

Abstract
Chronic Fatigue Syndrome (CFS) is a complex illness affecting 4 million Americans for which no characteristic lesion has been identified. Instead of searching for a deficiency in any single marker, we propose that CFS is associated with a profound imbalance in the regulation of immune function forcing a departure from standard preprogrammed responses. To identify these imbalances we apply network analysis to the co-expression of 16 cytokines in CFS subjects and healthy controls. Concentrations of IL-1a, 1b, 2, 4, 5, 6, 8, 10, 12, 13, 15, 17 and 23, IFN-γ, lymphotoxin-α (LT-α) and TNF-α were measured in the plasma of 40 female CFS and 59 case-matched controls. Cytokine co-expression networks were constructed from the pair-wise mutual information (MI) patterns found within each subject group. These networks differed in topology significantly more than expected by chance with the CFS network being more hub-like in design. Analysis of local modularity isolated statistically distinct cytokine communities recognizable as pre-programmed immune functional components. These showed highly attenuated Th1 and Th17 immune responses in CFS. High Th2 marker expression but weak interaction patterns pointed to an established Th2 inflammatory milieu. Similarly, altered associations in CFS provided indirect evidence of diminished NK cell responsiveness to IL-12 and LTα stimulus. These observations are consistent with several processes active in latent viral infection and would not have been uncovered by assessing marker expression alone. Furthermore this analysis identifies key subnetworks such as IL-2:IFNγ:TNFα that might be targeted in restoring normal immune function.
Keywords: cytokines, network theory, immune signaling, chronic fatigue, inflammation, latent viral infection
1. Background
Chronic Fatigue Syndrome (CFS) is characterized by persistent and unexplained fatigue resulting in severe impairment in daily function and is defined by symptoms, disability, and exclusion of medical and psychiatric conditions that could explain the fatigue (Fukuda et al., 1994; Reeves et al., 2003; Prins et al., 2006). The US Centers for Disease Control and Prevention (CDC) estimates that as many as 4 million people are affected with CFS in the US alone (Reeves et al. 2007; Chandler et al., 2008). Costs to the US economy are estimated at $9.1 billion in lost productivity (Reynolds et al., 2004) and up to $24 billion dollars in health care expenditures annually (Jason et al., 2008). Furthermore complications and co-morbidity can be severe. For example, CFS is associated with chronic and episodic cardiovascular and autonomic dysfunction (Gerrity et al., 2003). Therefore this illness has far-reaching consequences and constitutes a significant public health concern.
Evidence of chronic immune dysfunction in CFS has been reported by several groups (Klimas et al., 1990; Straus et al., 1993; Hilgers et al., 1994; Keller et al., 1994; Tirelli et al., 1996; Gupta et al., 1997; Patarca et al., 1997; Patarca-Montero et al., 2001; Seigel et al. 2006) though the exact nature of this dysfunction remains unclear (Maher et al., 2003). A principal avenue of investigation has been the measurement in blood of immune signals conducted by cytokines. Many of the symptoms experienced by CFS patients strongly resemble the “sickness behavior” that can be induced by the administration of pro-inflammatory cytokines. In particular decreased motor activity, altered food and water intake, sleep and cognition have been linked to increases in the levels of IL-1b, IL-6 and TNFα in the brain (Dantzer et al. 2008). Individual cytokines however are pleiotropic and their biological activities are known to be context specific. This becomes evident when considering the current body of work focused on immune dysfunction in CFS. While some studies have reported increased levels of anti-inflammatory cytokines such as IL-10 (ter Wolbeek et al., 2007) and IL-4 (Skowera et al., 2004), others have shown a correlation with pro-inflammatory signals TNF-α and IL-6 (Gaab et al., 2005; Carlo-Stella et al., 2006). Admittedly the heterogeneity of the CFS population (Vollmer-Conna et al., 2006; Aspler et al., 2008; Kerr et al. 2008b) has been an issue. However a major failing remains analytical. In particular immunological markers continue to be analyzed individually even though their expression is articulated as part of an integrated network. In addition to the numerical advantages of a combinatorial approach, for example the control of excessive measurement noise (Szymanska et al., 2007), it is becoming apparent that understanding complex disease will require more than a list of defective cells or genes. Because cellular and molecular components are highly inter-dependent it is necessary to understand the “wiring” via which they interact (Barabasi, 2007). Immune cells form a distributed network of diverse elements that exchange information through a complex web of interactions (Orosz, 2001). The architecture of such a networked system profoundly impacts its behavior (Klemm and Bornholdt, 2005) and the strategies that are available for adapting to change and maintaining homeostasis. Nonetheless, the formal analysis of biological networks in defining disease phenotypes has received relatively little attention. Recent attempts have focused on the visual comparison of relatively sparse collections of known pathway elements (Kerr et al., 2008a) or a broad description of shifts in overall structure (Emmert-Streib, 2007). We have extended this work in several important ways, introducing continuous metrics that quantify not only the degree of change but the type of change occurring in global and more importantly in local network structure. These metrics have allowed us to identify functional communities of markers within these networks as well as key elements driving disease-related changes in network structure (Fuite et al., 2008).
