

Abstract
Pheochromocytoma and paraganglioma are rare tumors of adrenals as well as the sympathetic and parasympathetic paraganglia. Clinical presentation of these tumors depends on localization, secretory profile and malignant potential. Four distinct syndromes - PGL1–4 - are related to mutations in the succinate dehydrogenase gene – mitochondrial complex involved in electron transfer and Krebs cycle. Here we describe etiology, genetics, as well as clinical aspects of SDH-related tumors. We also describe recent discoveries in HIF-related pathway of tumorigenesis and mutations in new SDH-related genes that have improved our understanding of this disease.
Keywords: Pheochromocytoma, Paraganglioma, Succinate dehydrogenase, Adrenal, Paragangia, Succinate dehydrogenase subunit A, Succinate dehydrogenase subunit B, Succinate dehydrogenase subunit C, Succinate dehydrogenase subunit D, Succinate dehydrogenase subunit 5, Malignant pheochromocytoma and paragangioma, Familial pheochromocytoma and paragangioma
Introduction
Pheochromocytomas (PHEOs) and paragangliomas (PGLs) are rare neuroendocrine tumors with an estimated prevalence of 1:4500 and 1:1700 and with an annual incidence of 3 to 8 cases per 1 million per year in the general population (1). PHEOs/PGLs arise from three anatomically and functionally distinct parts of the neural crest derived sympatho-adrenal system: adrenal medulla, sympathetic, and parasympathetic paraganglia. PHEOs are derived from the adrenal gland; PGLs arise from parasympathetic- (most commonly along the cranial nerves (e.g., glomus tumors, chemodectoma, and carotid body tumor) and from extra-adrenal sympathetic-associated chromaffin tissues (often designated as extra-adrenal PHEOs). The later are diffusely scattered throughout the human body explaining why these tumors may be found practically in any location. Historic use of chromaffin positivity as a universal defining feature of PHEOs/PGLs is somewhat misleading because parasympathetic PGLs are either weakly positive or negative on chromaffin staining. However, these tumors are histologically identical to adrenal medulla or sympathetic PGLs and, as recently described, are capable of catecholamine production (2).
Between a quarter and one third of PHEO/PGL cases have familial etiology (3, 4); this group is heterogeneous and includes following syndromes: von Hippel-Lindau (VHL), multiple endocrine neoplasia type 2 (MEN2), neurofibromatosis type 1 (NF1) and SDH mutation-related tumors. The two most recent additions to this group include mutations in SDH5 and propyl hydroxylase domain 2 (PDH2) genes (5, 6) that, although extremely rare, are also associated with PHEOs/PGLs. VHL, SDH and PDH-related PHEOs/PGLs seem to share the same pseudohypoxia/angiogenesis pathways throughout tumorigenesis (7). Despite the fact that different SDH mutations occur in single multi-unit enzyme, they express significant phenotype heterogeneity. Their prevalence is estimated to lie between 10–30% of PGLs (8), currently not well-known in apparently sporadic adrenal PHEOs. Historically, parasympathetic glomus tumors were not seen as part of catecholamine-secreting sympathetic PHEO/PGL syndrome, which caused some confusion with systematization of disease. Nevertheless, later unifying classification or these tumors and discovery of SDH-related PHEO/PGL syndrome propelled our understanding of tumorigenesis of PHEO and PGL in general.
Initial suggestion of genetic clustering of glomus tumors date back to the 1930s with Kroll et al (9) in 1964 being one of the first to suggest a genetic predisposition to glomus tumor development (10, 11). Studying a Dutch cohort with head and neck PGLs (HNPGL), in 1989 van der May et al found the mode of inheritance to be autosomal dominant with maternal imprinting in some cases (12). Later, several familial clusters of PHEO/PGL were described and defined as paraganglioma syndromes PGL1 through PGL4. Further testing revealed PGL1 to be related to mutation in the SDHD gene on 11q23 (Table 1), PGL2 to SDH5 on 11q13.1, PGL3 to mutation in SDHC on 1q21 and PGL4 to SDHB on 1p35–p36.1 (5, 13–15).
Table 1.
Characteristics of SDH-related PHEO/PGL
| SDH subunit | SDHA | SDHB | SDHC | SDHD | SDH5 |
|---|---|---|---|---|---|
| Chr/Gene | 5p15 | 1p35-p36.1 | 1q21 | 11q23 | 11q13.1 |
| Protein | 70 kDa/664 aa | 27 kDa/280 aa | 15 kDa/169 aa | 12 kDa/159 aa | 167 aa |
| Clinical features | Necrotizing encephalopathy | PHEO/PGL | PHEO/PGL | PHEO/PGL | PHEO/PGL |
| PGL Syndrome | Leigh | PGL4 | PGL3 | PGL1 | PGL2 |
| Function | Substrate binding, catalysis | Catalysis, electron transfer | Anchoring, ubiquinon binding | Anchoring, ubiquinon binding | Flavination of SDHA |
| Inheritance | Autosomal recessive | Autosomal dominant | Autosomal dominant | Maternal imprinting | Autosomal dominant |
| Malignancy risk | none | high | low | low | low |
| Multifocal disease | frequent | frequent |
Normal physiology of the SDH complex
Succinate dehydrogenase is a mitochondrial enzyme (also called as the mitochondrial complex II), carrying dual function in the process of mitochondrial energy generation. It is anchored to the inner mitochondrial membrane and consists of four functionally different subunits: A, B, C, and D (Fig. 1 and Table 1). SDHA is a hydrophilic flavoprotein that serves as substrate binding site and together with another hydrophilic SDHB forms the catalytic part of the enzyme (16). Hydrophobic SDHC and SDHD serve as membrane anchors and ubiquinone binding site. SDH differs significantly from other members of the mitochondrial electron transfer chain. It is the only non-transmembrane complex and has no ability to transfer hydrogen ions through the inner membrane into the intermembrane space. On the other hand, it is the only member of the chain that has an additional role that consists of active participation in the tricarboxylic acid (citric acid, Krebs) cycle. Putting these physiologic phenomena into practical perspective will generate following:
-
Oxidative phosphorylation
Electron transfer chain (ET, oxidative phosphorylation pathway) is represented by 5 protein complexes, responsible for electron transfer along the affinity gradient chain that allows H+ transport from mitochondrial matrix into the mitochondrial intermembrane space.
