

Abstract
Objective:
To determine the proton magnetic resonance spectroscopy (1H MRS) changes in carriers of microtubule-associated protein (MAPT) mutations in a case-control study.
Methods:
Patients with MAPT mutations (N279K, V337M, R406W, IVS9-10G>T, P301L) from 5 different families (n = 24) underwent MRI and single voxel 1H MRS from the posterior cingulate gyrus inferior precuneus at 3 T. Ten of the patients were symptomatic with median Clinical Dementia Rating sum of boxes score (CDR-SOB) of 6.5 and 14 patients were presymptomatic with CDR-SOB of 0. Age- and sex-matched controls (n = 24) were recruited.
Results:
Symptomatic MAPT mutation carriers were characterized by decreased N-acetylaspartate/creatine (NAA/Cr) ratio, an index of neuronal integrity, increased myoinositol (mI)/Cr ratio, a possible marker for glial activity, decreased NAA/mI, and hippocampal atrophy (p < 0.001). Whereas presymptomatic MAPT mutation carriers had elevated mI/Cr and decreased NAA/mI (p < 0.001), NAA/Cr levels and hippocampal volumes were not different from controls. Decrease in NAA/Cr (R2 = 0. 22; p = 0.021) and hippocampal volumes (R2 = 0.46; p < 0.001) were associated with proximity to the expected or actual age at symptom onset in MAPT mutation carriers.
Conclusion:
1H MRS metabolite abnormalities characterized by an elevated mI/Cr and decreased NAA/mI are present several years before the onset of symptoms in MAPT mutation carriers. The data suggest an ordered sequencing of the 1H MRS and MRI biomarkers. MI/Cr, a possible index of glial proliferation, precedes the decrease in neuronal integrity marker NAA/Cr and hippocampal atrophy. 1H MRS may be a useful inclusion biomarker for preventive trials in presymptomatic carriers of MAPT mutations and possibly other proteinopathies.
GLOSSARY
- AAL
= automated anatomic labeling;
- AD
= Alzheimer disease;
- CDR-SOB
= Clinical Dementia Rating sum of boxes score;
- Cr
= creatine;
- FTD
= frontotemporal dementia;
- FTLD
= frontotemporal lobar degeneration;
- GM
= gray matter;
- mI
= myoinositol;
- MNI
= Montreal Neurological Institute;
- MR
= magnetic resonance;
- MRS
= magnetic resonance spectroscopy;
- NAA
= N-acetylaspartate;
- ROI
= region of interest;
- SPM
= statistical parametric mapping;
- SV
= single voxel;
- WM
= white matter.
Frontotemporal dementia with parkinsonism linked to chromosome 17 is an autosomal dominant tauopathy that is linked to mutations in the gene encoding for the microtubule-associated protein tau (MAPT) on chromosome 17.1–4 Mutations in MAPT result in filamentous accumulation of hyperphosphorylated tau in neurons and glia leading to neurodegeneration and atrophy.5–7 Progressive accumulation of filamentous tau and subsequent neuronal death is central to the pathogenesis of many neurodegenerative diseases including Alzheimer disease (AD), and may begin years before the onset of clinical symptoms. Noninvasive biomarkers of this pathologic cascade would be valuable tools for early diagnosis and tracking disease progression in tauopathies.8
1H magnetic resonance spectroscopy (1H MRS) is a quantitative biochemical imaging technique and a sensitive marker for neurodegenerative pathology in AD9 and frontotemporal lobar degeneration (FTLD).10,11 The decrease in the neuronal integrity marker N-acetylaspartate (NAA) or NAA to creatine (NAA/Cr) ratio and the increase in the glial marker myoinositol (mI) or mI to creatine (mI/Cr) in a living person is associated with the clinical diagnosis of AD and FTLD,11 the likelihood of having AD pathology, the severity of the neurofibrillary pathology, and fibrillary tau density at autopsy.9,12
Symptomatic MAPT mutation carriers typically have severe anterior and medial temporal lobe atrophy.6,13 Our objective was to characterize the 1H MRS abnormalities in both symptomatic and presymptomatic carriers of the MAPT mutations. We hypothesized that 1H MRS abnormalities in MAPT mutation carriers precede the clinical symptoms and medial temporal lobe atrophy on MRI, and that the severity of 1H MRS abnormalities is associated with the proximity to the estimated age at disease onset in carriers of the MAPT mutations.
METHODS
Subjects.
We identified 24 MAPT mutation carriers who were recruited to the Mayo Clinic Alzheimer's Disease Research Center and participated in the 1H MRS study from 2007 through 2009. All subjects underwent a clinical examination at the time of magnetic resonance (MR) examination. The behavioral neurologist (B.F.B.) who examined all of the subjects was blinded to the MAPT mutation status and to the MRI and 1H MRS findings. None of the subjects had structural lesions that could cause cognitive impairment or dementia, such as cortical infarctions, tumor, or subdural hematoma, or had concurrent illnesses that would interfere with cognitive function at the time of the MR examination.
