were detected at their expected locations. The brown bar indicates the 147-bp 601 NPE. (c) Representative unzipping traces through two elongation complexes on a single DNA molecule after transcription for the indicated durations and after removal of histones. Each trace was from a different DNA molecule. Both the leading and trailing RNAPs were detected by their unzipping signatures. (d) Distribution of the leading RNAP active site location after 10 s transcription reaction.
The experimental procedures were similar to those described for single-promoter DNA template experiments (Methods). First, we examined PTCs that remained near the +20 nt position. Before the NTP chase, PTCs were allowed to equilibrate among their translocation states. Unzipping experiments showed clear force signatures for the two RNAPs stalled at their respective +20 nt loci (Fig. 4b). Upon NTP addition, a majority of the PTCs at each promoter escaped almost instantly. However, a small fraction escaped more slowly and then leveled off with time. For the trailing RNAP, the fraction remaining was clearly backtracked as indicated by the average location of remaining RNAPs relative to the expected RNAP location (Supplementary Figure 7b, dark yellow). Furthermore, the more extensive the backtracking, the longer it took for the RNAP to escape (Fig. 4c and Supplementary Fig. 7a, dark yellow). After 30 min of NTP chase, ~ 5% of trailing RNAP remained and they were backtracked by ~ 12 bp. These backtracked complexes were extremely stable and considered arrested on the experimental time scale. These properties were essentially identical to those exhibited by PTCs on the single-promoter template (Supplementary Fig. 7a and Supplementary Fig. 7b). This result provides direct evidence for nucleosome-independent backtracking. On the other hand, the leading RNAP escaped to completion in < 5 min (Fig. 4c, red). Given that both PTCs were identical, the different escape behaviors were a result of the interaction between the two RNAPs. This indicates that the trailing RNAP is capable of assisting the leading RNAP to escape from a backtracked state, rescuing it from an arrested state.
Second, we examined the RNAPs that escaped after NTP addition. Before the NTP chase, unzipping experiments showed clear force signatures for the two RNAPs stalled at their respective +20 nt loci followed by a nucleosome (Fig. 5b). As shown in the representative traces (Fig. 5c), upon the resumption of transcription the locations of both RNAPs were clearly discernable for each trace. Notice that the two RNAPs were not always found to be in immediate vicinity of each other. Although the interaction between two RNAPs would assist the leading RNAP elongation, this interaction would also possibly induce backtracking of the trailing RNAP. Thus a separation could be created between the two RNAPs.
The distribution of the leading RNAP location (Fig. 5d) shows that the peak location of the RNAP positions was shifted towards the nucleosome to –60 bp from the dyad, with a substantial fraction transcribing beyond the –60 bp pause site. As compared with the single RNAP experiments (Fig. 3c), the fraction elongating through the nucleosome was also increased.
Rate enhancement by a trailing RNAP at a nucleosome
In order to provide a quantitative measure of elongation rate enhancement of a leading RNAP due to a trailing RNAP, we examined the transcription runoff efficiency of each RNAP as a function of transcription time (Fig. 6a). Runoff efficiency was computed based on the percentage of DNA templates that showed an absence of RNAP during the DNA unzipping experiments since an RNAP did not dissociate until it reached the runoff end (Supplementary Fig. 8 and Supplementary Discussion). The runoff efficiencies are more concisely summarized using the initial transcription rate near zero transcription times and the rate to achieve 50% runoff to quantify the comparison (Fig. 6b). When a single RNAP transcribed through a mononucleosomal template of ~ 550 bp total transcript size, the transcription rate was reduced by a factor of ~20–35 relative to that of naked DNA. However, this rate was increased by a factor of 5 with the assistance of a trailing RNAP, a rate enhancement comparable to that achieved by using RNase T1. Even the trailing RNAP showed a rate enhancement by a factor of 2–3 compared with that from a single RNAP alone. This is consistent with at least partial eviction of histones by the leading RNAP as evidenced by the lack of pausing sites after RNAP moved beyond the dyad region of interactions (Fig. 2).
Figure 6.
