


Abstract
Objective:
Amyotrophic lateral sclerosis (ALS) is a progressive paralytic disorder caused by degeneration of motor neurons. Mutations in the FUS gene were identified in patients with familial ALS (FALS) and patients with sporadic ALS (SALS) from a variety of genetic backgrounds. This work further explores the spectrum of FUS mutations in patients with FALS and patients with FALS with features of frontotemporal dementia (FALS/FTD) or parkinsonism and dementia (FALS/PD/DE).
Methods:
All exons of the FUS gene were sequenced in 476 FALS index cases negative for mutations in SOD1 and TARDBP. A total of 561–726 controls were analyzed for genetic variants observed. Clinical data from patients with FUS mutations were compared to those of patients with known SOD1 and TARDBP mutations.
Results:
We identified 17 FUS mutations in 22 FALS families, 2 FALS/FTD families, and 1 FALS/PD/DE family from diverse genetic backgrounds; 11 mutations were novel. There were 4 frameshift, 1 nonsense, and 1 possible alternate splicing mutation. Patients with FUS mutations appeared to have earlier symptom onset, a higher rate of bulbar onset, and shorter duration of symptoms than those with SOD1 mutations.
Conclusions:
FUS gene mutations are not an uncommon cause in patients with FALS from diverse genetic backgrounds, and have a prevalence of 5.6% in non-SOD1 and non-TARDBP FALS, and ∼4.79% in all FALS. The pathogenicity of some of these novel mutations awaits further studies. Patients with FUS mutations manifest earlier symptom onset, a higher rate of bulbar onset, and shorter duration of symptoms.
GLOSSARY
- ALS
= amyotrophic lateral sclerosis;
- FALS
= familial amyotrophic lateral sclerosis;
- FALS/FTD
= familial amyotrophic lateral sclerosis with features of frontotemporal dementia;
- FALS/PD/DE
= familial amyotrophic lateral sclerosis with features of parkinsonism and dementia;
- SALS
= sporadic amyotrophic lateral sclerosis.
Amyotrophic lateral sclerosis (ALS) is a degenerative disease of motor neurons.1,2 Its etiologies are partially unknown. The most frequent known cause is mutations in the SOD1 gene, which are responsible for ∼20% of familial ALS (FALS) cases.3 To date, 21 mutations in the TARDBP genes have been reported in 643 FALS cases with varied frequencies.4–14 Mutations in ALS2, SET, VAPB, DCTN1, and ANG are very rare causes of ALS or ALS-like motor neuron diseases.2,15–17
Recently, FUS gene mutations were identified in a subset of patients with FALS and patients with sporadic ALS (SALS).18–22 The largest FALS sample set studied was 293 cases; however, 209 of them were only screened for exon 15.21 Most of the reported FALS cases with FUS mutations had no cognitive change.
ALS can overlap with frontotemporal dementia (FALS/FTD) or parkinsonism and dementia (FALS/PD/DE). FALS/FTD has been linked to chromosome 9q21-q2223 and 9p21.2-9p13.3,24–27 but no causative gene has been found. To further explore the spectrum of FUS mutations, we sequenced the FUS gene in a cohort of 476 FALS index cases and 41 SALS cases. Some of the FALS cases also had additional features like FTD or parkinsonism/dementia. We found 11 novel mutations including 4 frameshift, 1 nonsense, and 1 possible splicing site mutation in patients with FALS, FALS/FTD, and FALS/PD/DE from different ethnic backgrounds.
METHODS
Standard protocol approvals, registration, and patient consents.
Protocols were approved by the ethics committee on human experimentation of Northwestern University Feinberg School of Medicine. Blood samples were collected after patients gave written consent.
Participants.