H+ transport generates enough transmembrane gradient to allow ATP synthase to generate ATP from ADP.
This pathway generates significantly more ATP compared to glycolysis (34 versus 2 per each glucose molecule) and provides the majority of cell's energy requirements.
Malignant cells may preferably increase anaerobic glycolysis (Warburg effect) as an additional adaptation mechanism for prevention of relative tissue hypoxia in rapidly growing tumors.
-
SDH complex
SDH, on the other hand, converts succinate to fumarate (as part of Krebs cycle) and captures/transfers electron (as part of electron-transfer chain) and plays dual role in cellular energy homeostasis.
SDH functional duality is represented by the fact that it plays an important role in both the glycolytic pathway and electron transfer.
-
Apparent paradox
Although one could imagine the electron transfer chain (ETC) to represent an electron transfer consecutively through complexes I → II → III → IV → V, both complexes I and II provide complex III with a single electron that was acquired from different respective sources – NADH and FADH (see Figure 1). This phenomenon of “double electron supplier”, suggests an evolutionary need for crosstalk between oxidative phosphorylation and glycolysis. To not underestimate the need for apparently secondary importance of the SDH-related part of the chain, one should remember that inactivating mutation in SDHA causes the same necrotizing encephalopathy (Leigh syndrome) as mutation in any other complex of the ETC.
With this said, one can hardly not recognize a significant difference between SDH and other parts of the ETC. It is the only non transmembrane complex, the only non-proton pump, and while the main goal of electron transfer is to generate enough H+-driven gradient for ATP synthesis, the hydroxylation of succinate to fumarate actually generates intra-matrix H+ that can actually diminish that gradient.
Figure 1.

Q Ubiquinon
I–V ECT complexes
C Cytochrome c
H+ Hydrogen ion
ADP/ATP Adenosine di/triphosphate
S succinate
F fumarate
A, B, C, D Succinate dehydrogenase subunits
SDH-related genetics and tumorigenesis
Three out of four subunits of SDH complex are strongly associated with PHEO/PGL development. In contrast, mutation in the catalytic subunit SDHA causes necrotizing encephalopathy, which is caused by other mutations in genes involved in energy metabolism, such as the rest of the members of electron transfer chain and components of pyruvate dehydrogenase complex (17). Genes for SDHC, B, and D are nuclear and the mode of inheritance is autosomal dominant with maternal imprinting for SDHD and inactivating germline mutations result in loss of function of SDH. The precise mechanism of maternal imprinting in SDHD is still debated (11).
As noted above, SDH-related tumorigenesis is believed to associate with HIF/angiogenesis pathway, as has been suggested by Dahia (7, 18). Apart from classic VHL and SDH-mutation associated PHEO/PGL, tumorigenesis pathway includes recently discovered mutations along this pathway like PDH2 and possibly KIF1B (6, 19). This suggests that other proteins related to the HIF pathway may later be found to associate with PHEO/PGL. Thus, for SDH-related mutations, decrease in fumarate production and accumulation of succinate would be expected with a significant decrease in the amount of SDH subunits. This decrease of protein with resultant severe impairment of the SHD function and, therefore, an elevation in succinate levels were shown in SDHB and SDHD PHEOs/PGLs (20–22). The accumulated succinate diffuses back to the cytoplasm and inhibits prolyl hydroxylases (PHDs), resulting in further stabilization of the HIF (23). There seems to be even more complex metabolic relationship between SDH and HIF pathways (24). One would expect a phenotype similar to VHL, which is only partly true (e.g. noradrenergic phenotype). Another interesting observations on genotype-phenotype correlation show significant differences in clinical presentation among mutations in different SDH subunits. While all SDH-related mutations result in near abolished SDH complex activity (25), SDHB related tumors have a significant malignant potential and are commonly found in the abdomen compared to SDHD and SDHC tumors that are almost always benign and commonly found in the head and neck areas. One can argue that mutation in the catalytic subunit should prevent the complex from any enzymatic activity, while inability to anchor would retain some function within mitochondrial matrix. Suggestion that increased malignant potential of SDHB tumors might be related to the Warburg effect might not hold true because it seems to be pronounced in VHL rather than SDH related tumors (26). This could reflect a compensatory mechanism in cases of HIF-driven pseudohypoxia with intact machinery for both oxidative phosphorylation and electron transfer. On the other hand, Dahia showed in an animal model of heterozygous SDHD mutation that there is an early upregulation of glycolytic enzymes that differ from classic late Warburg effect (18). Favier et al. (26) found Warburg effect to be mostly expressed in VHL-related tumors, while SDH counterparts showed increased expression of glucose and hexose transporters.
