

Abstract
Point mutations within the CAP-gly domain of the p150Glued subunit of the dynactin complex have been identified in patients with distal Spinal Bulbar Muscular Atrophy (dSBMA) and Perry’s syndrome. Herein, we show by CD and NMR experiments that each mutated CAP-gly domain is folded, but less stable than the wildtype (WT) domain. We also demonstrate that the domains harboring these mutations bind to microtubules, but fail to bind to EB1. These data indicate that these disease-associated, point mutations affect the stability of this domain and inhibit their interaction with EB1 but do not inhibit their interaction with microtubules.
Dynein and dynactin concomitantly drive the bulk of MT-based, retrograde transport of membranous organelles and play a critical role in highly polarized cells (1). This is especially true for neurons, in which axons can extend over a meter in length and signaling between the synapse and cell body through vesicular transport is required for development and survival (2). Not surprisingly, mutations in dynein, dynactin or other dynein-interacting partners are commonly associated with neurological diseases. Recently, a new set of point mutations in the p150Glued subunit of dynactin have been identified in patients diagnosed with Perry’s syndrome, a severe neurological disease that shows symptoms of parkinsonism and weight loss and is accompanied by depression and suicidal attempts (3). These mutations, G71A, G71R, G71E, T72P and Q74P, map to the N-terminal CAP-gly domain of dynactin’s p150Glued subunit (Fig. 1). While this is the same domain that that harbors the G59S mutation identified in patients with dSBMA (4), the characteristics of the diseases are distinct (3).
Figure 1.
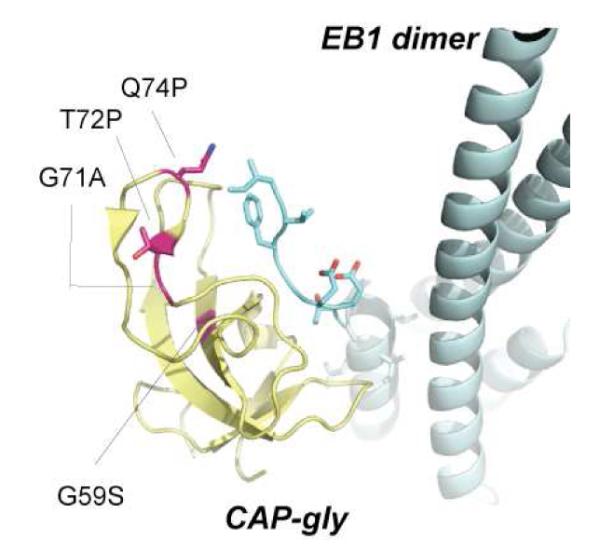
Ribbon diagram of the p150Glued CAP-gly domain (yellow) bound to EB1 (cyan). The point mutations identified in Perry’s syndrome are shown as sticks in magenta. These mutations are located in a long loop that connects β3 and β4 strands and part of the ‘GKNDG’ binding motif. The location of G59 is also shown.
The p150Glued CAP-gly domain weakly binds MTs and is proposed to anchor the dynactin complex to MTs to facilitate its interaction with dynein and increase the processivity of the dynein motor (5, 6). Recent studies, however, suggest the CAP-gly domain is necessary for MT organization, but not for dynein activity (7). In yeast, the CAP-gly domain was demonstrated to be essential for dynein-dependent spindle and nucleus positioning, suggesting that the CAP-gly domain is important for dynein-mediated processes under load (8).
The CAP-gly domain of p150Glued also binds to EB1, a MT plus-end tracking protein (9, 10). Recent in vivo studies suggest that this interaction is required for anchoring the minus-end of MTs to the centrosome (10). In vitro data also suggest that p150Glued recruits EB1 to the MT +TIP and modulates MT dynamics (11). While these studies mark the importance of this interaction, the pleiotropic effects of EB1 and p150Glued overexpression or depletion (as well as the addition of purification tags (12)) have made it difficult to confidently assign a specific function to this interaction.