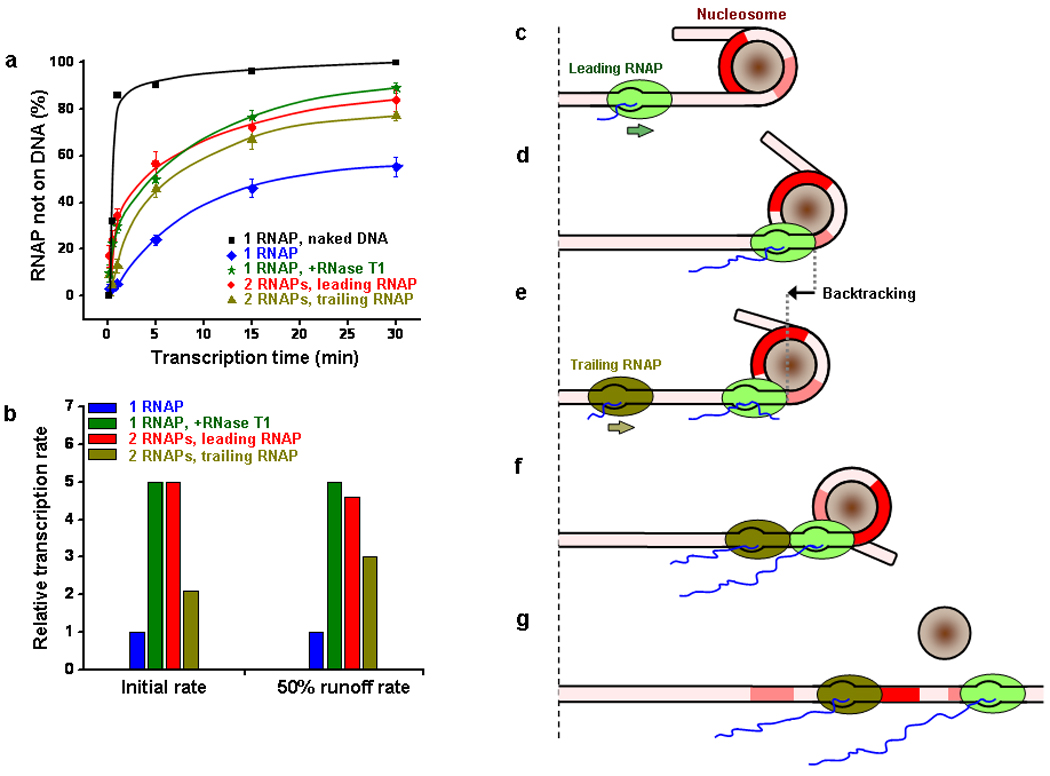
Transcription efficiency comparison and cartoon illustrating the mechanism of transcription through nucleosomal DNA. (a) Transcription runoff efficiencies vs. transcription time. A runoff efficiency was represented by the percentage of DNA template that showed an absence of RNAP during DNA unzipping experiments. The error bars are standard errors of the means. Smooth curves passing through the data points for each transcription condition were drawn for ease of comparison (not fits). Naked DNA runoff efficiency (black) was obtained from PAGE gel analysis and is shown for comparison. (b) Bar plot of relative transcription rates through nucleosomal DNA. The initial rate of a single RNAP transcribing through a nucleosomal template is used as a reference. The initial rates were estimated from the slopes of linear fits to the near zero transcription times (≤ 1 min). Note that since the trailing promoter is about 162 bp upstream of the leading promoter, a 10 s time delay was taken into account for the trailing RNAP transcription rate calculation. The 50% runoff rate is the reciprocal of the time to achieve 50% runoff. (c–g) Cartoon illustrations of the mechanism of transcription through a nucleosome. As an RNAP approaches a nucleosome (c), it encounters histone-DNA interactions in a nucleosome which induce RNAP pausing and likely backtracking (d and e). The arrival of a trailing RNAP (f) exerts an assisting force on the leading RNAP, rescuing the leading RNAP from its backtracked state. The two RNAPs, working synergistically, eventually evict downstream histones, resulting in the removal of the nucleosomal barrier and the resumption of efficient transcription (g). Regions of strong histone-DNA interactions in the nucleosomal DNA are indicated in red and pink.