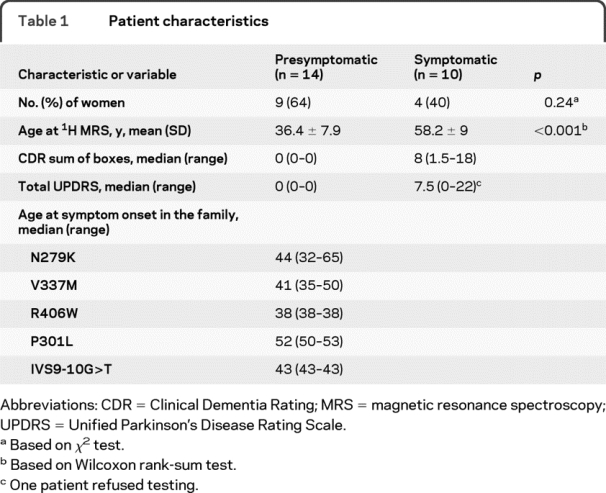
Table 1 lists characteristics of each of the MAPT mutation carriers. Ten patients explicitly declined to be informed about the results of the genetic testing. For this reason, demographic data of each of the individual patients are not presented to protect patient confidentiality. Of the 24 subjects, 14 had no clinical symptoms and had a Clinical Dementia Rating sum of boxes score of 0 (5 with N279K, 4 with V337M, 3 with R406W, and 2 with P301L mutations), which we refer to as presymptomatic patients. Ten patients were symptomatic: 8 patients were diagnosed with frontotemporal dementia (FTD) (4 with V337M, 3 with P301L, and 1 with IVS9-10G>T mutations), 1 patient was diagnosed with FTD with parkinsonism (R406W mutation), and 1 patient was diagnosed with pallidopontonigral degeneration (N279K mutation). Each of the N279K, V337M, R406W, P301L, and IVS9-10G>T mutation carriers were coming from 5 individual families. Median age at disease onset in each family/mutation type was estimated from the date of symptom onset in symptomatic patients of the family based on the clinical history and previous publications on these families.8,14,15
Table 1 Patient characteristics
We recruited 24 cognitively normal controls to the 1H MRS/MRI study, who did not have any neurologic or psychiatric disorders and who were matched to the mutation carriers on age and gender. This study was approved by the Institutional Review Board, and informed consent for participation was obtained from every subject or an appropriate surrogate.
1H MRS and MRI.
All subjects underwent 1H MRS and MRI studies within a week of the clinical evaluation. Single voxel (SV) 1H MRS and MRI studies were performed on a 3-Tesla scanner using an 8-channel phased array head coil (General Electric Medical Systems, Milwaukee, WI). A 3-dimensional high-resolution magnetization-prepared rapid gradient echo acquisition with repetition time/echo time/inversion time = 7/3/900 msec, flip angle 8 degrees, in-plane resolution of 1.0 mm, and a slice thickness of 1.2 mm was performed in sagittal plane for voxel positioning, anatomic segmentation, and labeling. 1H MRS studies were performed using the automated MRS package: Proton Brain Examination/SV.16 Point resolved spectroscopy sequence with repetition time = 2,000 msec, echo time = 30 msec, 2,048 data points, and 128 excitations was used for the examinations.
An 8 cm3 (2 × 2 × 2 cm) voxel, prescribed on a midsagittal T1-weighted image, included right and left posterior cingulate gyri and inferior precuneate gyri. The anterior border of splenium, the superior border of corpus callosum, and the cingulate sulcus were used as the anatomic landmarks to define the voxel.9 Metabolite intensity ratios calculated at the end of each PROBE/SV acquisition were analyzed. Quantifying metabolite intensities by referencing to an internal standard is preferred in clinical 1H MRS, because internal referencing does not require correction for coil loading, atrophy, and relaxation times and can readily be used in clinical practice with standard equipment and vendor-provided processing software.
We used the anatomic atlas labels from the in-house modified automated anatomic labeling (AAL) atlas template17,18 in order to derive the gray matter (GM) volumes from specific brain regions in statistical parametric mapping 5 (SPM5).19 For the current study, we analyzed the hippocampal volumes in both hemispheres. The hippocampal regions of interest (ROI) were chosen based on previous reports from our group showing significant medial temporal lobe atrophy in many symptomatic patients with MAPT mutations.6,13 Each subject's T1 MRI scan was spatially normalized and segmented into GM, white matter (WM), and CSF using the unified segmentation model of SPM5, giving a discrete cosine transformation, which normalizes each subject's MRI to the Montreal Neurological Institute (MNI) template space. Then for each subject, the inverse transformation was applied to the in-house modified AAL atlas in the MNI template space in order to warp the atlas labels to the subject's native anatomic space. The resulting subject-specific atlas was used to parcellate GM images into the ROI in the subject's T1 image space. The normalized volume of the hippocampus was computed by averaging the right and left hippocampal volumes and dividing by the total intracranial volume. Hippocampal volumes could not be analyzed in one of the symptomatic cases due to motion artifacts and poor scan quality.