Clinical presentation of SDH-related tumors
Numerous large multi-institutional or multi-national studies and reviews of national registries have concentrated on the relative prevalence of different SDH related PHEOs/PGLs, as well as their specific clinical features and malignant potential (8, 27–29). In a recent large prospective study, Burnichon et al. (30) supported previous findings of the high frequency of SDHD and SDHC mutations in head and neck paragangliomas, as well as higher frequency of abdominal and pelvic disease and overall malignancy in SDHB mutations. Both mediastinal PGLs and PGLs of the large para-aortic paraganglion, described by Emil Zuckerkandl, usually associate with SDHB or SDHD mutations (31, 32). Pediatric PHEOs/PGLs seem to be familial to greater degree than tumors in adults, mostly related to SDHB mutation and manifest with more aggressive course – higher rate of malignancy and more metastatic disease on presentation (Pacak et al. unpublished).
A. SDHB
SDHB mutations predispose to mainly extra-adrenal PHEOs with a high malignant potential and, less frequently, to benign parasympathetic head and neck PGLs (27, 28, 33–36). Despite the fact that 2/3 of SDHB patients present with extra-adrenal abdominal or thoracic disease, primary presentation with adrenal PHEOs is still evident in these patients (30). Another 1/3 of patients present with multifocal disease. The mean age at diagnosis of SDHB-related PGL is about 30 years of age, however, there are several studies reporting a significantly younger age (37, 38). By age 40, 45% of SDHB mutation carriers show clinical signs of the disease (29). Malignancy rates usually vary between 34–70% (27, 28, 35). If malignant sympathetic PGL arises from an extra-adrenal primary tumor, the prevalence of SDHB mutation can reach close to 50% (33).
Although most SDHB related tumors present with catecholamine excess, about 10% of SDHB related tumors are “biochemically silent” or produce only dopamine (35, 39). Morbidity of the disease is most likely related to metastatic disease, rather than hypercatecholaminemia per se. Younger age or metastatic disease at presentation, larger tumor size, and, to lesser extend, dopaminergic secretory profile and misssense mutation seem to associate with more aggressive tumor behavior, but there is no clear phenotype/genotype correlation (34, 35).
B. SDHD
SDHD related PHEO/PGL was first SDH-associate mutation to be found, in part due to the large carrier cohort with Dutch founder mutation (13). SDHD mutations are typically associated with multifocal parasympathetic head and neck PGLs and usually benign extra-adrenal and adrenal PHEOs (27, 34). A German-Polish cohort of 34 patients with SDHD mutation showed that 53% of carriers had adrenal PHEO, while 79% had head and neck PGLs (27). Metastatic pheochromocytoma is rare in SDHD mutation carriers for both sympathetic and parasympathetic disease, but can occur (36, 40). For example, the D92Y cofounder mutation was described to be associated with malignant course of the disease, but additional mutations with malignant potential have been described (41). SDHD-related PHEO/PGL express more genotype-phenotype correlation, with reports of nonsense and splicing mutations being associated with earlier disease development and the presence of PHEO in addition to head and neck PGLs (42). One can argue that these mutations generate truncated protein or no protein, while a missense mutation produces proteins with some remaining function.
C. SDHC
SDHC mutations are rare, and are almost exclusively associated with parasympathetic head and neck PGL (8), although rare cases of SDHC-associated extra-adrenal PHEO have been reported (43, 44). Although sympathetic head and neck PGLs usually clinically behave like benign sporadic counterparts, Niemann et al (45) described a patient with malignant catecholamine-producing PGL of carotid bifurcation.
D. SDH5 (SDHAF2)
Recent developments in PGL2 represent a great finale for two seemingly independent stories. PGL2 was described in 1980s in a large Dutch kindred with head and neck paragangliomas (46). It was clearly familial and initially mapped to chromosome 11, but as other PGL syndromes were successfully found to be related to specific gene mutations, one thing seem to hold in relation to PGL2: it was not related to mutation in SDHA subunit – the only subunit left without mutation causing PHEO/PGL. On the other hand, while a Utah group was working on characterization of mammalian mitochondrial proteome, they found in yeast model that SDH5 protein is heavily involved in flavination of SDHA subunit and its' mutation dramatically affects SDHA function (15). Low and behold, when they decided to test the PGL2 kindred, mutation in SHD5 at G78R was found in 45 members of this lineage with disease haplotype and none in controls or lineage members without disease and without disease-specific haplotype. Furthemore, mutation in SDH5 gene results in about 95% decrease in flavination (attachment of FAD) of the SDHA and significant decrease in the overall SDH activity. Out of 45, 33 subjects developed disease, while 11 did not. Of those 11, 7 have signs of SDHD-type maternal imprinting, while the other 5 with paternal mutation might not developed disease yet, probably because of age-related penetrance.
Despite excitement following this discovery and the fact that it advanced further our understanding of the SDH complex, overall SDH5 mutation are very rare, with only one additional family described thus far (47, 48).