DISCUSSION
This work provides a coherent picture of transcription through a nucleosome (Fig. 6c–g). As an RNAP encounters a nucleosome barrier, it must sequentially overcome the histone-DNA interactions within the nucleosome. The locations and strengths of these interactions dictate the pausing pattern of the RNAP, yielding pausing behaviors that are characteristic of these interactions. Pauses occur approximately every 10 bp (when RNAP encounters DNA minor groove interactions with the core histone surface), with the strongest pausing at around −60 bp before the dyad (upon encountering the first off-dyad region of strong interactions) and at around −30 bp before the dyad (upon encountering the dyad region of strong interactions), but no pausing occurs once the leading edge of the RNAP passes the dyad region (possibly due to histone dissociation). At each pause site prior to reaching the dyad region, RNAP may backtrack to a variable distance and the mean backtracking distance is ~ 10–15 bp. Such a large backtracking distance makes it difficult for RNAP to resume active elongation. Thus any mechanism that would reduce backtracking should facilitate the escape of RNAP from a nucleosome-induced backtracking pause. A trailing RNAP, which initiates from the same or a different promoter, may then catch up with a leading RNAP and interact with it to facilitate its exit from the backtracked state and entry into productive elongation. Once the leading RNAP overcomes the dyad region of interactions, it may then proceed forward with little resistance.
The current work employed E. coli RNAP but many findings here may also be more generally applicable to Pol II.
First, we showed that E. coli RNAP displayed a characteristic 10 bp periodic pausing pattern when encountering the promoter-proximal half of the nucleosome. Such periodicity has not been explicitly reported for Pol II or E. coli RNAP and the apparent lack of reported periodicity may be due to nucleosome positioning heterogeneity. The 5s rRNA NPE generates several major and minor nucleosome positions40 and in previous studies where it was used 4–6, nucleosome-specific pauses might have been masked by multiple sequence-specific pause sites enhanced by the presence of the nucleosome. 601 and 603 NPEs can position a nucleosome more uniquely, but the positioning accuracy may still be influenced by the length of DNA template used and the position of a nucleosome relative to the DNA ends7, 8. Nonetheless there have been interesting hints of the presence of a 10 bp pausing periodicity of Pol II from previous studies that used 601 and 603 NPEs7, 8. Also, the 10 bp pausing periodicity was observed for Pol III3 but was interpreted as a restricted rotation of Pol III due to DNA loop formation. Our work offers an alternative and much simpler explanation. Despite the evidence discussed above, we can not fully exclude the possibility that the lack of strong 10-bp pausing periodicity by Pol II transversal of a nucleosome could be due to a difference between bacterial and eukaryotic RNA polymerases.
Second, we found that the strongest pause sites occurred at around −60 bp, and then −30 bp before the dyad. Essentially identical pausing regions were identified for Pol II albeit with a lack of distinct, or less pronounced, periodicity6–8. This again suggests a high degree of similarity in the nature of the nucleosome barrier encountered by E. coil RNAP and Pol II as has been previously reported5.
Third, we have provided direct evidence for E. coli RNAP backtracking upon encountering a nucleosome barrier and shown that the mean backtracking distance is ~10–15 bp, and that RNase T1 can facilitate transcription through a nucleosome. These findings are consistent with previous work that showed cleavage sensitivity of transcripts to TFIIS for Pol II6. However, the current work has provided a more direct method to quantitatively determine the extent of backtracking.
The nucleosome barrier encourages the RNAP to extensively backtrack and the backtracked state may be further stabilized or “trapped” by the histones due to the exposed 3′-RNA interaction with histones41. Although nucleosome-induced backtracking has been identified as an important mechanism of the nucleosome barrier, transcript cleavage factors such as GreB and TFIIS that rapidly rescue backtracked complexes reduce but do not eliminate the nucleosome barrier4, 5, 7. These results argue for the existence of pausing mechanisms other than backtracking at the nucleosome. During elongation, RNA polymerase rapidly shifts between the pre-translocation and the post-translocation states at each template position. A physical blockage imposed by the nucleosome should increase the dwell time at the pre-translocated state, leading to pausing42. At the nucleosome barrier, pre-translocation pausing is poised to occur during each elongation cycle and thus may be an important mechanism of polymerase pausing at a nucleosome. Cleavage factors would have no effect on this type of pausing.