To determine whether point mutations in the CAP-gly domain found in Perry’s syndrome affect the stability, MT binding or EB1 interaction, each point mutation was generated by site directed mutagenesis in a construct that spanned residues 19-107. The domains with point mutations were expressed in E. coli and purified to homogeneity (see Supporting Information). Each mutated domain required expression at 18 °C to obtain a soluble protein. In addition, to test whether the point mutations associated with Perry’s syndrome behave differently than the point mutation associated with dSBMA, we also generated the G59S point mutation. This mutation was not soluble despite attempts to express it at 15 °C, the induction of bacterial chaperones by cold shock (mid-log phase cells were cooled to 10 °C for 20 min), and addition of ethanol to the medium (2% v/v added at mid-log growth). Because G59 is located in the hydrophobic core of the CAP-gly domain and simple modeling of the mutation (using the structure 2HKQ.pdb) indicated that the hydroxyl group of the Ser mutation produces significant steric clashes irrespective of the rotamer chosen, we generated a domain with the mutation, G59A, to reduce (but not eliminate) these clashes. This construct, G59A, was soluble.
Next, we collected CD and NMR spectra to characterize the effect of these mutations on the overall structure of the CAP-gly domain. Previously, we showed that the CD spectra of the WT CAP-gly domain exhibited a positive inflection at 223 nm, and we attributed this anomalous spectrum to the unusual packing of aromatics in the core (13). The CD spectra of the point mutations deviated from the WT spectrum, indicating that each mutation affects the overall fold (13) (see Supporting Information).
We collected 1H-15N HSQC spectra to measure the extent of the perturbations due to the G71A, Q74P and G59A mutations in the WT CAP-gly domain (Fig. 2A; Supporting Information). First, we noted that each domain with a point mutation displayed well-resolved spectra, indicating that each is folded. We also observed that a large number of peaks for the domains with point mutations were shifted relative to the WT spectrum and that spectra of the Perry’s syndrome point mutations differed from the G59A mutation. These data suggest the disease-associated point mutations perturb the fold globally.
Figure 2.

Characterization of the CAP-gly point mutations: A) NMR spectrum of the G71P point mutation indicates the domain is folded, but perturbed globally compared to WT. B) Thermal denaturation of each point mutation and WT CAP-gly. The closed symbols represent the ellipticity of the sample at 223 nm upon heating (e.g., 4 to 80 °C), the open symbols represent cooling (e.g., 80 to 4 °C).
To address the extent of these perturbations, we characterized the thermal properties of each point mutation by CD (Fig. 2B). WT CAP-gly showed a single and reversible transition at 60 °C, while the mutation, Q74P, also produced a single transition at 45 °C and was reversible. The remaining point mutations, G59A, G71A and T72P, produced a broad, irreversible transition with a midpoint at 45 °C. These data further suggest that the point mutations affect the CAP-gly domain globally.
Next, we asked whether these point mutations affect the MT-binding properties of the CAP-gly domain. To do this, we used a standard MT precipitation assay; taxol-stabilized MTs (5 μM) were incubated with each point mutation (75 μM) and precipitated by centrifugation at 200,000 × G. SDS-PAGE analysis of the supernatant and the pelleted MTs revealed that each point mutation could be precipitated with MTs. We also observed minor precipitation of the domains with point mutations in the absence of MTs as compared to WT; however, each construct harboring a mutation bound MTs better than the WT construct (Fig. 3A).
Figure 3.

Characterization of CAP-gly binding interactions: A) SDS-PAGE gel of the pelleted fraction of CAP-gly constructs in the presence and absence of MTs (see Supporting Information). B) Quantification of the intensities of precipitated fractions indicate that the mutated domains bind to taxol stabilized MTs better than the WT construct (grey bars; n=3). The amount of protein precipitated in the absence of MTs is shown as white bars. C) SEC analysis of the CAP-gly interaction with EB1. D) SDS-PAGE analysis of fractions 1 (first peak) and 2 (second peak) shown in panel C).