Genetic analysis.
Analysis of MAPT exons 1, 7, and 9–13 was performed using primers and conditions that were previously described.2 PCR amplicons were purified using the Multiscreen system (Millipore, Billerica, MA) and then sequenced in both directions using Big Dye chemistry following the manufacturer's protocol (Applied Biosystems, Foster City, CA). Sequence products were purified using the Montage system (Millipore) before being run on an Applied Biosystems 3730 DNA Analyzer. Sequence data were analyzed using either SeqScape (Applied Biosystems) or Sequencher software (Gene Codes, Ann Arbor, MI).
Statistical analysis.
We compared the demographic aspects in presymptomatic and symptomatic MAPT mutation carriers to the controls using Wilcoxon rank sum test and χ2 tests. Between-group comparisons of 1H MRS metabolite ratios and hippocampal volumes were performed using Wilcoxon rank sum test. Symptomatic patients were older than the presymptomatic subjects (p < 0.001). Because age may influence metabolite ratios, we used the data from the control group to estimate the age effects on metabolite ratios and adjusted the metabolite ratios in the MAPT mutation carriers for age. Therefore, between-group comparisons of MR markers were performed after adjusting for age. The association between 1H MRS data and time to expected age at onset was tested using linear regression analysis after adjusting for age.
RESULTS
The 1H MRS metabolite and hippocampal volume differences among the control, presymptomatic, and symptomatic patients are listed in table 2. Representative spectra from the 3 clinical groups are shown in figure 1. Symptomatic patients had higher mI/Cr, lower NAA/Cr and NAA/mI ratios, and smaller hippocampal volumes compared to the control group after adjusting for age (p < 0.001). Similarly, the presymptomatic patients on average had higher mI/Cr and lower NAA/mI levels compared to the controls (p < 0.001), but the neuronal marker NAA/Cr ratio and the hippocampal volumes were not different from normal in these patients (p > 0.09) after adjusting for age. The decrease in NAA/mI appeared to be mainly driven by the elevation in mI in the presymptomatic patients. Only one presymptomatic patient had hippocampal atrophy. This patient also had elevated mI/Cr and decreased NAA/mI levels. Hippocampal volumes in all of the other presymptomatic subjects were within the control range. The NAA/Cr (p = 0.003) and hippocampal volumes (p = 0.002) were lower in symptomatic patients compared to the presymptomatic patients; however, mI/Cr levels were not different among the symptomatic and presymptomatic cases (p = 0.22), suggesting that while the elevation of mI/Cr is an early marker for the tau-related pathology, elevation in mI/Cr appears to plateau once an individual becomes symptomatic (figure 2).
Table 21H MRS metabolite ratios and hippocampal volumes in clinical groups
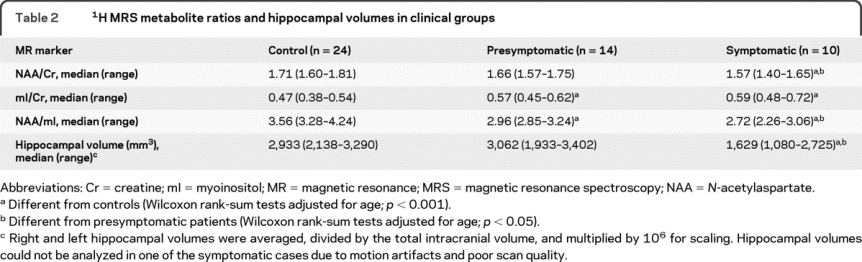

Figure 1 Representative 1H magnetic resonance spectra from the posterior cingulate gyrus and inferior precuneus region
Control subject (A), presymptomatic patient (B), and a patient with frontotemporal dementia (FTD) (C) with MAPT mutations. The spectra are scaled to the creatine (Cr) peak as indicated with the dotted red line. The myoinositol (mI) peak is elevated in the presymptomatic patient (B) and the patient with FTD (C). The N-acetylaspartate (NAA) peak is decreased only in the patient with FTD (C).

Figure 2 Box plots show the hippocampal volumes (corrected for the total intracranial volume) and 1H magnetic resonance spectroscopy metabolite ratios
Controls (n = 24), presymptomatic (n = 14), and symptomatic (n = 10) MAPT mutation carriers. Cr = creatine; mI = myoinositol; NAA = N-acetylaspartate.