ALS was diagnosed according to the El Escorial criteria.28 Some ALS cases had additional features of dementia and parkinsonism. The diagnosis of FTD was based on the revised criteria by Neary et al.29 Pedigrees and clinical data were collected through specialists in neuromuscular diseases and were verified by medical records to establish diagnosis. A total of 476 FALS index cases without SOD1 and TARDBP gene mutations and 41 SALS cases were sequenced for the FUS gene. Of the 476 FALS cases, 393 cases had only ALS, 76 cases had FALS/FTD, and 7 cases had FALS/PD/DE. Self-reporting ethnicity showed that 93.9% of the cases were white (European American), 2.5% were Asian, 1.9% were African American, and 1.3% were Latino. The ethnicity of 2 cases was unknown. Control DNA samples were primarily collected by our laboratory. A majority of the controls were white (97.6%), 0.56% were African American, 0.98% were Latino, and 0.84% were Asian. Less than 1% of the patients with FALS were on respiratory support. SPSS software (release 16.0.0) was used for statistical analysis. Differences in age at symptom onset were obtained with Kaplan-Meier analyses; percentage of site of symptom onset was analyzed with Pearson χ2 test. Mean duration of symptoms was obtained by the t test.
Sequencing analysis of the FUS gene.
Genomic DNA was extracted from transformed lymphoblastoid cell lines, whole blood, or brain tissues by standard methods (Qiagen, Valencia, CA). Intronic primers for PCR and sequencing covered the coding sequence as reported.21 Genomic DNA was amplified with high-fidelity TaKaRa LA Taq™ polymerase (Takara, Japan). Unconsumed dNTPs and primers were digested with exonuclease I and shrimp alkaline phosphatase (ExoSAP-IT, USB, Cleveland, OH). Fluorescent dye–labeled single-strand DNA was amplified using Beckman Coulter sequencing reagents (GenomeLab DTCS Quick Start Kit) followed by bidirectional sequencing with a CEQ™ 8000 Genetic Analysis System (Beckman Coulter, CA). When a variant was identified, a large number of control DNA samples (561–726) were analyzed to exclude the possibility of a rare polymorphism. Splicing site changes were predicted with SpliceView (http://bioinfo.itb.cnr.it/oriel/splice-view.html).
RESULTS
We sequenced the FUS gene for all 15 exons in a cohort of 476 non-SOD1 and non-TARDBP FALS index cases and 41 SALS cases; cosegregation analysis was performed if DNA samples were available from additional family members. We found no FUS mutations in SALS cases, but 17 heterozygous nucleotide mutations were found in 25 index cases; 11 are novel mutations (table 1).
Table 1FUS mutations in a cohort of 461 patients with FALS
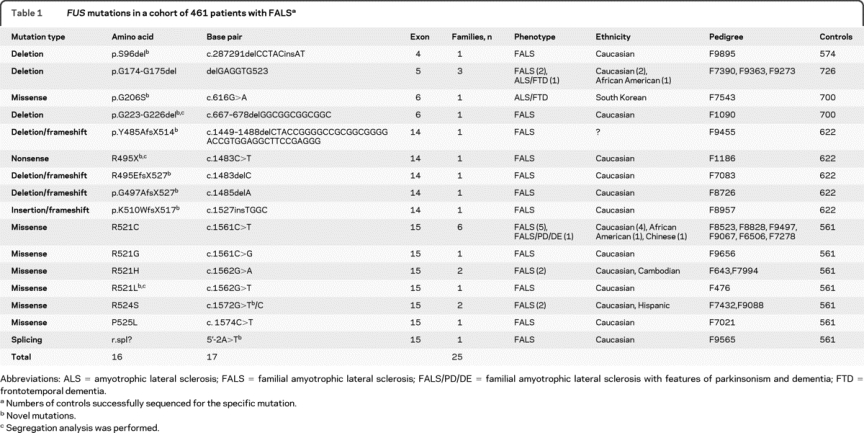
One nonsense and 4 frameshift mutations were found, all in exon 14 (table 1). One patient with G497AfsX527 mutation in F8726 developed left leg weakness at 12.5 years of age, and died in 18 months. Seven patients with nonsense and frameshift mutations also showed short duration of symptoms (table 2). Patients with FUS mutations showed remarkable variation at symptom onset. Obligate carriers in F1186 and F476 were symptom-free even after their offspring became affected (figure 1, table 2).