Diagnosis and treatment of SDH-related tumors
SDH-related PHEO/PGL represent a significant diagnostic challenge, because of several disease-specific issues. SDH PGL is different from PHEO in general in the fact that tissue expression of PMNT in these tumors is minimal, which means that the preferential catecholamine production is norepinephrine or dopamine (35, 49, 50). SDHB tumors may also present as biochemically silent due to the lack of tyrosine hydroxylase (51). On the other hand, parasympathetic head and neck PGLs may present with space-occupying complaints as a leading symptom although recently, a frequent production of dopamine has been described in these tumors, nevertheless with no clinical consequences (see above). Biochemical diagnosis of SDH-related PHEO/PGL follows expert recommendations from the International Symposium on Pheochromocytoma for initial biochemical testing that includes measurements of fractionated metanephrines in urine or plasma, or both, as available (52, 53). For non-secreting parasympathetic, as well as secreting sympathetic tumors, measurement of chromogranin A can be used as secondary diagnostic and follow up marker. Genetic testing should be carried out in family members with SDH-related tumors, all cases of malignant PHEO/PGL, early onset or multifocal disease, as well as in all cases of head and neck paragangliomas.
SDH-related tumor localization studies should be carried out after biochemical diagnosis suggested existing disease in agreement with diagnostic approach to other PHEOs/PGLs. It should be stressed that in non-secreting PGLs, imaging should be initiated whenever PGL is suspected (e.g. palpable nodule in the neck, family history). Both CT scan and MRI techniques are sensitive anatomical imaging modalities (54), but lack specificity. Functional imaging techniques, on the other hand, are able to show functional tissue based on following physiologic phenomena: a] 123I-metaiodobenzylguanidine (MIBG) and 18F-fluorodopamine scintigraphy – images abnormal tissue through isotope uptake by cell membrane norepinephrine transporters; b] 18F-fluorodihydroxyphenylalanine positron emission tomography (PET) that images cells through catecholamine-production-related amino acid uptake; and c] 18F-fluorodeoxyglucose PET shows tumors through isotope uptake through glucose transporters and hexokinase activity. The last imaging modality, although it lacks tissue specificity, is efficient in showing metabolically active tissues and is especially efficient in aggressive malignant disease such as SDHB-related PGL (so called “flip-flop” phenomenon) (35). Although preferential aerobic glucolysis (Warburg effect) was thought to explain this phenomenon, a recent study showed it to be preferentially expressed in VHL compared to SDH-related tumors (26), which suggests that increased glucose uptake in the later may be related to increases in glucose and hexose transporters rather than to changes in glycolysis. Other imaging modalities include 111In-pentetreotide scintigraphy, another less specific technique, based on cellular expression of somatostatin receptors, which may be useful in patients with metastatic disease (27, 34). Differential expression of different subtypes of somatostatin receptor may improve sensitivity of this technique in the future. Another major development in somatostatin receptor scintigraphy is development of new chelating agents that allow use of novel isotopes. Two major restrictions, on the other hand, are availability of strong nuclear radiochemist and state/facility restrictions on use of particular isotopes.
The mainstay in the treatment any PHEO/PGL is surgical resection, which can be done either conventionally or through laparoscopy. Pre-operative management is aimed at the prevention of catecholamine-induced complications like hypertrnsive crisis and cardiac arrythmias. It is imperative to remember that head and neck PGLs may be either syptathetic or parasympathetic and ratain ability to synthesize catecholamines, which makes adequate testing and appropriate pre-oprative preparation absolutely necessary. With adequate pharmacological preparation, operative mortality is down to below 1% (55). For sympathetic PHEO/PGL authors prefer to combine an α-adrenoceptor blocking agent (phenoxybenzamine, doxazosin, prazosin, and others), a β-adrenoceptor blocking agent (propranolol, atenolol), and α-methyl-paratyrosine, an inhibitor of catecholamine synthesis. Life-expectancy after surgery of benign PGL, in general is either normal (56) or mildly decreased (57) as compared with the general population. Post-operative follow-up with bi-annual measurement of blood pressure and annual plasma or urine metanephrines, as well as annual MRI or CT of the neck, thorax, abdomen, and pelvis should continue indefinitely. Patients with larger tumors (≥ 5 cm) should initially be followed every 3 months. The value of chromogranin A as a follow up marker is still debated, but in cases with significant preoperative values, it could probably represent initial tumor burden and could turn out to be of good sensitivity.
There is no effective treatment for malignant tumors. The usefulness of surgical debulking of tumor tissue has not been established. Treatment with therapeutic doses of 131I-MIBG or combination chemotherapy (cyclophosphamide, vincristine, and darcabazine) may induce (partial) responses (58–60). High dose of 131I-MIBG High doses of 131I-MIBG are associated with higher remission and cure rates than lower doses (60). In addition, a new highly specific activity Ultratrace™ [131I]-iobenguane (Azedra, Molecular Insight Pharmaceuticals Inc.) is undergoing a Phase II clinical trial and preliminary data is promising (61). External beam irradiation can be useful in the treatment of local tumor complications. Symptomatic treatment by decreasing catecholamine burden can be obtained with α-adrenergic blockers and α-methyl-paratyrosine. The prognosis after diagnosis of metastatic PGL is highly variable, with an estimated 5-year survival rate of 50% (62). Specific data on SDHB-associated malignant PGL are lacking. In our experience, survival among these patients is highly variable as well. In one case, prolonged survival exceeding 30 years has been reported (63).
Summary.
SDH-related PHEO/PGL are rare, but potentially lethal conditions
SDH is a mitochondrial complex participating in both electron transfer and the Krebs cycle
There are 4 PGL syndromes, related to mutations of different succinate dehydrogenase subunits
Biochemically these tumors can be secretory or silent
Some of these tumors – especially SDHB-related tumors – carry significant malignant potential
Figure 2.