Median age at symptom onset in the 4 families/mutation types ranged from 38 to 52 years (table 1). Three presymptomatic carriers of the MAPT mutations were past the median age at symptom onset in their family by 5 and 8 years. Symptomatic patients were experiencing symptoms for 6 to 44 years. We found a significant association between the estimated proximity to symptom onset and the 1H MRS metabolite ratios and hippocampal volumes in the entire group of presymptomatic and symptomatic carriers of MAPT mutations. The NAA/Cr (R2 = 0.22; p = 0.021) and hippocampal volumes (R2 = 0.46; p < 0.001) decreased as the ages of MAPT mutation carriers approached and passed the estimated age at symptom onset (figure 3A). The NAA/mI ratios in all 6 presymptomatic patients with MAPT mutations who had 5 years to reach or who were past estimated age at symptom onset were lower than the lowest value in the control range (NAA/mI < 3.28). In contrast, the NAA/mI ratios in 3 presymptomatic patients who had more than 5 years to reach the estimated age at symptom onset were within the normal range (figure 3B).

Figure 3 N-acetylaspartate (NAA)/myoinositol (mI) levels and the estimated proximity to symptom onset
NAA/mI ratios in the entire group of MAPT mutation carriers (A) and in only presymptomatic carriers of MAPT mutations (B) compared to the NAA/mI levels in the controls (C). In the horizontal axis, 0 indicates the estimated age at symptom onset based on the median age at symptom onset in symptomatic patients from the same family in presymptomatic patients and the age at actual symptom onset in symptomatic patients. (B) All of the presymptomatic MAPT mutation carriers who are estimated to reach the age at symptom onset in less than 5 years (subjects to the right of the black line on B) or who have passed the estimated age at symptom onset have lower NAA/mI levels than the lowest value in the control range.
DISCUSSION
The results of this study demonstrate 1H MRS metabolite abnormalities in presymptomatic carriers of mutations in the gene encoding for MAPT on chromosome 17. The severity of 1H MRS and MRI abnormalities were associated with the proximity to the estimated age at symptom onset. NAA/mI ratio was fully outside of the control range in presymptomatic MAPT mutation carriers who had 5 years to reach or who were past the estimated age at symptom onset, indicating presence of biochemical abnormalities related to neurodegeneration, years before the onset of symptoms in MAPT mutation carriers.
Our findings in MAPT mutation carriers show similarities to the NAA/mI abnormalities reported in 7 presymptomatic carriers of the presenilin 1 (PS1) and amyloid precursor protein gene (APP) mutations who are destined to develop AD,20 and the elevation of mI/Cr in the 2 asymptomatic carriers of the P102L mutation in the prion protein gene who are destined to develop inherited prion disease.21 A relationship between the decrease in NAA/mI and the proximity of expected age at onset was identified in the PS1 and APP mutation carriers.20 In the current study, there was an association between the proximity to the age at symptom onset and the NAA/Cr ratio and hippocampal volumes but not the mI/Cr ratio in the entire cohort of presymptomatic and symptomatic carriers of MAPT mutations. Our interpretation is that the elevation of mI/Cr is an early event that plateaus once an individual becomes symptomatic. In contrast, NAA/Cr and hippocampal volumes are later biomarkers that decrease progressively during the entire disease course, including the clinically symptomatic period. Agreement between the metabolite abnormalities we encountered in the MAPT mutation carriers and the abnormalities in asymptomatic patients with PS1 and APP mutations and P102L mutation in the prion protein gene suggest that the 1H MRS changes in the brain may be early markers of the neurodegenerative pathology in familial neurodegenerative dementias caused by a variety of different mutations leading to proteinopathies.