In this work, we have provided direct evidence for the synergistic actions of multiple RNAPs working in concert to overcome the nucleosome barrier. In the presence of a trailing RNAP, a leading RNAP was found to transcribe through a nucleosome with a rate enhancement by a factor of 5. The trailing RNAP is capable of assisting the leading RNAP, likely by exerting an assisting force on it43, and facilitating the leading RNAP to exit the backtracked state and resume elongation. Indeed RNAPs are known to be powerful molecular motors that can exert forces and work against resistance. E. coli RNAP is able to generate ~ 27 pN of force27, and Pol II at least ~ 8 pN of force16. Forces of such magnitude have been shown to significantly speed active elongation rates on naked DNA44, 45. Alternatively, the trailing RNAP can form a steric hindrance to prevent the leading one from entering the backtracked state
In vivo, multiple initiation is common among highly expressed genes. It has been demonstrated that the rates and efficiencies of transcription elongation in various eukaryotic and prokaryotic cells are directly proportional to the rates of transcriptional initiation26, 46. Although transcription elongation factors have been found to associate with coding regions in vivo, there is also evidence that many transcription factors that travel along with Pol II, do not affect the Pol II elongation rate21, 47. Remarkably, cleavage factors, that have been suggested to reactivate backtracked RNAP and contribute to the rapid progression of RNAP elongation, are dispensable in vivo under physiological conditions48, 49. Therefore, it is likely that multiple initiation may serve as an alternative mechanism to remove roadblocks, such as nucleosomes and other DNA binding proteins, during transcription. In addition, it has been increasingly evident that promoter-proximal pausing is a common feature in the expression of many genes 50–55. It is possible that if a second Pol II initiates, it may collide with the leading one, and thus this collision may function as a control of Pol II escape at these pause sites.
It has recently been suggested that during multiple initiation the leading RNAP that first encounters nucleosomes might be a specialized “pioneer” polymerase equipped with additional factors to open unmodified, fully repressed chromatin56. However there is little evidence that such a pioneer RNAP differs from its trailing RNAPs. Then how does a pioneer RNAP work so effectively? Our study suggests a much simpler explanation without invoking a pioneer RNAP of unique properties. The initial few RNAPs may together function as a group effectively acting as pioneer RNAPs so that their additive force is sufficient to evict histones and thereby establish a more accessible chromatin for trailing RNAPs.
METHODS
Methods and any associated references are available in the online version of the paper at http://www.nature.com/nsmb/.
METHODS
Nucleosomal DNA templates for transcription
We prepared nucleosomal DNA templates using methods similar to those previously described 31, 32, 57, except that these templates contained either one or two promoters. Briefly, each DNA construct consisted of an anchoring and an unzipping segment (Supplementary Fig. 1a). An ~1.3 kbp anchoring segment was labeled with digoxigenin at one end and a ligatable DraIII overhang at the other end. Two unzipping segments were constructed. The single-promoter segment was 792 bp long and composed of one T7A1 promoter followed by one 601 NPE (Fig. 1b). The two-promoter segment was 850 bp long and contained two T7A1 promoters 162 bp apart, followed by a 601 NPE (Fig. 5a). This was achieved by inserting a second T7A1 promoter upstream of the original one shown in Figure 1b. Both segments were synthesized by PCR using a biotin-labeled primer. The PCR products were then digested by the restriction enzyme DraIII to generate a ligatable end and dephosphorylated using CIP (NEB) to introduce a nick into the final DNA templates. Nucleosomes were assembled onto the unzipping segments using purified HeLa histones by a well established salt dialysis method. The anchoring and unzipping segments were joined by ligation immediately prior to use. This produced a complete template that was labeled with a single dig tag on one end and a biotin tag located 5 bp away from the nick in one DNA strand.
Bulk transcription assays
Transcription was first initiated by incubation of 20 nM E. coli RNAP, 4 nM transcription DNA template, 250 µM ApU initiating dinucleotide, 50 µM ATP/GTP, and α-[32P] CTP [5 µCi (1 µCi = 37 GBq) at 3,000 Ci mmol−1] in transcription buffer (TB: 25 mM Tris•Cl, pH 8.0, 100 mM KCl, 4 mM MgCl2, 1 mM DTT, 3% (v/v) glycerol, 0.15 mg ml−1 acetylated BSA) for 20 min at 37°C to form PTCs which contained DNA, RNAP and 20 nt RNA transcript. PTCs were then diluted by a factor of 10 in TB and transcription was resumed at room temperature (23°C ± 1°C) by addition of 1 mM of all four unlabeled NTPs. To prevent re-initiation, competitor DNA was added to 15 nM to serve as an RNAP sink immediately before the resumption of transcription (Supplementary Fig. 3a). Transcription reactions were quenched at predetermined time points by addition of EDTA to 10 mM. Transcripts were analyzed on polyacrylamide sequencing gels and imaged with PhosphorImager (Molecular Dynamics)28.