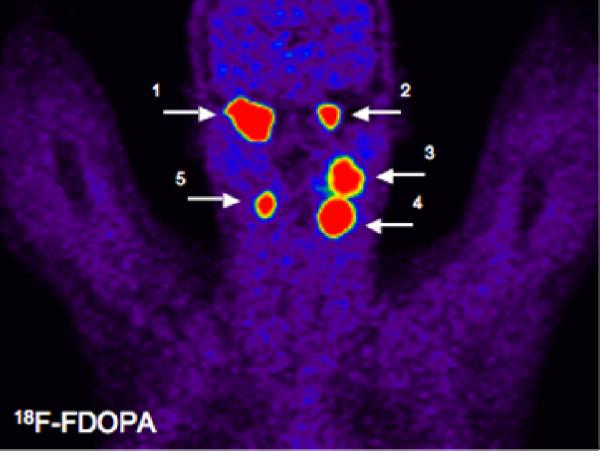
18F-FDOPA was used to localize five head and neck paragangliomas: Right (1) and left (2) glomus jugulare tumors; a left glomus vagale tumor (3); and left (4) and right (5) carotid body tumors.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Pacak K, Chrousos GP, Koch CA, Lenders JW, Eisenhofer G. Pheochromocytoma: progress in diagnosis, therapy, and genetics. In: Margioris A, Chrousos GP, editors. Adrenal Disorders. 1 ed. Vol 1. Humana Press; Totowa: 2001. pp. 479–523. [Google Scholar]
- 2.van Duinen N, Steenvoorden D, Kema IP, et al. Increased urinary excretion of 3-methoxytyramine in patients with head and neck paragangliomas. The Journal of clinical endocrinology and metabolism. 2010 Jan;95(1):209–214. doi: 10.1210/jc.2009-1632. [DOI] [PubMed] [Google Scholar]
- 3.Neumann HP, Bausch B, McWhinney SR, et al. Germ-line mutations in nonsyndromic pheochromocytoma. The New England journal of medicine. 2002 May 9;346(19):1459–1466. doi: 10.1056/NEJMoa020152. [DOI] [PubMed] [Google Scholar]
- 4.Erlic Z, Rybicki L, Peczkowska M, et al. Clinical predictors and algorithm for the genetic diagnosis of pheochromocytoma patients. Clin Cancer Res. 2009 Oct 15;15(20):6378–6385. doi: 10.1158/1078-0432.CCR-09-1237. [DOI] [PubMed] [Google Scholar]
- 5.Niemann S, Muller U. Mutations in SDHC cause autosomal dominant paraganglioma, type 3. Nat Genet. 2000;26(3):268–270. doi: 10.1038/81551. [DOI] [PubMed] [Google Scholar]
- 6.Ladroue C, Carcenac R, Leporrier M, et al. PHD2 mutation and congenital erythrocytosis with paraganglioma. The New England journal of medicine. 2008 Dec 18;359(25):2685–2692. doi: 10.1056/NEJMoa0806277. [DOI] [PubMed] [Google Scholar]
- 7.Dahia PL, Hao K, Rogus J, et al. Novel pheochromocytoma susceptibility loci identified by integrative genomics. Cancer Res. 2005 Nov 1;65(21):9651–9658. doi: 10.1158/0008-5472.CAN-05-1427. [DOI] [PubMed] [Google Scholar]
- 8.Schiavi F, Boedeker CC, Bausch B, et al. Predictors and prevalence of paraganglioma syndrome associated with mutations of the SDHC gene. Jama. 2005 Oct 26;294(16):2057–2063. doi: 10.1001/jama.294.16.2057. [DOI] [PubMed] [Google Scholar]
- 9.Kroll AJ, Alexander B, Cochios F, Pechet L. Hereditary Deficiencies of Clotting Factors Vii and X Associated with Carotid-Body Tumors. The New England journal of medicine. 1964 Jan 2;270:6–13. doi: 10.1056/NEJM196401022700102. [DOI] [PubMed] [Google Scholar]
- 10.Eng C, Kiuru M, Fernandez MJ, Aaltonen LA. A role for mitochondrial enzymes in inherited neoplasia and beyond. Nature reviews. 2003 Mar;3(3):193–202. doi: 10.1038/nrc1013. [DOI] [PubMed] [Google Scholar]
- 11.Pigny P, Cardot-Bauters C. Genetics of pheochromocytoma and paraganglioma: New developments. Annales d'endocrinologie. 2009 Dec 21; doi: 10.1016/j.ando.2009.11.004. [DOI] [PubMed] [Google Scholar]
- 12.van der Mey AG, Maaswinkel-Mooy PD, Cornelisse CJ, Schmidt PH, van de Kamp JJ. Genomic imprinting in hereditary glomus tumours: evidence for new genetic theory. Lancet. 1989 Dec 2;2(8675):1291–1294. doi: 10.1016/s0140-6736(89)91908-9. [DOI] [PubMed] [Google Scholar]
- 13.Baysal BE, Ferrell RE, Willett-Brozick JE, et al. Mutations in SDHD, a mitochondrial complex II gene, in hereditary paraganglioma. Science (New York, NY. 2000;287(5454):848–851. doi: 10.1126/science.287.5454.848. [DOI] [PubMed] [Google Scholar]
- 14.Astuti D, Latif F, Dallol A, et al. Gene mutations in the succinate dehydrogenase subunit SDHB cause susceptibility to familial pheochromocytoma and to familial paraganglioma. American journal of human genetics. 2001 Jul;69(1):49–54. doi: 10.1086/321282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hao HX, Khalimonchuk O, Schraders M, et al. SDH5, a gene required for flavination of succinate dehydrogenase, is mutated in paraganglioma. Science (New York, NY. 2009 Aug 28;325(5944):1139–1142. doi: 10.1126/science.1175689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yankovskaya V, Horsefield R, Tornroth S, et al. Architecture of succinate dehydrogenase and reactive oxygen species generation. Science (New York, NY. 2003 Jan 31;299(5607):700–704. doi: 10.1126/science.1079605. [DOI] [PubMed] [Google Scholar]