The early increase in mI/Cr in presymptomatic patients, followed by a decrease in the neuronal integrity marker NAA/Cr and hippocampal atrophy when patients became symptomatic, implies a temporal sequence to the 1H MRS changes in MAPT mutation carriers. This is consistent with the temporal sequence of 1H MRS findings in other tauopathies such as AD and Down syndrome.22–24 For example, mI/Cr is elevated in patients with amnestic mild cognitive impairment, many of whom have early AD pathology,16,25 in patients at the intermediate stage of neurofibrillary pathology at autopsy,9 and in patients with Down syndrome who develop dementia in the future.22,23 These conditions are characterized by an initial elevation in mI/Cr without a significant decrease in NAA/Cr in the parietal lobe regions, and later by decreased NAA/Cr in patients with dementia and in patients with high levels of neurofibrillary pathology at autopsy.9,25
The metabolite mI is mainly located in glial cells,26 and is regarded as a possible marker for glial activation. mI is elevated in glial tumors27 and mI levels are associated with glial activation in inflammatory CNS demyelination.28 Similarly, elevation of mI/Cr in patients with FTLD and carriers of MAPT mutations may be related to the microglial activation and gliosis encountered in these patients.8,10,11,29–31 There is evidence that activated microglia may be harmful to axons and dendrites by interfering with neuronal transport.32,33 A mechanistic link between early microglial activation and progression of tau pathology has been demonstrated in the P301S mutant transgenic tau mice.34 This study showed that microglial activation is one of the earliest pathologic findings in these transgenic tau mice, present at 6 months of age, followed by hippocampal neuronal loss and atrophy detected after 9 months of age.34 The increase in mI/Cr earlier than the decrease in the neuronal integrity marker NAA/Cr and hippocampal atrophy on MRI in MAPT mutation carriers is consistent with the temporal course of microglial activation preceding neuronal loss and hippocampal atrophy in transgenic tau mice, and supports the hypothesis that microglial activation is driving neuronal damage downstream.32
Although symptomatic patients with MAPT mutations have striking medial temporal lobe atrophy, we study the posterior cingulate gyrus region with 1H MRS in our standard MRS protocol for several important reasons relating to spectral quality. The quality and reliability 1H MR spectra from the posterior cingulate is significantly superior to the spectra from the medial temporal lobe, owing to the close proximity of the medial temporal lobe to the magnetic susceptibility artifacts at the skull base. This is particularly true for short echo time spectra, and quantification of mI requires a short echo time 1H MRS acquisition.35–37 In fact, the unreliability of mI measurements from the medial temporal lobe region have been demonstrated in a multicenter study.38 Furthermore, the posterior cingulate voxel location can be identified by clear anatomic landmarks by trained technicians, which is critical for longitudinal serial measurements. Because we found a significant decrease in NAA/mI in patients with FTLD in the posterior cingulate voxel in a previous study,11 we felt confident in this acquisition for the current analysis. However, significant abnormalities may also be found in the frontal lobe, which is also involved in symptomatic carriers of MAPT mutations but less significantly than the medial temporal lobe.
The median age at symptom onset in symptomatic members of the family was used for estimating the time to symptom onset in presymptomatic mutation carriers. The clinical presentation of FTLD-17 is heterogenous among various mutations, within a single mutation, and even among members of the same family.5 The age at symptom onset may also vary among the members of a single family.8 This was evident in the 2 presymptomatic carriers of the same MAPT mutation who were past the expected age at symptom onset by 5 and 8 years but had decreased NAA/mI ratios suggesting preclinical neurodegenerative changes. Interestingly, another presymptomatic patient in this family, who was 13 years younger than the expected age at symptom onset, had a similarly low NAA/mI level (3.19) as the presymptomatic MAPT mutation carrier who was past the expected age at symptom onset by 5 years (figure 3). Another presymptomatic patient who was 13 years younger than the estimated age at symptom onset had NAA/mI levels as low as some of the symptomatic patients (2.89). However, this decrease in NAA/mI was mainly due to an elevated mI/Cr (0.59) because NAA/Cr level (1.71) in this patient was within the control range. A relatively slow progression of the neurodegenerative pathology may be responsible for the reduced variability in NAA/mI relative to the estimated time to symptom onset in both of these families. Longitudinal follow-up is necessary to determine the actual age at symptom onset in the presymptomatic patients.
Metabolite abnormalities detected in MAPT mutation carriers give insights into disease pathogenesis in tauopathies. The neurodegenerative changes that are characterized by an elevation in the possible glial marker mI/Cr begin years before the onset of symptoms, and the decrease in the neuronal marker NAA/Cr and hippocampal atrophy appear to follow shortly before dementia ensues and become progressively more abnormal as dementia worsens. This sequence of 1H MRS and volumetric MRI changes in MAPT mutation carriers show similarities to the 1H MRS findings in sporadic and familial AD, and is in agreement with microglial activation observed prior to neuronal loss and hippocampal atrophy in tau transgenic mice. 1H MRS is a noninvasive acquisition technique that is present on all modern clinical MR scanners and can implemented in multicenter projects.38 These early findings suggest the possibility that 1H MRS may be used as a noninvasive imaging marker for neuroprotective interventions,39,40 before there is significant loss of neuronal integrity in MAPT mutation carriers as well as in other proteinopathies such as AD. Validating 1H MRS abnormalities as biomarkers of tau-mediated pathology will require longitudinal follow-up.
AUTHOR CONTRIBUTIONS
Statistical analysis was conducted by Dr. Kejal Kantarci.
ACKNOWLEDGMENT
The authors thank Stephen D. Weigand, Division of Biostatistics, Mayo Clinic, Rochester, MN, for input on statistical analysis, and Audrey Strongosky, Department of Neurology, Mayo Clinic, Jacksonville, FL, for assistance in research subject recruitment.