Single molecule transcription assays
Transcription reactions were typically performed using identical protocols as in bulk transcription assays except that 50 µM unlabeled CTP was used instead of α-[32P] CTP during PTC formation. After the transcription reactions were quenched, 4 mg ml−1 heparin was used to chemically dissociate histone proteins. Single molecule sample preparation was then immediately performed using protocols similar to those previously described57. In the experiments where RNase T1 was needed, 5 units µl−1 was added right before the addition of NTPs. For experiments described in Figure 1c, PTCs at +20 nt were formed by incubating 2 nM RNAP, 0.4 nM DNA template, and 1mM ApUTP and ATP/GTP/CTP in transcription buffer for 2 min at room temperature before the reaction was quenched by EDTA.
Single molecule DNA unzipping experiments
The experimental configuration for optical trapping was similar to that previously described (Supplementary Fig. 1b)57. Briefly, one end of an anchoring segment was attached to a microscope coverslip via a digoxigenin/anti-digoxigenin connection. The 5′ nicked unzipping segment was attached to a 0.48 µm-diameter microsphere via a biotin-strepavidin connection. A single-molecule optical trapping setup was used to unzip the DNA template by moving the coverslip horizontally away from the optical trap. When a bound protein was encountered, a computer-controlled feedback loop increased the applied load linearly with time (8 pN s−1) as necessary to unzip through the protein-DNA interactions. Data were digitized at 12 kHz and boxcar-averaged to 60 Hz. The acquired data signals were converted into force and number of base pairs unzipped as described. Additionally, the force-versus-base pair unzipped curves were aligned as previously described to achieve high precision position detection32.
Supplementary Material
ACKNOWLEDGEMENTS
We thank members of the Wang laboratory for critical reading of the manuscript, J. Widom for the plasmid containing the 601 NPE, and the Jeff Roberts lab for help with the phosphorescence gel scanner. MDW wishes to acknowledge support from the NIH (GM059849), NSF (MCB-0820293), the Keck Foundation Distinguished Young Scholar in Medical Research Award, and the Cornell Nanobiotechnology Center.
Footnotes
SUPPLEMENTARY INFORMATION
Supplementary information includes eight items, and can be found with this article online at http://www.nature.com/nsmb.
AUTHOR CONTIBUTIONS
J.J. designed and constructed transcription templates, designed and performed bulk and single molecule transcription experiments, analyzed the data, participated in all discussions and proposal of the model, and also wrote the manuscript. L.B, D.S.J and M.L.K offered suggestions and technical advice and helped troubleshoot the experiments. R.M.F purified E.coli RNAP and histones, and revised the manuscript. M.K helped with the design of the project and offered advice on biochemical studies, and contributed significantly to the manuscript revision. M.D.W supervised the study throughout all stages of the project and wrote the manuscript.
REFERENCES
- 1.Izban MG, Luse DS. Transcription on nucleosomal templates by RNA polymerase II in vitro: inhibition of elongation with enhancement of sequence-specific pausing. Genes Dev. 1991;5:683–696. doi: 10.1101/gad.5.4.683. [DOI] [PubMed] [Google Scholar]
- 2.Studitsky VM, Clark DJ, Felsenfeld G. Overcoming a nucleosomal barrier to transcription. Cell. 1995;83:19–27. doi: 10.1016/0092-8674(95)90230-9. [DOI] [PubMed] [Google Scholar]
- 3.Studitsky VM, Kassavetis GA, Geiduschek EP, Felsenfeld G. Mechanism of transcription through the nucleosome by eukaryotic RNA polymerase. Science. 1997;278:1960–1963. doi: 10.1126/science.278.5345.1960. [DOI] [PubMed] [Google Scholar]
- 4.Kireeva ML, et al. Nucleosome remodeling induced by RNA polymerase II: loss of the H2A/H2B dimer during transcription. Mol Cell. 2002;9:541–552. doi: 10.1016/s1097-2765(02)00472-0. [DOI] [PubMed] [Google Scholar]