Table 2 Phenotype of selected ALS families with additional features
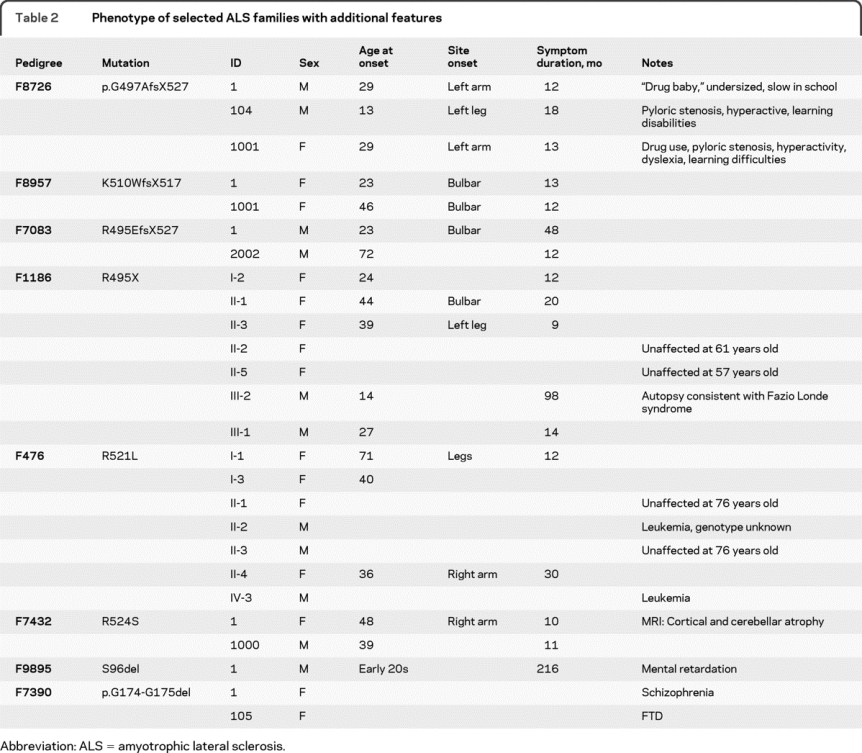

Figure 1 Cosegregation of FUS mutations in representative familial amyotrophic lateral sclerosis pedigrees
Affected patients are marked by a black symbol, unaffected with an open symbol. A slashed symbol indicates a deceased patient. + = With the mutation; − = without the mutation; (+) = genotype inferred. Gender and birth order have been omitted for reasons of confidentiality.
FUS mutations were also found in patients with ALS with parkinsonism or FTD. F8828 had the most frequent R521C mutation. The proband developed ALS with gait and speech difficulty at age 85. The proband's brother was diagnosed with Parkinson features and dementia, and her mother and sister had dementia. F7543 carried G206S, a novel mutation in exon 6. The index patient developed ALS at age 54; his 2 brothers had behavior problems in their 40s consistent with the diagnosis of FTD. The proband of F7390 was diagnosed with schizophrenia, and then developed ALS. No detailed medical records were available for the patient's diagnosis of schizophrenia, so the possibility of dementia could not be excluded. His sister had FTD with behavioral changes. The family had p.G174-G175del in exon 5 (table 1).
Unusual clinical features were also noted in patients with ALS with FUS mutations. A patient from F7432 with R524S developed right arm weakness at age 48, and MRI showed significant cortical and cerebellar atrophy (table 2). The R524S mutation due to c.1572G>C change has been previously reported, but R524S due to c.1572G>T in F7432 was novel. The proband of F9895 with S96del mutation in exon 4 had mental retardation (table 2). Individual IV-3 of F476 with R521L mutation had leukemia. II-2 also had leukemia, but his genotype was not known (figure 1). This is interesting as FUS was first identified as part of a fusion gene with DNA-damage-inducible transcript 3 (CHOP) from tissues of liposarcoma30,31 and the transcription factor ERG-1 in human myeloid leukemias.32
Eight nucleotide mutations were located in exon 15 in 14 families. Four mutations involved residue R521 and were seen in 10 out of 25 FALS families. The R521C mutation was identified in 6 families of diverse ethnic background, including European American, Chinese, and African American. An A>T change in intron 14, 2 base pairs away from the acceptor site, was found in F9565 of Italian origin. This changes the typical acceptor site sequence from AG to TG, which was expected to eliminate the splicing site between intron 14 and exon 15. This possibility was also supported by an analysis in silico with SpliceView.
We obtained clinical data of 101 patients from these 25 families with FUS gene mutations. Fifty-five of the 101 cases were male (54.5%). The average age at symptom onset was 43.6 ± 15.8 years (n = 54) for FUS mutations, 47.7 ± 13.0 (n = 164) for SOD1, and 54.7 ± 15.3 (n = 34) for TARDBP mutations. The average age at symptom onset in patients with FUS gene mutations was remarkably earlier than patients with TARDBP mutations and the distribution of age at symptom onset was significantly different among the patients with FUS, SOD1, or TARDBP mutations (figure 2).