- 17.Dahl HH. Getting to the nucleus of mitochondrial disorders: identification of respiratory chain-enzyme genes causing Leigh syndrome. American journal of human genetics. 1998 Dec;63(6):1594–1597. doi: 10.1086/302169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Qin Y, Buddavarapu K, Dahia PL. Pheochromocytomas: from genetic diversity to new paradigms. Hormone and metabolic research = Hormon- und Stoffwechselforschung = Hormones et metabolisme. 2009 Sep;41(9):664–671. doi: 10.1055/s-0029-1215590. [DOI] [PubMed] [Google Scholar]
- 19.Schlisio S, Kenchappa RS, Vredeveld LC, et al. The kinesin KIF1Bbeta acts downstream from EglN3 to induce apoptosis and is a potential 1p36 tumor suppressor. Genes & development. 2008 Apr 1;22(7):884–893. doi: 10.1101/gad.1648608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gimenez-Roqueplo AP, Favier J, Rustin P, et al. The R22X mutation of the SDHD gene in hereditary paraganglioma abolishes the enzymatic activity of complex II in the mitochondrial respiratory chain and activates the hypoxia pathway. American journal of human genetics. 2001 Dec;69(6):1186–1197. doi: 10.1086/324413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gimenez-Roqueplo AP, Favier J, Rustin P, et al. Functional consequences of a SDHB gene mutation in an apparently sporadic pheochromocytoma. The Journal of clinical endocrinology and metabolism. 2002 Oct;87(10):4771–4774. doi: 10.1210/jc.2002-020525. [DOI] [PubMed] [Google Scholar]
- 22.Pollard PJ, Briere JJ, Alam NA, et al. Accumulation of Krebs cycle intermediates and over-expression of HIF1alpha in tumours which result from germline FH and SDH mutations. Human molecular genetics. 2005 Aug 1;14(15):2231–2239. doi: 10.1093/hmg/ddi227. [DOI] [PubMed] [Google Scholar]
- 23.Selak MA, Armour SM, MacKenzie ED, et al. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-alpha prolyl hydroxylase. Cancer cell. 2005 Jan;7(1):77–85. doi: 10.1016/j.ccr.2004.11.022. [DOI] [PubMed] [Google Scholar]
- 24.Bayley JP, Devilee P. Warburg tumours and the mechanisms of mitochondrial tumour suppressor genes. Barking up the right tree? Current opinion in genetics & development. 2010 Mar 19; doi: 10.1016/j.gde.2010.02.008. [DOI] [PubMed] [Google Scholar]
- 25.van Nederveen FH, Gaal J, Favier J, et al. An immunohistochemical procedure to detect patients with paraganglioma and phaeochromocytoma with germline SDHB, SDHC, or SDHD gene mutations: a retrospective and prospective analysis. The lancet oncology. 2009 Aug;10(8):764–771. doi: 10.1016/S1470-2045(09)70164-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Favier J, Briere JJ, Burnichon N, et al. The warburg effect is genetically determined in inherited pheochromocytomas. PloS one. 2009;4(9):e7094. doi: 10.1371/journal.pone.0007094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Neumann HP, Pawlu C, Peczkowska M, et al. Distinct clinical features of paraganglioma syndromes associated with SDHB and SDHD gene mutations. JAMA. 2004 Aug 25;292(8):943–951. doi: 10.1001/jama.292.8.943. [DOI] [PubMed] [Google Scholar]
- 28.Amar L, Bertherat J, Baudin E, et al. Genetic testing in pheochromocytoma or functional paraganglioma. J Clin Oncol. 2005 Dec 1;23(34):8812–8818. doi: 10.1200/JCO.2005.03.1484. [DOI] [PubMed] [Google Scholar]
- 29.Benn DE, Richardson AL, Marsh DJ, Robinson BG. Genetic testing in pheochromocytoma- and paraganglioma-associated syndromes. Annals of the New York Academy of Sciences. 2006 Aug;1073:104–111. doi: 10.1196/annals.1353.011. [DOI] [PubMed] [Google Scholar]
- 30.Burnichon N, Rohmer V, Amar L, et al. The succinate dehydrogenase genetic testing in a large prospective series of patients with paragangliomas. The Journal of clinical endocrinology and metabolism. 2009 Aug;94(8):2817–2827. doi: 10.1210/jc.2008-2504. [DOI] [PubMed] [Google Scholar]
- 31.Van Nederveen FH, Dinjens WN, Korpershoek E, De Krijger RR. The occurrence of SDHB gene mutations in pheochromocytoma. Annals of the New York Academy of Sciences. 2006 Aug;1073:177–182. doi: 10.1196/annals.1353.018. [DOI] [PubMed] [Google Scholar]