- 5.Walter W, Kireeva ML, Studitsky VM, Kashlev M. Bacterial polymerase and yeast polymerase II use similar mechanisms for transcription through nucleosomes. J Biol Chem. 2003;278:36148–36156. doi: 10.1074/jbc.M305647200. [DOI] [PubMed] [Google Scholar]
- 6.Kireeva ML, et al. Nature of the nucleosomal barrier to RNA polymerase II. Mol Cell. 2005;18:97–108. doi: 10.1016/j.molcel.2005.02.027. [DOI] [PubMed] [Google Scholar]
- 7.Bondarenko VA, et al. Nucleosomes can form a polar barrier to transcript elongation by RNA polymerase II. Mol Cell. 2006;24:469–479. doi: 10.1016/j.molcel.2006.09.009. [DOI] [PubMed] [Google Scholar]
- 8.Ujvari A, Hsieh FK, Luse SW, Studitsky VM, Luse DS. Histone N-terminal tails interfere with nucleosome traversal by RNA polymerase II. J Biol Chem. 2008;283:32236–32243. doi: 10.1074/jbc.M806636200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hodges C, Bintu L, Lubkowska L, Kashlev M, Bustamante C. Nucleosomal fluctuations govern the transcription dynamics of RNA polymerase II. Science. 2009;325:626–628. doi: 10.1126/science.1172926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kulaeva OI, et al. Mechanism of chromatin remodeling and recovery during passage of RNA polymerase II. Nat Struct Mol Biol. 2009;16:1272–1278. doi: 10.1038/nsmb.1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.O'Brien T, Lis JT. Rapid changes in Drosophila transcription after an instantaneous heat shock. Mol Cell Biol. 1993;13:3456–3463. doi: 10.1128/mcb.13.6.3456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tennyson CN, Klamut HJ, Worton RG. The human dystrophin gene requires 16 hours to be transcribed and is cotranscriptionally spliced. Nat Genet. 1995;9:184–190. doi: 10.1038/ng0295-184. [DOI] [PubMed] [Google Scholar]
- 13.Darzacq X, et al. In vivo dynamics of RNA polymerase II transcription. Nat Struct Mol Biol. 2007;14:796–806. doi: 10.1038/nsmb1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Awrey DE, et al. Yeast transcript elongation factor (TFIIS), structure and function. II: RNA polymerase binding, transcript cleavage, and read-through. J Biol Chem. 1998;273:22595–22605. doi: 10.1074/jbc.273.35.22595. [DOI] [PubMed] [Google Scholar]
- 15.Weilbaecher RG, Awrey DE, Edwards AM, Kane CM. Intrinsic transcript cleavage in yeast RNA polymerase II elongation complexes. J Biol Chem. 2003;278:24189–24199. doi: 10.1074/jbc.M211197200. [DOI] [PubMed] [Google Scholar]
- 16.Galburt EA, et al. Backtracking determines the force sensitivity of RNAP II in a factor-dependent manner. Nature. 2007;446:820–823. doi: 10.1038/nature05701. [DOI] [PubMed] [Google Scholar]
- 17.Brown CE, Lechner T, Howe L, Workman JL. The many HATs of transcription coactivators. Trends Biochem Sci. 2000;25:15–19. doi: 10.1016/s0968-0004(99)01516-9. [DOI] [PubMed] [Google Scholar]
- 18.Anderson JD, Lowary PT, Widom J. Effects of histone acetylation on the equilibrium accessibility of nucleosomal DNA target sites. J Mol Biol. 2001;307:977–985. doi: 10.1006/jmbi.2001.4528. [DOI] [PubMed] [Google Scholar]
- 19.Roth SY, Denu JM, Allis CD. Histone acetyltransferases. Annu Rev Biochem. 2001;70:81–120. doi: 10.1146/annurev.biochem.70.1.81. [DOI] [PubMed] [Google Scholar]
- 20.Belotserkovskaya R, et al. FACT facilitates transcription-dependent nucleosome alteration. Science. 2003;301:1090–1093. doi: 10.1126/science.1085703. [DOI] [PubMed] [Google Scholar]
- 21.Schwabish MA, Struhl K. The Swi/Snf complex is important for histone eviction during transcriptional activation and RNA polymerase II elongation in vivo. Mol Cell Biol. 2007;27:6987–6995. doi: 10.1128/MCB.00717-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim TH, et al. A high-resolution map of active promoters in the human genome. Nature. 2005;436:876–880. doi: 10.1038/nature03877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee CK, Shibata Y, Rao B, Strahl BD, Lieb JD. Evidence for nucleosome depletion at active regulatory regions genome-wide. Nat Genet. 2004;36:900–905. doi: 10.1038/ng1400. [DOI] [PubMed] [Google Scholar]