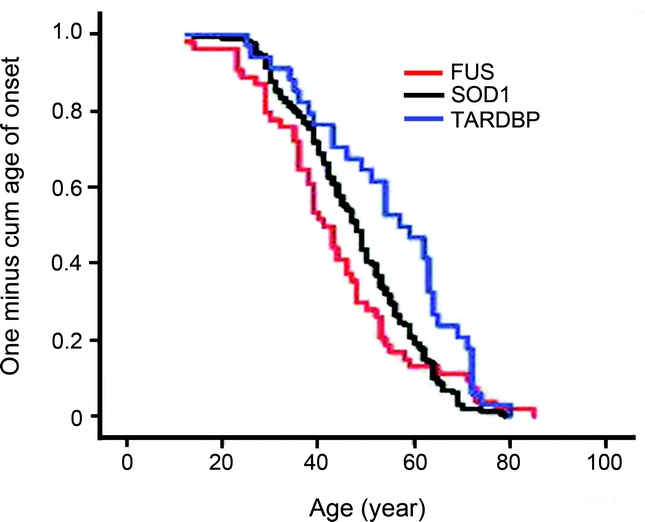
Figure 2 Comparison of age at symptom onset
Kaplan-Meier analysis. Y-axis: proportion of patients with symptom onset. X-axis: age. Trend difference was analyzed with log rank (p = 0.012), Breslow (p = 0.02), and Tarone-Ware (p = 0.02) tests. FUS, n = 54; SOD1, n = 164; TARDBP, n = 34.
The average duration of symptoms was 3.4 ± 5.7 years (n = 44) for patients with FUS mutations, 4.1 ± 4.9 years (n = 144) with SOD1 mutations, and 3.3 ± 2.3 years (n = 30) with TARDBP mutations. The duration of symptoms varied widely: 1 patient with the FUS p.S96del mutation survived for 18 years. However, 88.6% of FUS mutated patients died in less than 4 years, significantly higher than patients with SOD1 (70.1%) and TARDBP (70.0%) mutations.
Patients with FUS, SOD1, and TARDBP gene mutations showed increased incidence of spinal onset vs bulbar onset, but the percentage of bulbar onset of patients with FUS (33.3%) and TARDBP (32.1%) was significantly higher than that of patients with SOD1 mutations (7.6%) (appendix e-1 on the Neurology® Web site at www.neurology.org).
DISCUSSION
The genetic causes of FALS are partly known. In this study, we found that 22 out of 393 FALS index cases without FTD or PD, 2 out of 76 patients with FALS/FTD, and 1 out of 7 FALS/PD/DE index cases had FUS gene mutations. The FUS gene mutation frequency of non-SOD1 and non-TARDBP cases of FALS was 5.6%, and of all cases of FALS was about 4.79%. It was reported that 4.5% of 198 index cases without SOD1, VAPB, ANG, DYNACTIN, CHMP2B, or TARDBP mutations22 and 5.3% of 94 cases of FALS without SOD1, TARDBP, and ANG mutations had FUS mutations.20 Both reports estimated the FUS mutational frequency of the general FALS population to be ∼4%. FUS mutation frequency was found to be 5.8% in 293 FALS, but it was unclear which gene mutations were excluded, and 209 cases of the 293 cases of FALS were screened only for exon 15.21 FUS and TARDBP are both DNA/RNA binding proteins.33 The prevalence of mutations of TARDBP in ALS varied from 0.65% to 4.85% in studies with 80 to 154 patients with FALS examined and 0% to 5% in studies with 86 to 541 patients with SALS screened4–14; the variation might be related to clinical heterogeneity of patients. To date, 21 mutations in the TARDBP genes were found in 643 cases of FALS with a mutational frequency of 3.27%. FUS mutations are more frequent than TARDBP mutations, and appear to be the second most frequent cause of disease after SOD1 mutations in FALS. For SALS, we found no FUS gene mutations in 41 cases; however, the sample size was very small. One report found no FUS mutation in 293 patients with SALS,21 but another found 3 out of 405 SALS cases had FUS gene mutation.18
We found that 1 FALS/PD/DE index case out of 7 and 2 ALS/FTD index cases out of 76 had FUS gene mutations. The R521C mutation in the index case of F8828 with FALS/PD/DE was the most frequent mutation documented in FALS.21,22 The G206S mutation in F7543 with ALS/FTD has not been reported. G174-G175 mutation in exon 5 was first reported in a FALS pedigree (F213) with a screening of 176 controls.21 We found it in 1 ALS/FTD and 2 FALS index cases, and it was not present in the sequencing of 726 controls. No patients with FALS with cognitive deficiency were reported in the first 2 FUS mutation reports,21,22 but FUS immunoreactive neuronal inclusions have been reported in FTD.34 However, a G156E mutation was found in an Italian FALS index case who developed FTD in his fourth decade.20 The proband with S96del had mental retardation, the proband with G174-G175 del in F7390 had the diagnosis of schizophrenia, and the proband with R524S had cerebellar atrophy on MRI, but no additional clinical records were available for further characterization. All the affected individuals in F8726 with G497AfsX527 mutation had learning disabilities; 1 died at age 14; the 2 adults were drug users. It was suggested that FUS mutations might result in cognitive dysfunction, which merits further study.