- 32.Ghayee HK, Havekes B, Corssmit EP, et al. Mediastinal paragangliomas: association with mutations in the succinate dehydrogenase genes and aggressive behavior. Endocrine-related cancer. 2009 Mar;16(1):291–299. doi: 10.1677/ERC-08-0214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brouwers FM, Eisenhofer G, Tao JJ, et al. High Frequency of SDHB Germline Mutations in Patients with Malignant Catecholamine-Producing Paragangliomas: Implications for Genetic Testing. The Journal of clinical endocrinology and metabolism. 2006 Nov;91(11):4505–4509. doi: 10.1210/jc.2006-0423. [DOI] [PubMed] [Google Scholar]
- 34.Benn DE, Gimenez-Roqueplo AP, Reilly JR, et al. Clinical presentation and penetrance of pheochromocytoma/paraganglioma syndromes. The Journal of clinical endocrinology and metabolism. 2006 Mar;91(3):827–836. doi: 10.1210/jc.2005-1862. [DOI] [PubMed] [Google Scholar]
- 35.Timmers HJ, Kozupa A, Eisenhofer G, et al. Clinical presentations, biochemical phenotypes, and genotype-phenotype correlations in patients with succinate dehydrogenase subunit B-associated pheochromocytomas and paragangliomas. The Journal of clinical endocrinology and metabolism. 2007 Mar;92(3):779–786. doi: 10.1210/jc.2006-2315. [DOI] [PubMed] [Google Scholar]
- 36.Havekes B, Corssmit EP, Jansen JC, van der Mey AG, Vriends AH, Romijn JA. Malignant paragangliomas associated with mutations in the succinate dehydrogenase D gene. The Journal of clinical endocrinology and metabolism. 2007 Apr;92(4):1245–1248. doi: 10.1210/jc.2006-1993. [DOI] [PubMed] [Google Scholar]
- 37.Pham TH, Moir C, Thompson GB, et al. Pheochromocytoma and paraganglioma in children: a review of medical and surgical management at a tertiary care center. Pediatrics. 2006 Sep;118(3):1109–1117. doi: 10.1542/peds.2005-2299. [DOI] [PubMed] [Google Scholar]
- 38.Prodanov T, Havekes B, Nathanson KL, Adams KT, Pacak K. Malignant paraganglioma associated with succinate dehydrogenase subunit B in an 8-year-old child: the age of first screening? Pediatr Nephrol. 2009 Feb 3; doi: 10.1007/s00467-008-1111-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Timmers H, Gimenez-Roqueplo AP, Mannelli M, Pacak K. Clinical aspects of SDHx-related pheochromocytoma and paraganglioma. Endocrine-related cancer. 2009 Feb 3; doi: 10.1677/ERC-08-0284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Timmers HJ, Pacak K, Bertherat J, et al. Mutations associated with succinate dehydrogenase d-related malignant paragangliomas. Clinical endocrinology. 2007 Oct 31; doi: 10.1111/j.1365-2265.2007.03086.x. [DOI] [PubMed] [Google Scholar]
- 41.Timmers HJ, Pacak K, Bertherat J, et al. Mutations associated with succinate dehydrogenase D-related malignant paragangliomas. Clinical endocrinology. 2008 Apr;68(4):561–566. doi: 10.1111/j.1365-2265.2007.03086.x. [DOI] [PubMed] [Google Scholar]
- 42.Astrom K, Cohen JE, Willett-Brozick JE, Aston CE, Baysal BE. Altitude is a phenotypic modifier in hereditary paraganglioma type 1: evidence for an oxygen-sensing defect. Human genetics. 2003 Aug;113(3):228–237. doi: 10.1007/s00439-003-0969-6. [DOI] [PubMed] [Google Scholar]
- 43.Mannelli M, Ercolino T, Giache V, Simi L, Cirami C, Parenti G. Genetic screening for pheochromocytoma: should SDHC gene analysis be included? J Med Genet. 2007 Sep;44(9):586–587. doi: 10.1136/jmg.2007.051045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Peczkowska M, Cascon A, Prejbisz A, et al. Extra-adrenal and adrenal pheochromocytomas associated with a germline SDHC mutation. Nat Clin Pract Endocrinol Metab. 2008 Feb;4(2):111–115. doi: 10.1038/ncpendmet0726. [DOI] [PubMed] [Google Scholar]
- 45.Niemann S, Muller U, Engelhardt D, Lohse P. Autosomal dominant malignant and catecholamine-producing paraganglioma caused by a splice donor site mutation in SDHC. Human genetics. 2003 Jul;113(1):92–94. doi: 10.1007/s00439-003-0938-0. [DOI] [PubMed] [Google Scholar]
- 46.van Baars F, Cremers C, van den Broek P, Geerts S, Veldman J. Genetic aspects of nonchromaffin paraganglioma. Human genetics. 1982;60(4):305–309. doi: 10.1007/BF00569208. [DOI] [PubMed] [Google Scholar]
- 47.Bayley JP, Kunst HP, Cascon A, et al. SDHAF2 mutations in familial and sporadic paraganglioma and phaeochromocytoma. The lancet oncology. 2010 Apr;11(4):366–372. doi: 10.1016/S1470-2045(10)70007-3. [DOI] [PubMed] [Google Scholar]