- 24.Varv S, Kristjuhan K, Kristjuhan A. RNA polymerase II determines the area of nucleosome loss in transcribed gene loci. Biochem Biophys Res Commun. 2007;358:666–671. doi: 10.1016/j.bbrc.2007.05.003. [DOI] [PubMed] [Google Scholar]
- 25.Epshtein V, Nudler E. Cooperation between RNA polymerase molecules in transcription elongation. Science. 2003;300:801–805. doi: 10.1126/science.1083219. [DOI] [PubMed] [Google Scholar]
- 26.Epshtein V, Toulme F, Rahmouni AR, Borukhov S, Nudler E. Transcription through the roadblocks: the role of RNA polymerase cooperation. EMBO J. 2003;22:4719–4727. doi: 10.1093/emboj/cdg452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang MD, et al. Force and velocity measured for single molecules of RNA polymerase. Science. 1998;282:902–907. doi: 10.1126/science.282.5390.902. [DOI] [PubMed] [Google Scholar]
- 28.Shundrovsky A, Santangelo TJ, Roberts JW, Wang MD. A single-molecule technique to study sequence-dependent transcription pausing. Biophys J. 2004;87:3945–3953. doi: 10.1529/biophysj.104.044081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ebright RH. RNA polymerase: structural similarities between bacterial RNA polymerase and eukaryotic RNA polymerase II. J Mol Biol. 2000;304:687–698. doi: 10.1006/jmbi.2000.4309. [DOI] [PubMed] [Google Scholar]
- 30.Korzheva N, Mustaev A. Transcription elongation complex: structure and function. Curr Opin Microbiol. 2001;4:119–125. doi: 10.1016/s1369-5274(00)00176-4. [DOI] [PubMed] [Google Scholar]
- 31.Shundrovsky A, Smith CL, Lis JT, Peterson CL, Wang MD. Probing SWI/SNF remodeling of the nucleosome by unzipping single DNA molecules. Nat Struct Mol Biol. 2006;13:549–554. doi: 10.1038/nsmb1102. [DOI] [PubMed] [Google Scholar]
- 32.Hall MA, et al. High-resolution dynamic mapping of histone-DNA interactions in a nucleosome. Nat Struct Mol Biol. 2009;16:124–129. doi: 10.1038/nsmb.1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jiang J, et al. Detection of high-affinity and sliding clamp modes for MSH2–MSH6 by single-molecule unzipping force analysis. Mol Cell. 2005;20:771–781. doi: 10.1016/j.molcel.2005.10.014. [DOI] [PubMed] [Google Scholar]
- 34.Nudler E, Gusarov I, Avetissova E, Kozlov M, Goldfarb A. Spatial organization of transcription elongation complex in Escherichia coli. Science. 1998;281:424–428. doi: 10.1126/science.281.5375.424. [DOI] [PubMed] [Google Scholar]
- 35.Korzheva N, et al. A structural model of transcription elongation. Science. 2000;289:619–625. doi: 10.1126/science.289.5479.619. [DOI] [PubMed] [Google Scholar]
- 36.Zaychikov E, Denissova L, Heumann H. Translocation of the Escherichia coli transcription complex observed in the registers 11 to 20: "jumping" of RNA polymerase and asymmetric expansion and contraction of the "transcription bubble". Proc Natl Acad Sci U S A. 1995;92:1739–1743. doi: 10.1073/pnas.92.5.1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lowary PT, Widom J. New DNA sequence rules for high affinity binding to histone octamer and sequence-directed nucleosome positioning. J Mol Biol. 1998;276:19–42. doi: 10.1006/jmbi.1997.1494. [DOI] [PubMed] [Google Scholar]
- 38.Samkurashvili I, Luse DS. Translocation and transcriptional arrest during transcript elongation by RNA polymerase II. J Biol Chem. 1996;271:23495–23505. doi: 10.1074/jbc.271.38.23495. [DOI] [PubMed] [Google Scholar]
- 39.Luger K, Mader AW, Richmond RK, Sargent DF, Richmond TJ. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature. 1997;389:251–260. doi: 10.1038/38444. [DOI] [PubMed] [Google Scholar]