The deletion of 4 glycine residues (G223–G226del) in exon 6 occurred in a 10-glycine stretch from amino acid residue 222 to 231. Six out of the 10 glycine residues were coded by 6 GGC repeats. A loss of 3 glycine residues was found in a SALS case and in 1 out of 190 controls. A glycine insertion in the same region was found in a patient with schizophrenia and autism.18 The 4-glycine residue deletion in F1090 segregated with the patients and was not present in 700 controls (appendix e-1). In this study, the controls were mainly European Americans, and we had no autopsy tissue for pathologic evaluation; the glycine stretch changes could be polymorphism, and pathogenicity of some of the novel mutations awaits further verification.
Identification of 4 novel frameshift mutations and 1 nonsense mutation in exon 14 suggests that the c-terminal amino acid residues may be of key biologic relevance if the pathogenesis of FUS mutation was due to loss of function. It may also imply that a shorter N-terminal peptide of 494 amino acids is sufficient to cause neuronal toxicity via a gain of function mechanism (figure e-1). Such a phenomenon was previously observed in truncated SOD1-mediated ALS in humans and transgenic mice.35 A nonsense change at Y374X in the TARDBP gene resulting in a truncated C-terminal has also been reported in SALS.5 The nonsense R495X and frameshift mutations in FUS may suggest that haploinsufficiency rather than a gain of function of FUS could cause ALS. Truncated transcript and protein would be degraded or functionless, nevertheless, FUS knockout mice had perinatal mortality, sterility, and radiation sensitivity, but had no obvious neurologic manifestation.36,37
The distribution of FUS mutations reported to date (figure e-1) may delineate 2 major mutation clusters in the FUS gene: 1 in exon 4 to 6 and the other in exon 14 to 15. The apparent grouping of mutations in these 2 clusters may imply the functional importance of these regions in triggering motor neuron degeneration. It may also indicate regions of interest for genetic screening of FUS mutations in patients with ALS.
In previous studies, FUS mutations were found in ALS cases of European American and Cape Verdean Island origins.21,22 We identified FUS mutations in additional ethnic groups, including European American, African American, Asian (Chinese, Korean, and Cambodian), and Latino. Most FUS mutations identified in European Americans were also identified in patients from other ethnicity except G206S, which was found in a family of South Korean origin. Although this study is not a population study, it suggests that FUS mutations may be a globally distributed genetic cause of FALS in patients of different genetic backgrounds.