Here we use network constructs such as these to examine how patterns in the coordinated expression of cytokines might differ in CFS subjects. In a recent publication we introduced the multiplex method to simultaneously measure a broad spectrum of 16 cytokines in order to assess their use as biomarkers for_CFS (Fletcher et al., 2009). Using this same experimental data we have now constructed separate networks describing co-expression of these 16 cytokines in a group of CFS subjects and in a group of healthy controls respectively. Pair-wise mutual information (MI), estimated from the biological variability within each group, was used as a robust measure of association between cytokines. These networks were then analyzed using quantitative metrics rooted in graph theory to assess the importance and nature of architectural changes related to illness. In particular we assessed local changes in the degree of connectivity at cytokine nodes and the redistribution of these connections as they form distinct and more locally centered communities. Consistent with our previous work (Fuite et al., 2008) we found that these cytokine networks differed significantly in architecture between diagnostic groups emphasizing that the organizational attributes of the immune response in addition to the activation level of individual markers constitute a unique characteristic of CFS. Of note distinct modules emerged in both healthy control and CFS networks that were recognizable as components of Th1, Th2 and Th17 responses. In CFS we found consistent but significantly attenuated patterns of Th1 and Th17 response occurring in the context of a well-established Th2 inflammatory environment. These patterns would have escaped detection had the analysis focused solely on differential expression of individual cytokines. Interestingly the cytokine co-expression patterns described in this study, though not uniquely assignable to a viral pathology, were at least consistent with the disruptive effects of latent viral infection by pathogens such as Epstein-Barr virus (EBV) (Samanta and Takada, 2009; Tsuge et al., 2001).
2. Materials and Methods
2.1. Sample collection and processing
2.1.1 Subject cohort
Female CFS patients (n=40; mean age 50) were from the CFS and Related Disorders Clinic at the University of Miami. A diagnosis of CFS was made using the International Case Definition (Fukuda et al., 1994; Reeves et al., 2003). Healthy female controls (n=59; mean age 53) were from a NIH funded study. All CFS study subjects had a SF-36 summary physical score (PCS) below the 50th percentile, based on population norms. Exclusion criteria for CFS included all of those listed in the current Centers for Disease Control (CDC) CFS case definition, including the listed psychiatric exclusions, as clarified in the International CFS Working Group (Reeves et al., 2003). All CFS subjects were assessed for psychiatric diagnosis at the time of recruitment with the Composite International Diagnostic Instrument (World Health Organization, 1997). Based on this assessment, we excluded subjects with DSM IV diagnoses for psychotic or melancholic depression, panic attacks, substance dependency, or psychoses as well as any subjects currently suicidal. We also excluded subjects with Borderline or Antisocial Personality Disorder. Subjects had no history of heart disease, COPD, malignancy, or other systemic disorders that would be exclusionary, as clarified by Reeves et al. (2003). Subjects were excluded for the following reasons: less than 18 yrs of age, active smoking or alcohol history, history of significant inability to keep scheduled clinic appointments in past.
Ethics statement
All subjects signed an informed consent approved by the Institutional Review Board of the University of Miami. Ethics review and approval for data analysis was also obtained by the IRB of the University of Alberta.