DISCLOSURE
Dr. Kantarci receives research support from the NIH (K23 AG030935 [PI], P50 AG16574/Project1 [PI], R21 NS066147 [PI], and R01 AG11378 [Co-I]). Dr. Boeve has served as a consultant to GE Healthcare; receives royalties from the publication of Behavioral Neurology of Dementia (Cambridge Medicine, 2009); and receives research support from Myriad Genetics Inc., Cephalon, Inc., the NIH (P50 AG16574 [Co-I], UO1 AG06786 [Co-I, and RO1 AG15866 [Co-I]), the Alzheimer's Association, and the Center for Inherited Disease Research (U24 AG026395 [Co-I]). Dr. Wszolek serves as Co-Editor-in-Chief of Parkinsonism and Related Disorders, Regional Editor of the European Journal of Neurology, and on the editorial boards of Neurologia i Neurochirurgia Polska, Advances in Rehabilitation, the Medical Journal of the Rzeszow University, and Clinical and Experimental Medical Letters; holds and has contractual rights for receipt of future royalty payments from patents re: A novel polynucleotide involved in heritable Parkinson's disease; receives royalties from publishing Parkinsonism and Related Disorders (Elsevier, 2007, 2008, 2009) and the European Journal of Neurology (Wiley-Blackwell, 2007, 2008, 2009); receives research support from Allergan, Inc., the NIH (NIA P01AG017216-1 [coinvestigator], NIA R01AG015866-1 [coinvestigator], and NINDS P50NS 40256 [coinvestigator]), the Pacific Alzheimer Research Foundation (Canada), and the CIHR. Dr. Rademakers holds patents re: Methods and Materials for Detecting and Treating Dementia and receives research support from the NIH (P50-AG16574 [PI on Project 2], RO1 NS065782-01 [PI], and R56 AG26251-03A1 [coinvestigator]), the Pacific Alzheimer Research Foundation (Canada), the Association for Frontotemporal Dementia, and the Amyotrophic Lateral Sclerosis Association. Dr. Whitwell receives research support from the Dana Foundation. M.L. Senjem has received research support from Pfizer Inc. M.C. Baker holds patents re: Methods and Materials for Detecting and Treating Dementia. A.R. Samikoglu reports no disclosures. Dr. Knopman serves as Deputy Editor of Neurology®; has served on data safety monitoring boards for Sanofi-Aventis, GlaxoSmith Kline, and Eli Lilly and Company; is an investigator in clinical trials sponsored by Elan Corporation, Baxter International Inc., and Forest Laboratories, Inc.; and receives research support from the NIH (R01-AG023195 [PI], R01-AG11378 [Co-I], P50 AG16574 [Co-I], U01 AG 06786 [Co-I], and R01 HL70825 [Co-I]). Dr. Petersen serves on scientific advisory boards for Elan Pharmaceuticals, Wyeth Pharmaceuticals, and GE Healthcare; receives royalties from publishing Mild Cognitive Impairment (Oxford University Press, 2003); and receives research support from the NIH (P50-AG16574 [PI] and U01-AG06786 [PI], R01-AG11378 [Co-I], and U01-24904 [Co-I]). Dr. Jack serves as a consultant for Elan Corporation; receives research support from Pfizer Inc., the NIH/NIA (R01-AG11378 [PI], P50-AG16574 [Co-I], and U01 AG024904-01 [Co-I]), and the Alexander Family Alzheimer's Disease Research Professorship of the Mayo Foundation; and holds stock in GE Healthcare and Johnson & Johnson.
Address correspondence and reprint requests to Dr. Kejal Kantarci, Mayo Clinic, 200 First Street SW, Rochester, MN 55905 kantarci.kejal@mayo
Study funding: Supported by NIH K23 AG030935, P50 AG16574/P1, P50 AG16574/P2, R01 AG11378, R01 NS065782, the Robert H. and Clarice Smith and Abigail Van Buren Alzheimer's Disease Research Program of the Mayo Foundation, and the NIH Construction Grant C06 RR018898.
Disclosure: Author disclosures are provided at the end of the article.
Received February 2, 2010. Accepted in final form May 12, 2010.