When comparing patients with ALS with SOD1 and TARDBP mutations, patients with FUS mutations had earlier symptom onset, a higher rate of bulbar onset, and a shorter duration of symptoms in general. No report has compared the phenotype of FUS, SOD1, or TARDBP mutations, but the average age at symptom onset and duration of symptoms of patients with FUS mutations from previous studies were close to those of this study.21,22 The reported average age at symptom onset of patients with TARDBP mutations was 55.6 years (n = 8),6 close to the 54.7 years observed in the 34 cases in this study. The variability in survival noted in FUS families may pose difficulties in assessing response to treatment. We found a patient with the p.S96del mutation who survived for 18 years, suggesting that patients with certain specific FUS mutations may have a better prognosis than others. This variation in survival has also been observed in some mutant SOD1-mediated ALS cases. Patients with the H46R mutation have a much longer survival (>17 years) than those with the A4V mutation (1 year) in SOD1.38,39 We also noted that patients in the same pedigree may have significant differences in age and site of symptom onset. Some individuals with FUS mutations had no symptoms even as their children were getting affected. This phenomenon suggests that other factors, including genetic background and environmental exposure, may modulate the clinical course. Differences in age at onset between FUS and TARDBP mutations are interesting as these 2 molecules share similar domains, and both are RNA/DNA binding proteins.33 Our study has enlarged the spectrum of neurodegenerative phenotype associated with mutations in FUS.
AUTHOR CONTRIBUTIONS
Patients with FALS were identified and their clinical findings verified by Teepu Siddique or Benjamin Brooks and W. King Engel. Clinical data were complied by Nailah Siddique and Sandra Donkervoort. DNA samples were prepared by Jian Guo Zheng. Sequencing was done by Jianhua Yan, Faisal Fecto, Wenjie Chen, Erdong Liu, Yi Yang, and Yong Shi, and verified by Teepu Siddique and Han-Xiang Deng. Statistical analysis was done by Kreshnik Ahmeti and Jianhua Yan. Drafting the manuscript, including responding to reviewers' comments, editing, copyediting, and word processing, was done by Teepu Siddique, Han-Xiang Deng, and Jianhua Yan. All authors participated in the review of the manuscript at each of these stages and approved the final draft.
DISCLOSURE
Dr. Yan and Dr. Deng report no disclosures. N. Siddique receives salary support from the NIH (NINDS NS050641 [research nurse]) and the Les Turner ALS Foundation. Dr. Fecto, Dr. Chen, Y. Yang, and Dr. Liu report no disclosures. S. Donkervoort receives research support from the NIH (NINDS NS050641). Dr. Zheng, Dr. Shi, and K.B. Ahmeti report no disclosures. Dr. Brooks serves/has served on scientific advisory boards for Avanir Pharmaceuticals and Sanofi-Aventis and receives research support from Avanir Pharmaceuticals, Carolinas ALS Research Fund, and Harris Research Fund–Carolinas Healthcare Foundation. Dr. Engel reports no disclosures. Dr. Siddique serves on the scientific advisory board of NIH: Skeletal Muscle and Exercise Physiology (SMEP) Study Section; serves on the editorial boards of Neurogenetics and Amyotrophic Lateral Sclerosis; holds a patent re: Human alpha-tocopherol transport protein: compositions and methods; and receives research support from the NIH (NINDS RO1 NS046535 [PI], NINDS RO1 NS050641 [PI], NIEHS-RO1 ES014469 [PI], NIEHS PO1 ES016742 [PI]), the Harold Post ALS Research Fund, the Les Turner ALS Foundation/Herbert C. Wenske Foundation, Vena Schaaf, Frank White ALS Research Fund, the Spastic Paraplegia Foundation, Inc., the Amyotrophic Lateral Sclerosis Association, the CVS/ALS Therapy Alliance, and the Blazeman Foundation for ALS.
Supplementary Material
Address correspondence and reprint requests to Dr. Teepu Siddique, Davee Department of Neurology and Clinical Neurosciences, Northwestern University Feinberg School of Medicine, Tarry Building 13-715, 303 E. Chicago Ave., Chicago, IL 60611 t-siddique@northwestern.edu
See page 815
Supplemental data at www.neurology.org
e-Pub ahead of print on July 28, 2010, at www.neurology.org.
Study funding: Supported by The National Institute of Neurological Disorders and Stroke (NS050641), Les Turner ALS Foundation, Vena E. Schaff ALS Research Fund, Harold Post Research Professorship, Herbert and Florence C. Wenske Foundation, The David C. Asselin MD Memorial Fund, Help America Foundation, and Les Turner ALS Foundation/Herbert C. Wenske Foundation Professor.
Disclosure: Author disclosures are provided at the end of the article.
Received August 25, 2009. Accepted in final form March 22, 2010.