- 40.Dong F, Hansen JC, van Holde KE. DNA and protein determinants of nucleosome positioning on sea urchin 5S rRNA gene sequences in vitro. Proc Natl Acad Sci U S A. 1990;87:5724–5728. doi: 10.1073/pnas.87.15.5724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Peng HF, Jackson V. Measurement of the frequency of histone displacement during the in vitro transcription of nucleosomes: RNA is a competitor for these histones. Biochemistry. 1997;36:12371–12382. doi: 10.1021/bi971046g. [DOI] [PubMed] [Google Scholar]
- 42.Bai L, Shundrovsky A, Wang MD. Sequence-dependent kinetic model for transcription elongation by RNA polymerase. J Mol Biol. 2004;344:335–349. doi: 10.1016/j.jmb.2004.08.107. [DOI] [PubMed] [Google Scholar]
- 43.Saeki H, Svejstrup JQ. Stability, flexibility, and dynamic interactions of colliding RNA polymerase II elongation complexes. Mol Cell. 2009;35:191–205. doi: 10.1016/j.molcel.2009.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shaevitz JW, Abbondanzieri EA, Landick R, Block SM. Backtracking by single RNA polymerase molecules observed at near-base-pair resolution. Nature. 2003;426:684–687. doi: 10.1038/nature02191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bai L, Fulbright RM, Wang MD. Mechanochemical kinetics of transcription elongation. Phys Rev Lett. 2007;98:068103. doi: 10.1103/PhysRevLett.98.068103. [DOI] [PubMed] [Google Scholar]
- 46.Yankulov K, Blau J, Purton T, Roberts S, Bentley DL. Transcriptional elongation by RNA polymerase II is stimulated by transactivators. Cell. 1994;77:749–759. doi: 10.1016/0092-8674(94)90058-2. [DOI] [PubMed] [Google Scholar]
- 47.Mason PB, Struhl K. Distinction and relationship between elongation rate and processivity of RNA polymerase II in vivo. Mol Cell. 2005;17:831–840. doi: 10.1016/j.molcel.2005.02.017. [DOI] [PubMed] [Google Scholar]
- 48.Archambault J, Lacroute F, Ruet A, Friesen JD. Genetic interaction between transcription elongation factor TFIIS and RNA polymerase II. Mol Cell Biol. 1992;12:4142–4152. doi: 10.1128/mcb.12.9.4142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Orlova M, Newlands J, Das A, Goldfarb A, Borukhov S. Intrinsic transcript cleavage activity of RNA polymerase. Proc Natl Acad Sci U S A. 1995;92:4596–4600. doi: 10.1073/pnas.92.10.4596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Strobl LJ, Eick D. Hold back of RNA polymerase II at the transcription start site mediates down-regulation of c-myc in vivo. EMBO J. 1992;11:3307–3314. doi: 10.1002/j.1460-2075.1992.tb05409.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Benjamin LR, Gilmour DS. Nucleosomes are not necessary for promoter-proximal pausing in vitro on the Drosophila hsp70 promoter. Nucleic Acids Res. 1998;26:1051–1055. doi: 10.1093/nar/26.4.1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Conaway JW, Shilatifard A, Dvir A, Conaway RC. Control of elongation by RNA polymerase II. Trends Biochem Sci. 2000;25:375–380. doi: 10.1016/s0968-0004(00)01615-7. [DOI] [PubMed] [Google Scholar]
- 53.Cheng C, Sharp PA. RNA polymerase II accumulation in the promoter-proximal region of the dihydrofolate reductase and gamma-actin genes. Mol Cell Biol. 2003;23:1961–1967. doi: 10.1128/MCB.23.6.1961-1967.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Saunders A, Core LJ, Lis JT. Breaking barriers to transcription elongation. Nat Rev Mol Cell Biol. 2006;7:557–567. doi: 10.1038/nrm1981. [DOI] [PubMed] [Google Scholar]
- 55.Core LJ, Lis JT. Transcription regulation through promoter-proximal pausing of RNA polymerase II. Science. 2008;319:1791–1792. doi: 10.1126/science.1150843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Orphanides G, Reinberg D. RNA polymerase II elongation through chromatin. Nature. 2000;407:471–475. doi: 10.1038/35035000. [DOI] [PubMed] [Google Scholar]
- 57.Koch SJ, Shundrovsky A, Jantzen BC, Wang MD. Probing protein-DNA interactions by unzipping a single DNA double helix. Biophys J. 2002;83:1098–1105. doi: 10.1016/S0006-3495(02)75233-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.