2.1.2. Cytokine profiles
Morning blood samples were collected into ethylene diamine tetra acetic acid. Plasma was separated within 2 hours of collection and stored at −80°C until assayed. We measured 16 cytokines in plasma using Quansys reagents and instrument (Quansys Biosciences, Logan, Utah). The Quansys Imager, driven by an 8.4 megapixel Canon 20D digital SLR camera, supports 96 well plate based chemiluminescent imaging. The Q-Plex™ Human Cytokine - Screen (16-plex) is a quantitative enzyme-linked immunoabsorbent assay (ELISA)-based test where sixteen distinct capture antibodies have been absorbed to each well of a 96-well plate in a defined array. Manipulation of the range of the standard curves and exposure time allowed reliable comparisons between CFS patients and controls of both low and high level cytokine concentrations in plasma. For the standard curves, we used the second order (k=2) polynomial regression model (parabolic curve): Yp=b0+b1X1+b2X2....+bkXk, where Yp is the predicted outcome value for the polynomial model with regression coefficients b1 to k for each degree and y intercept b0. Quadruplicate determinations were made, i.e., each sample was run in duplicate in two separate assays. Statistics reported in Table S3 show an average coefficient of variability (CV) of 0.20 for inter-assay and 0.09 for intra-assay repeatability. Also reported in Table S3 are the lower limits of detection (LLD) for each cytokine estimated from the standard calibration curve. In many cases the standard curve yielded a negative intercept value indicating that the modified assay produced a background optical signal at zero concentration. In the case of cytokines with positive intercept values very few samples produced results below the LLD with the exception of IL-17. While the LLD for IL-17 was lower with the modified protocol roughly one quarter of the CFS patients, and 1 in 10 control subjects, registered average expression values below detection.
2.2 Statistical Analysis
Association networks were constructed using mutual information criteria (MI) implemented in the ARACNe software (Margolin et al., 2006a, b). The mutual information MI(X; Y) shared by X and Y corresponds to the total entropy H(X) and H(Y) of these variables minus their joint entropy H(X, Y) (Eq. 1–3). In order to use this metric the continuous scale for the concentration of each cytokine was divided into bins defined by a set of Gaussian kernels. The optimal choice of kernel width is dependent on the sample size and the distribution statistics of the data. The algorithm used by the ARACNe platform is based on a computationally efficient estimation algorithm (Beirlant et al., 1997) and described in detail in Margolin et al. (2006a) and the Supplementary Technical Report in Margolin et al. (2006b). The null probability of each MI value was computed by sub-sampling with replacement. Subsets of 30 observations were repeatedly constructed by sampling each subject group separately. Samples were not removed from the candidate list if selected thereby making them available again for the next iteration. The final aggregate networks for each diagnostic group were generated from a consensus of 300 sub-sampled networks. Networks were stable in size over a wide range of MI significance thresholds (supplementary Figure S1) and p≤0.001 was used in all subsequent computations. This was used as the threshold for MI confidence in all subsequent computations. This consensus averaging across sub-sampled data sets and the fact that MI assigns equal influence to each measured value makes this approach quite robust to outliers (Craddock et al., 2006; Butte and Kohane, 2000). Nonetheless for additional detail we have included the values for conventional Spearman rank-based cross-correlation of cytokines in Tables S4 and S5 for the healthy controls (HC) and CFS patient groups respectively.
| (1) |
| (2) |
| (3) |
Indirect associations were removed using data processing inequality (DPI) (Cover and Thomas, 2006). DPI states that if X and Z interact only through a third variable Y then Eq. 4 applies. Thus the smallest MI value can only come from indirect interaction. ARACNe removes this edge.
| (4) |
Topological differences in networks were evaluated using a weighted graph edit distance (Bunke, 2000) corresponding to the minimum summed “cost” associated with the removal and insertion of edges transforming one graph into the other (Dickinson et al., 2004; Harper et al., 2004). Herein we make the costs of these edit operations directly proportional to the changes in edge MI. The weighted graph edit distance, dGED, between two undirected networks of order N with adjacency matrices, A and B, is computed as follows where aij=MIij if P(MIij>0) ≥ 0.001, else aij=0 and similarly for bij:
| (5) |
Significance of this edit distance was estimated (i) using reference networks generated by random sub-sampling of HC subjects, (ii) from equal-sized random networks conserving edge weight distribution (Milo et al., 2004) and (iii) through multi-graphs conserving node degree distribution (Newman, 2004b).