- 48.Yao L, Barontini M, Niederle B, Jech M, Pfragner R, Dahia PL. Mutations of the metabolic genes IDH1, IDH2, and SDHAF2 are not major determinants of the pseudohypoxic phenotype of sporadic pheochromocytomas and paragangliomas. The Journal of clinical endocrinology and metabolism. 2010;95(3):1469–1472. doi: 10.1210/jc.2009-2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Eisenhofer G, Lenders JW, Goldstein DS, et al. Pheochromocytoma Catecholamine Phenotypes and Prediction of Tumor Size and Location by Use of Plasma Free Metanephrines. Clin Chem. 2005 Feb 17;51:735–744. doi: 10.1373/clinchem.2004.045484. [DOI] [PubMed] [Google Scholar]
- 50.Eisenhofer G, Goldstein DS, Sullivan P, et al. Biochemical and clinical manifestations of dopamine-producing paragangliomas: utility of plasma methoxytyramine. The Journal of clinical endocrinology and metabolism. 2005 Jan 11;90:2086–2075. doi: 10.1210/jc.2004-2025. [DOI] [PubMed] [Google Scholar]
- 51.Timmers HJ, Pacak K, Huynh TT, et al. Biochemically silent abdominal paragangliomas in patients with mutations in the succinate dehydrogenase subunit B gene. The Journal of clinical endocrinology and metabolism. 2008 Dec;93(12):4826–4832. doi: 10.1210/jc.2008-1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pacak K, Eisenhofer G, Ahlman H, et al. Pheochromocytoma: recommendations for clinical practice from the First International Symposium. Nat Clin Pract Endocrinol Metab. 2007 Feb;3(2):92–102. doi: 10.1038/ncpendmet0396. [DOI] [PubMed] [Google Scholar]
- 53.Grossman A, Pacak K, Sawka A, et al. Biochemical diagnosis and localization of pheochromocytoma: can we reach a consensus? Annals of the New York Academy of Sciences. 2006 Aug;1073:332–347. doi: 10.1196/annals.1353.038. [DOI] [PubMed] [Google Scholar]
- 54.Maurea S, Cuocolo A, Reynolds JC, Neumann RD, Salvatore M. Diagnostic imaging in patients with paragangliomas. Computed tomography, magnetic resonance and MIBG scintigraphy comparison. Q J Nucl Med. 1996;40(4):365–371. [PubMed] [Google Scholar]
- 55.Niemann U, Hiller W, Behrend M. 25 years experience of the surgical treatment of phaeochromocytoma. Eur J Surg. 2002;168(12):716–719. doi: 10.1080/00000000000000009. [DOI] [PubMed] [Google Scholar]
- 56.Stenstrom G, Ernest I, Tisell LE. Long-term results in 64 patients operated upon for pheochromocytoma. Acta Med Scand. 1988;223(4):345–352. doi: 10.1111/j.0954-6820.1988.tb15883.x. [DOI] [PubMed] [Google Scholar]
- 57.Khorram-Manesh A, Ahlman H, Nilsson O, et al. Long-term outcome of a large series of patients surgically treated for pheochromocytoma. J Intern Med. 2005 Jul;258(1):55–66. doi: 10.1111/j.1365-2796.2005.01504.x. [DOI] [PubMed] [Google Scholar]
- 58.Averbuch SD, Steakley CS, Young RC, et al. Malignant pheochromocytoma: effective treatment with a combination of cyclophosphamide, vincristine, and dacarbazine. Ann Intern Med. 1988;109(4):267–273. doi: 10.7326/0003-4819-109-4-267. [DOI] [PubMed] [Google Scholar]
- 59.Fitzgerald PA, Goldsby RE, Huberty JP, et al. Malignant pheochromocytomas and paragangliomas: a phase II study of therapy with high-dose 131I-metaiodobenzylguanidine (131I-MIBG) Annals of the New York Academy of Sciences. 2006 Aug;1073:465–490. doi: 10.1196/annals.1353.050. [DOI] [PubMed] [Google Scholar]
- 60.Gonias S, Goldsby R, Matthay KK, et al. Phase II study of high-dose [131I]metaiodobenzylguanidine therapy for patients with metastatic pheochromocytoma and paraganglioma. J Clin Oncol. 2009 Sep 1;27(25):4162–4168. doi: 10.1200/JCO.2008.21.3496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Coleman RE, Stubbs JB, Barrett JA, de la Guardia M, Lafrance N, Babich JW. Radiation dosimetry, pharmacokinetics, and safety of ultratrace Iobenguane I-131 in patients with malignant pheochromocytoma/paraganglioma or metastatic carcinoid. Cancer biotherapy & radiopharmaceuticals. 2009 Aug;24(4):469–475. doi: 10.1089/cbr.2008.0584. [DOI] [PubMed] [Google Scholar]
- 62.Eisenhofer G, Bornstein SR, Brouwers FM, et al. Malignant pheochromocytoma: current status and initiatives for future progress. Endocrine-related cancer. 2004 Sep;11(3):423–436. doi: 10.1677/erc.1.00829. [DOI] [PubMed] [Google Scholar]
- 63.Young AL, Baysal BE, Deb A, Young WF., Jr. Familial malignant catecholamine-secreting paraganglioma with prolonged survival associated with mutation in the succinate dehydrogenase B gene. The Journal of clinical endocrinology and metabolism. 2002 Sep;87(9):4101–4105. doi: 10.1210/jc.2002-020312. [DOI] [PubMed] [Google Scholar]