To test whether the point mutations affected the interaction of the CAP-gly domain with EB1, we generated, expressed and purified an EB1 fragment (residues 194 to 268) that binds to the WT CAP-gly domain (9, 10, 14). We used SEC (size exclusion chromatography) to characterize their hydrodynamic properties and interaction. Each CAP-gly mutation and the WT CAP-gly eluted at 12.5 mL, while the EB1 construct eluted at 10.0 mL (Superdex G75 in PBS) (Fig. 3C). We note that EB1 is an elongated coiled coil (2 × MW = 17.4 kDa) and therefore elutes earlier than the CAP-gly domain (MW = 9.5 kDa). We observed a significant shift to an earlier elution volume for an admixture of the WT CAP-gly and EB1, as well as for the admixture of G59A and EB1. The elution profiles for admixtures of the remaining CAP-gly mutations and EB1 (where the CAP-gly domain was added at a 1.2-fold excess) appeared as a sum of the individual components. SDS-PAGE of both peaks (Fig. 3D) confirmed that WT and G59A co-eluted with EB1 while the other CAP-gly mutations did not (Fig. 3D). Although we could not test the dSBMA point mutation, G59S, point mutations identified in patients with Perry’s syndrome failed to bind to EB1.
Taken together, the results of this study show that each point mutation identified in patients with Perry’s syndrome affects the EB1 interaction and the stability of the CAP-gly domain. These results also indicate that the mutations increase MT binding. Although we were unable to produce a soluble form of G59S to obtain a quantitative measure of its instability and to differentiate the dSBMA and Perry’s syndrome mutations, we did show that the mutations G59A (which we propose is an intermediate step to G59S sterically) is less stable than the WT domain and could bind to EB1. We infer that the additional steric clashes stemming from the G59S mutation produce unfolded protein under physiological conditions.
The role of these point mutations that lead to or compound the pathogenesis of these distinct diseases remains unclear. Previous studies showed that the G59S mutation leads to formation of inclusion bodies, suggesting that the loss of stability promotes aggregation (15). The point mutations found in Perry’s Syndrome also reduce the stability of the CAP-gly domain, but to a lesser degree. Relevant to the difference in stability, expression of these Perry’s syndrome point mutations in HEK293T cells also produced inclusion bodies, but to a lesser extent than the G59S mutation (3).
Recent findings also indicate that the CAP-gly domain in p150Glued is necessary for processes requiring greater dynein force production (e.g., spindle positioning) and microtubule organization, but not dynein-mediated, retrograde transport (7, 8, 16). In fact, deletion of the EB1 homolog in yeast phenocopied the G59S mutation as well as the deletion of the CAP-gly domain, indicating that they belong to the same pathway (8). The p150Glued G59S mutation that was expressed in mammalian cells also failed to colocalize with EB1 at the MT +TIP (15). Taken together, these observations also suggest that the loss of the CAP-gly:EB1 interaction could play an important role in disease progression.
Finally, although we observed that the Perry’s syndrome mutations increased MT binding, the role of the p150Glued CAP-gly domain in dynein-mediated cargo transport (e.g., processivity) appears to be minimal (7, 8, 16). Thus, this increase in MT binding is unlikely to be responsible for compounding the pathology of these diseases.
In summary, we provide biochemical and spectroscopic evidence using homogenous samples that these disease-related mutations reduce the stability of the CAP-gly domain and affect EB1 and MT binding. These studies also suggest future experiments need to differentiate the loss of EB1 binding, stability and potentially MT binding to better understand the roles of these point mutations in disease progression.