Node degree centrality or direct connectivity of each node i to its immediate neighborhood Ni was computed as . Eigenvector centrality xi was also computed for each node i as a measure of that node’s connectivity to its remote neighbors. For the ith node the eigenvector centrality score xi is proportional to the sum of xj for all nodes connected to it such that:
| (6) |
where Ni is the neighborhood of i, λ is some constant and N is the order of the network. Constraining all ai,j and xi to real positive values implies, by the Perron-Frobenius theorem, that only the largest principal eigenvalue solution to Eq. 6 is accepted (Kleinberg, 1999). Finally we have also scaled the principal eigenvector X to adjust for network size as follows:
| (7) |
where X̂ is the normalized principal eigenvector and ||X|| is the norm. This scaling is based on a maximum of xi = 1 for the center node of a star network (Ruhnau, 2000). The two node centralities, degree and eigenvector, are among the common numerical values that measure network connectedness to imply node reach, control, and influence within groups.
The overall degree of centralization for any network of order N and normalized principal eigenvector X̂ is the centrality index C:
| (8) |
Modularity, Q, is a measure of community structure within a network (Girvan and Newman, 2002; Newman, 2004a), Q = (fraction of edges within modules) − (fraction of edges expected within modules), such that (Newman and Girvan, 2004),
| (9) |
where m is the graph size,
| (10) |
n is the graph order, Ai,j is a component of the symmetric weighted adjacency matrix describing the network, and gi is the community to which node i is a member. The expected probability an edge randomly falls between two nodes is
| (11) |
where is the degree of node i.
To split any network or sub-network on the basis of maximizing modularity, a modularity matrix, B, is established having elements (Newman, 2006a),
| (12) |
Elements of the leading eigenvector of the modularity matrix are used to direct a splitting of the network into two modules and to assign corresponding node membership based on sign (+/−) and magnitude (Newman, 2006b). This process was iterated and modules sequentially identified until a maximum modularity for the overall network was reached or until further cuts increased modularity insignificantly (p > 0.05).
Graphical rendering was performed using a “spring-electrical” embedding (Pemmaraju and Skiena, 2003) where nodes are idealized as similarly charged objects that repel each other. Edges are imagined as springs adhering to Hooke’s law with spring-constants proportional to their MI weights. The network is relaxed iteratively to a minimum energy embedding, which naturally reveals modular structure.
3. Results
3.1. Cytokines undergo widespread differential expression in CFS
Results of the nonparametric Wilcoxon rank-sum test comparing the difference in median expression for each cytokine in CFS versus healthy control (HC) have been presented previously (Fletcher et al., 2009) and are summarized in supplemental Table S1. Briefly these show that 10 of the 16 of the cytokines surveyed had significantly different median expression levels (p ≤ 0.05) across groups. Circulating concentrations of interleukins (IL) IL-1a, 1b (p ≤ 0.05) as well as 4, 5, 6, 12 and lymphotoxin-alpha (LTα) (p ≤ 0.01) were markedly higher. Conversely, CFS patients exhibited lower expression of IL-8, 13, and 15 (p ≤ 0.01). Levels of IL-2, 10, 17, and 23, interferon-gamma (IFNγ), and tumor necrosis factor-alpha (TNFα) showed little difference in expression between groups. Increased levels of IL-1b and IL-6 in CFS align with experimental results showing the induction of “sickness behavior” from increased levels of pro-inflammatory cytokines (Dantzer et al., 2008) in the brain.
3.2 Altered associations are pervasive among cytokines in CFS
In order to verify the relative homogeneity of subject groups with regard to their cytokine signatures we first used a transpose of the experimental data to construct an analogous MI association network where each subject was represented by a node. The topology of the resulting network, when viewed as a low energy embedding, showed a natural separation of subjects into two non-overlapping regions consistent with the diagnostic assignment (Figure 1 inset). As a result all subjects in each group were used in the construction of the cytokine co-expression networks for CFS and HC respectively. Individual networks were then constructed for HC and CFS subjects using the within-group variability to estimate the pair-wise MI or shared information linking the expression of these 16 cytokines (Figure 1). Random sub-sampling of the subject groups was conducted to establish confidence intervals for the graph edit distance between phenotypes (Figure S1). The narrow distribution of edit distance values separating within-group networks further supported the assessment that each diagnostic group was relative homogeneous in composition.
Fig. 1.