REFERENCES
- 1.Pasinelli P, Brown RH. Molecular biology of amyotrophic lateral sclerosis: insights from genetics. Nat Rev Neurosci 2006;7:710–723. [DOI] [PubMed] [Google Scholar]
- 2.Valdmanis PN, Rouleau GA. Genetics of familial amyotrophic lateral sclerosis. Neurology 2008;70:144–152. [DOI] [PubMed] [Google Scholar]
- 3.Deng HX, Hentati A, Tainer JA, et al. Amyotrophic lateral sclerosis and structural defects in Cu, Zn superoxide dismutase. Science 1993;261:1047–1051. [DOI] [PubMed] [Google Scholar]
- 4.Corrado L, Ratti A, Gellera C, et al. High frequency of TARDBP gene mutations in Italian patients with amyotrophic lateral sclerosis. Hum Mutat 2009;30:688–694. [DOI] [PubMed] [Google Scholar]
- 5.Daoud H, Valdmanis PN, Kabashi E, et al. Contribution of TARDBP mutations to sporadic amyotrophic lateral sclerosis. J Med Genet 2009;46:112–114. [DOI] [PubMed] [Google Scholar]
- 6.Kabashi E, Valdmanis PN, Dion P, et al. TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat Genet 2008;40:572–574. [DOI] [PubMed] [Google Scholar]
- 7.Kuhnlein P, Sperfeld AD, Vanmassenhove B, et al. Two German kindreds with familial amyotrophic lateral sclerosis due to TARDBP mutations. Arch Neurol 2008;65:1185–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Van Deerlin VM, Leverenz JB, Bekris LM, et al. TARDBP mutations in amyotrophic lateral sclerosis with TDP-43 neuropathology: a genetic and histopathological analysis. Lancet Neurol 2008;7:409–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yokoseki A, Shiga A, Tan CF, et al. TDP-43 mutation in familial amyotrophic lateral sclerosis. Ann Neurol 2008;63:538–542. [DOI] [PubMed] [Google Scholar]
- 10.Guerreiro RJ, Schymick JC, Crews C, Singleton A, Hardy J, Traynor BJ. TDP-43 is not a common cause of sporadic amyotrophic lateral sclerosis. PLoS ONE 2008;3:e2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gitcho MA, Baloh RH, Chakraverty S, et al. TDP-43 A315T mutation in familial motor neuron disease. Ann Neurol 2008;63:535–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sreedharan J, Blair IP, Tripathi VB, et al. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 2008;319:1668–1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Winton MJ, Van Deerlin VM, Kwong LK, et al. A90V TDP-43 variant results in the aberrant localization of TDP-43 in vitro. FEBS Lett 2008;582:2252–2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rutherford NJ, Zhang YJ, Baker M, et al. Novel mutations in TARDBP (TDP-43) in patients with familial amyotrophic lateral sclerosis. PLoS Genet 2008;4:e1000193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yang Y, Hentati A, Deng HX, et al. The gene encoding alsin, a protein with three guanine-nucleotide exchange factor domains, is mutated in a form of recessive amyotrophic lateral sclerosis. Nat Genet 2001;29:160–165. [DOI] [PubMed] [Google Scholar]
- 16.Chen YZ, Bennett CL, Huynh HM, et al. DNA/RNA helicase gene mutations in a form of juvenile amyotrophic lateral sclerosis (ALS4). Am J Hum Genet 2004;74:1128–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Greenway MJ, Andersen PM, Russ C, et al. ANG mutations segregate with familial and ‘sporadic’ amyotrophic lateral sclerosis. Nat Genet 2006;38:411–413. [DOI] [PubMed] [Google Scholar]
- 18.Belzil VV, Valdmanis PN, Dion PA, et al. Mutations in FUS cause FALS and SALS in French and French Canadian populations. Neurology 2009;73:1176–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chio A, Restagno G, Brunetti M, et al. Two Italian kindreds with familial amyotrophic lateral sclerosis due to FUS mutation. Neurobiol Aging 2009;30:1272–1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ticozzi N, Silani V, Leclerc AL, et al. Analysis of FUS gene mutation in familial amyotrophic lateral sclerosis within an Italian cohort. Neurology 2009;73:1180–1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kwiatkowski TJ Jr, Bosco DA, Leclerc AL, et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 2009;323:1205–1208. [DOI] [PubMed] [Google Scholar]