Supplementary Material
Footnotes
Support provided in part by National Institutes of Health NCRR 5P20RR017716-07 (T.P) and The American Heart Association 0715196U (A.E.S)
SUPPORTING INFORMATION AVAILABLE : Materials, experimental procedures and additional spectra. This material is available free of charge via the Internet at http://pubs.acs.org
REFERENCES
- (1).Vallee RB, Williams JC, Varma D, Barnhart LE. Dynein: An ancient motor protein involved in multiple modes of transport. J Neurobiol. 2004;58:189–200. doi: 10.1002/neu.10314. [DOI] [PubMed] [Google Scholar]
- (2).Gunawardena S, Goldstein LS. Cargo-carrying motor vehicles on the neuronal highway: transport pathways and neurodegenerative disease. J Neurobiol. 2004;58:258–271. doi: 10.1002/neu.10319. [DOI] [PubMed] [Google Scholar]
- (3).Farrer MJ, Hulihan MM, Kachergus JM, Dachsel JC, Stoessl AJ, Grantier LL, Calne S, Calne DB, Lechevalier B, Chapon F, Tsuboi Y, Yamada T, Gutmann L, Elibol B, Bhatia KP, Wider C, Vilarino-Guell C, Ross OA, Brown LA, Castanedes-Casey M, Dickson DW, Wszolek ZK. DCTN1 mutations in Perry syndrome. Nat Genet. 2009;41:163–165. doi: 10.1038/ng.293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Puls I, Jonnakuty C, LaMonte BH, Holzbaur EL, Tokito M, Mann E, Floeter MK, Bidus K, Drayna D, Oh SJ, Brown RH, Jr., Ludlow CL, Fischbeck KH. Mutant dynactin in motor neuron disease. Nat Genet. 2003;33:455–456. doi: 10.1038/ng1123. [DOI] [PubMed] [Google Scholar]
- (5).Schroer TA, Sheetz MP. Two activators of microtubule-based vesicle transport. J. Cell Biol. 1991;115:1309–1318. doi: 10.1083/jcb.115.5.1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).King SJ, Schroer TA. Dynactin increases the processivity of the cytoplasmic dynein motor. Nat Cell Biol. 2000;2:20–24. doi: 10.1038/71338. [DOI] [PubMed] [Google Scholar]
- (7).Kim H, Ling SC, Rogers GC, Kural C, Selvin PR, Rogers SL, Gelfand VI. Microtubule binding by dynactin is required for microtubule organization but not cargo transport. J Cell Biol. 2007;176:641–651. doi: 10.1083/jcb.200608128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Moore JK, Sept D, Cooper JA. Neurodegeneration mutations in dynactin impair dynein-dependent nuclear migration. Proc Natl Acad Sci U S A. 2009;106:5147–5152. doi: 10.1073/pnas.0810828106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Honnappa S, Okhrimenko O, Jaussi R, Jawhari H, Jelesarov I, Winkler FK, Steinmetz MO. Key interaction modes of dynamic +TIP networks. Mol Cell. 2006;23:663–671. doi: 10.1016/j.molcel.2006.07.013. [DOI] [PubMed] [Google Scholar]
- (10).Askham JM, Vaughan KT, Goodson HV, Morrison EE. Evidence that an interaction between EB1 and p150(Glued) is required for the formation and maintenance of a radial microtubule array anchored at the centrosome. Mol Biol Cell. 2002;13:3627–3645. doi: 10.1091/mbc.E02-01-0061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Manna T, Honnappa S, Steinmetz MO, Wilson L. Suppression of microtubule dynamic instability by the +TIP protein EB1 and its modulation by the CAP-Gly domain of p150glued. Biochemistry. 2008;47:779–786. doi: 10.1021/bi701912g. [DOI] [PubMed] [Google Scholar]
- (12).Zhu ZC, Gupta KK, Slabbekoorn AR, Paulson BA, Folker ES, Goodson HV. Interactions between EB1 and microtubules: dramatic effect of affinity tags and evidence for cooperative behavior. J Biol Chem. 2009;284:32651–32661. doi: 10.1074/jbc.M109.013466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Sun S, Siglin A, Williams JC, Polenova T. Solid-state and solution NMR studies of the CAP-Gly domain of mammalian dynactin and its interaction with microtubules. J Am Chem Soc. 2009;131:10113–10126. doi: 10.1021/ja902003u. [DOI] [PubMed] [Google Scholar]
- (14).Hayashi I, Wilde A, Mal TK, Ikura M. Structural basis for the activation of microtubule assembly by the EB1 and p150Glued complex. Mol Cell. 2005;19:449–460. doi: 10.1016/j.molcel.2005.06.034. [DOI] [PubMed] [Google Scholar]
- (15).Levy JR, Sumner CJ, Caviston JP, Tokito MK, Ranganathan S, Ligon LA, Wallace KE, LaMonte BH, Harmison GG, Puls I, Fischbeck KH, Holzbaur EL. A motor neuron disease-associated mutation in p150Glued perturbs dynactin function and induces protein aggregation. J Cell Biol. 2006;172:733–745. doi: 10.1083/jcb.200511068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Culver-Hanlon TL, Lex SA, Stephens AD, Quintyne NJ, King SJ. A microtubule-binding domain in dynactin increases dynein processivity by skating along microtubules. Nat Cell Biol. 2006;8:264–270. doi: 10.1038/ncb1370. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.