REFERENCES
- 1.Boeve BF, Hutton M. Refining frontotemporal dementia with parkinsonism linked to chromosome 17: introducing FTDP-17 (MAPT) and FTDP-17 (PGRN). Arch Neurol 2008;65:460–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hutton M, Lendon CL, Rizzu P, et al. Association of missense and 5′-splice-site mutations in tau with the inherited dementia FTDP-17. Nature 1998;393:702–705. [DOI] [PubMed] [Google Scholar]
- 3.Rademakers R, Cruts M, van Broeckhoven C. The role of tau (MAPT) in frontotemporal dementia and related tauopathies. Hum Mutat 2004;24:277–295. [DOI] [PubMed] [Google Scholar]
- 4.Foster NL, Wilhelmsen K, Sima AA, Jones MZ, D'Amato CJ, Gilman S. Frontotemporal dementia and parkinsonism linked to chromosome 17: a consensus conference. Ann Neurol 1997;41:706–715. [DOI] [PubMed] [Google Scholar]
- 5.Ingram EM, Spillantini MG. Tau gene mutations: dissecting the pathogenesis of FTDP-17. Trends Mol Med 2002;8:555–562. [DOI] [PubMed] [Google Scholar]
- 6.Whitwell JL, Jack CR Jr, Boeve BF, et al. Voxel-based morphometry patterns of atrophy in FTLD with mutations in MAPT or PGRN. Neurology 2009;72:813–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bunker JM, Kamath K, Wilson L, Jordan MA, Feinstein SC. FTDP-17 mutations compromise the ability of tau to regulate microtubule dynamics in cells. J Biol Chem 2006;281:11856–11863. [DOI] [PubMed] [Google Scholar]
- 8.Arvanitakis Z, Witte RJ, Dickson DW, et al. Clinical-pathologic study of biomarkers in FTDP-17 (PPND family with N279K tau mutation). Parkinsonism Relat Disord 2007;13:230–239. [DOI] [PubMed] [Google Scholar]
- 9.Kantarci K, Knopman DS, Dickson DW, et al. Alzheimer disease: postmortem neuropathologic correlates of antemortem 1H MR spectroscopy metabolite measurements. Radiology 2008;248:210–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ernst T, Chang L, Melchor R, Mehringer CM. Frontotemporal dementia and early Alzheimer disease: differentiation with frontal lobe H-1 MR spectroscopy. Radiology 1997;203:829–836. [DOI] [PubMed] [Google Scholar]
- 11.Kantarci K, Petersen RC, Boeve BF, et al. 1H MR spectroscopy in common dementias. Neurology 2004;63: 1393–1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Murray ME, Dickson D, Jack CR, Kantarci K. Aberrant tau pathology underlies 1H MR spectroscopic alterations in Alzheimer's disease. Neurology 2009;72(suppl 3):A173. [Google Scholar]
- 13.Cordes M, Wszolek ZK, Calne DB, Rodnitzky RL, Pfeiffer RF. Magnetic resonance imaging studies in rapidly progressive autosomal dominant parkinsonism and dementia with pallido-ponto-nigral degeneration. Neurodegeneration 1992;1:217–224. [DOI] [PubMed] [Google Scholar]
- 14.Malkani R, D'Souza I, Gwinn-Hardy K, Schellenberg GD, Hardy J, Momeni P. A MAPT mutation in a regulatory element upstream of exon 10 causes frontotemporal dementia. Neurobiol Dis 2006;22:401–403. [DOI] [PubMed] [Google Scholar]
- 15.Wszolek ZK, Pfeiffer RF, Bhatt MH, et al. Rapidly progressive autosomal dominant parkinsonism and dementia with pallido-ponto-nigral degeneration. Ann Neurol 1992;32:312–320. [DOI] [PubMed] [Google Scholar]
- 16.Webb PG, Sailasuta N, Kohler SJ, Raidy T, Moats RA, Hurd RE. Automated single-voxel proton MRS: technical development and multisite verification. Magn Reson Med 1994;31:365–373. [DOI] [PubMed] [Google Scholar]
- 17.Tzourio-Mazoyer N, Landeau B, Papathanassiou D, et al. Automated anatomical labeling of activations in SPM using a macroscopic anatomical parcellation of the MNI MRI single-subject brain. Neuroimage 2002;15:273–289. [DOI] [PubMed] [Google Scholar]
- 18.Jack CR Jr, Lowe VJ, Senjem ML, et al. 11C PiB and structural MRI provide complementary information in imaging of Alzheimer's disease and amnestic mild cognitive impairment. Brain 2008;131:665–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ashburner J, Friston KJ. Unified segmentation. Neuroimage 2005;26:839–851. [DOI] [PubMed] [Google Scholar]
- 20.Godbolt AK, Waldman AD, MacManus DG, et al. MRS shows abnormalities before symptoms in familial Alzheimer disease. Neurology 2006;66:718–722. [DOI] [PubMed] [Google Scholar]