- 22.Vance C, Rogelj B, Hortobagyi T, et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 2009;323:1208–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hosler BA, Siddique T, Sapp PC, et al. Linkage of familial amyotrophic lateral sclerosis with frontotemporal dementia to chromosome 9q21-q22. JAMA 2000;284:1664–1669. [DOI] [PubMed] [Google Scholar]
- 24.Morita M, Al-Chalabi A, Andersen PM, et al. A locus on chromosome 9p confers susceptibility to ALS and frontotemporal dementia. Neurology 2006;66:839–844. [DOI] [PubMed] [Google Scholar]
- 25.Valdmanis PN, Dupre N, Bouchard JP, et al. Three families with amyotrophic lateral sclerosis and frontotemporal dementia with evidence of linkage to chromosome 9p. Arch Neurol 2007;64:240–245. [DOI] [PubMed] [Google Scholar]
- 26.Vance C, Al-Chalabi A, Ruddy D, et al. Familial amyotrophic lateral sclerosis with frontotemporal dementia is linked to a locus on chromosome 9p13.2–21.3. Brain 2006;129:868–876. [DOI] [PubMed] [Google Scholar]
- 27.Yan J, Siddique N, Slifer S, et al. A major novel locus for ALS/FTD on chromosome 9p21 and its pathological correlates. Neurology 2006;67:186. [Google Scholar]
- 28.Brooks BR, Subcommittee on Motor Neuron Diseases/Amyotrophic Lateral Sclerosis of the World Federation of Neurology Research Group on Neuromuscular Diseases and the El Escorial “Clinical limits of amyotrophic lateral sclerosis” workshop contributors. El Escorial World Federation of Neurology criteria for the diagnosis of amyotrophic lateral sclerosis. J Neurol Sci 1994;124(suppl):96–107. [DOI] [PubMed]
- 29.Neary D, Snowden JS, Gustafson L, et al. Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology 1998;51:1546–1554. [DOI] [PubMed] [Google Scholar]
- 30.Crozat A, Aman P, Mandahl N, Ron D. Fusion of CHOP to a novel RNA-binding protein in human myxoid liposarcoma. Nature 1993;363:640–644. [DOI] [PubMed] [Google Scholar]
- 31.Rabbitts TH, Forster A, Larson R, Nathan P. Fusion of the dominant negative transcription regulator CHOP with a novel gene FUS by translocation t(12;16) in malignant liposarcoma. Nat Genet 1993;4:175–180. [DOI] [PubMed] [Google Scholar]
- 32.Pereira DS, Dorrell C, Ito CY, et al. Retroviral transduction of TLS-ERG initiates a leukemogenic program in normal human hematopoietic cells. Proc Natl Acad Sci USA 1998;95:8239–8244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lagier-Tourenne C, Cleveland DW. Rethinking ALS: the FUS about TDP-43. Cell 2009;136:1001–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Neumann M, Roeber S, Kretzschmar HA, Rademakers R, Baker M, Mackenzie IR. Abundant FUS-immunoreactive pathology in neuronal intermediate filament inclusion disease. Acta Neuropathol 2009;118:605–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Deng HX, Shi Y, Furukawa Y, et al. Conversion to the amyotrophic lateral sclerosis phenotype is associated with intermolecular linked insoluble aggregates of SOD1 in mitochondria. Proc Natl Acad Sci USA 2006;103:7142–7147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hicks GG, Singh N, Nashabi A, et al. Fus deficiency in mice results in defective B-lymphocyte development and activation, high levels of chromosomal instability and perinatal death. Nat Genet 2000;24:175–179. [DOI] [PubMed] [Google Scholar]
- 37.Kuroda M, Sok J, Webb L, et al. Male sterility and enhanced radiation sensitivity in TLS(−/−) mice. EMBO J 2000;19:453–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Juneja T, Pericak-Vance MA, Laing NG, Dave S, Siddique T. Prognosis in familial amyotrophic lateral sclerosis: progression and survival in patients with glu100gly and ala4val mutations in Cu, Zn superoxide dismutase. Neurology 1997;48:55–57. [DOI] [PubMed] [Google Scholar]
- 39.Aoki M, Ogasawara M, Matsubara Y, et al. Mild ALS in Japan associated with novel SOD mutation. Nat Genet 1993;5:323–324. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.