- 21.Waldman AD, Cordery RJ, MacManus DG, Godbolt A, Collinge J, Rossor MN. Regional brain metabolite abnormalities in inherited prion disease and asymptomatic gene carriers demonstrated in vivo by quantitative proton magnetic resonance spectroscopy. Neuroradiology 2006;48:428–433. [DOI] [PubMed] [Google Scholar]
- 22.Huang W, Alexander GE, Daly EM, et al. High brain myo-inositol levels in the predementia phase of Alzheimer's disease in adults with Down's syndrome: a 1H MRS study. Am J Psychiatry 1999;156:1879–1886. [DOI] [PubMed] [Google Scholar]
- 23.Shonk T, Ross BD. Role of increased cerebral myo-inositol in the dementia of Down syndrome. Magn Reson Med 1995;33:858–861. [DOI] [PubMed] [Google Scholar]
- 24.Kantarci K. 1H magnetic resonance spectroscopy in dementia. Br J Radiol 2007;80:S146–152. [DOI] [PubMed] [Google Scholar]
- 25.Kantarci K, Jack CR Jr, Xu YC, et al. Regional metabolic patterns in mild cognitive impairment and Alzheimer's disease: a 1H MRS study. Neurology 2000;55:210–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Glanville NT, Byers DM, Cook HW, Spence MW, Palmer FB. Differences in the metabolism of inositol and phosphoinositides by cultured cells of neuronal and glial origin. Biochim Biophys Acta 1989;1004:169–179. [DOI] [PubMed] [Google Scholar]
- 27.Hattingen E, Raab P, Franz K, Zanella FE, Lanfermann H, Pilatus U. Myo-inositol: a marker of reactive astrogliosis in glial tumors? NMR Biomed 2008;21:233–241. [DOI] [PubMed] [Google Scholar]
- 28.Bitsch A, Bruhn H, Vougioukas V, et al. Inflammatory CNS demyelination: histopathologic correlation with in vivo quantitative proton MR spectroscopy. Am J Neuroradiol 1999;20:1619–1627. [PMC free article] [PubMed] [Google Scholar]
- 29.Reed LA, Schmidt ML, Wszolek ZK, et al. The neuropathology of a chromosome 17-linked autosomal dominant parkinsonism and dementia (“pallido-ponto-nigral degeneration”). J Neuropathol Exp Neurol 1998;57:588–601. [DOI] [PubMed] [Google Scholar]
- 30.Cooper PN, Siddons CA, Mann DM. Patterns of glial cell activity in fronto-temporal dementia (lobar atrophy). Neuropathol Appl Neurobiol 1996;22:17–22. [PubMed] [Google Scholar]
- 31.Slowinski J, Dominik J, Uitti RJ, Ahmed Z, Dickson DD, Wszolek ZK. Frontotemporal dementia and Parkinsonism linked to chromosome 17 with the N279K tau mutation. Neuropathology 2007;27:73–80. [DOI] [PubMed] [Google Scholar]
- 32.McGeer PL, McGeer EG. The inflammatory response system of brain: implications for therapy of Alzheimer and other neurodegenerative diseases. Brain Res Brain Res Rev 1995;21:195–218. [DOI] [PubMed] [Google Scholar]
- 33.Takeuchi H, Mizuno T, Zhang G, et al. Neuritic beading induced by activated microglia is an early feature of neuronal dysfunction toward neuronal death by inhibition of mitochondrial respiration and axonal transport. J Biol Chem 2005;280:10444–10454. [DOI] [PubMed] [Google Scholar]
- 34.Yoshiyama Y, Higuchi M, Zhang B, et al. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron 2007;53:337–351. [DOI] [PubMed] [Google Scholar]
- 35.Ebel A, Soher BJ, Maudsley AA. Assessment of 3D proton MR echo-planar spectroscopic imaging using automated spectral analysis. Magn Reson Med 2001;46:1072–1078. [DOI] [PubMed] [Google Scholar]
- 36.Soher BJ, Vermathen P, Schuff N, et al. Short TE in vivo (1)H MR spectroscopic imaging at 1.5 T: acquisition and automated spectral analysis. Magn Reson Imaging 2000;18:1159–1165. [DOI] [PubMed] [Google Scholar]
- 37.Kantarci K, Reynolds G, Petersen RC, et al. Proton MR spectroscopy in mild cognitive impairment and Alzheimer disease: comparison of 1.5 and 3 T. AJNR Am J Neuroradiol 2003;24:843–849. [PMC free article] [PubMed] [Google Scholar]
- 38.Jessen F, Gur O, Block W, et al. A multicenter (1)H-MRS study of the medial temporal lobe in AD and MCI. Neurology 2009;72:1735–1740. [DOI] [PubMed] [Google Scholar]
- 39.Ludolph AC, Kassubek J, Landwehrmeyer BG, et al. Tauopathies with parkinsonism: clinical spectrum, neuropathologic basis, biological markers, and treatment options. Eur J Neurol 2009;16:297–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Trojanowski JQ, Duff K, Fillit H, et al. New directions for frontotemporal dementia drug discovery. Alzheimers Dement 2008;4:89–93. [DOI] [PMC free article] [PubMed] [Google Scholar]