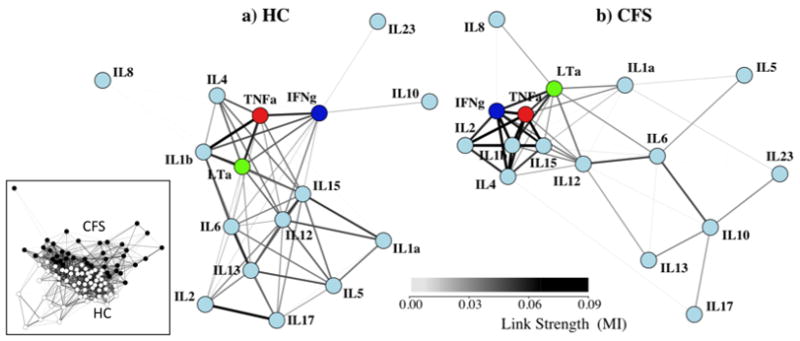
Networks for HC and CFS have visibly different topologies. A weighted spring-electrical embedding structurally reveals the subject-subject (inset) and cytokine-cytokine associations based on measurements in 59 healthy control subjects (a) and 40 CFS patients (b). All edge weights are significant at p ≤ 0.01. Separation of subjects was consistent with their assignment to diagnostic groups supporting the use of within-group variation in the estimation of mutual information for cytokine-cytokine associations.
Summary statistics describing basic properties of the CFS and HC networks are shown in Table 1. Interestingly while the average number of links per node differed between networks the overall mutual information supported by these connections did not. An average node was connected to its neighbors by one additional link in the HC network, namely 5.9 versus 5.1 links in the CFS network (p<0.01). Nonetheless the mutual information carried to the average node by these connections was essentially the same if we compare the cumulated link weight of 0.236 in HC to that of 0.240 in CFS (p ≫ 0.05). Although these networks were similar in terms of their overall mutual information (cumulated edge weight) or network size, they differed significantly in how this mutual information was distributed. The CFS network had a significantly higher centrality index, 0.448 versus 0.331 in HC (p ≪ 0.01), suggesting a greater reliance on a minority of highly connected hubs. Accordingly a quantitative comparison of overall network topology showed that HC and CFS networks were separated by a weighted edit distance of ~1.96 (Eq. 5) as a result of this re-structuring (Figure S1). This corresponded to more than 10 standard deviations (0.13) above the expected distance between two networks constructed from randomly sampled HC subjects (dedit ~0.18) and 3 standard deviations (~0.50) above the expected separation between two randomly assembled multi-graphs (dedit ~0.88) conserving node degree (data not shown).
Table 1. Connectivity Patterns Differ Significantly between Groups.
Summary of network-wide descriptive metrics with associated standard error ( ) for the HC and CFS networks as well as for sub-networks I+ and II−.
| Network | Metric | HC | CFS | p-value |
|---|---|---|---|---|
| Overall network | Order (total number of nodes) | 16 | 16 | |
| Mean links per node a | 5.9 (0.2) | 5.1 (0.2) | 0.009 | |
| Mean node degree b | 0.236 (0.007) | 0.240 (0.007) | 0.689 | |
| Centrality Index | 0.331 (0.011) | 0.448 (0.006) | 0.000 | |
| Modularity Index | 0.398 (0.019) | 0.394 (0.020) | 0.978 | |
| Cluster I+ | Order (total number of nodes) | 8 | 10 | |
| Mean links per node | 5.3 (0.2) | 2.6 (0.2) | 0.000 | |
| Mean node degree | 0.217 (0.008) | 0.088 (0.005) | 0.000 | |
| Centrality Index | 0.187 (0.011) | 0.609 (0.016) | 0.000 | |
| Cluster II− | Order (total number of nodes) | 8 | 6 | |
| Mean links per node | 2.8 (0.2) | 5.0 (0.0) | 0.000 | |
| Mean node degree | 0.121 (0.005) | 0.332 (0.002) | 0.000 | |
| Centrality Index | 0.562 (0.012) | 0.112 (0.002) | 0.000 |
Mean links per node counts all links with non-zero weight as 1 link.
Mean node degree uses the link weight or MI value
The spring-mass representations shown in Figure 1 confirm that these networks were visibly different in topology. This increase in overall network centrality in CFS was driven primarily by a few interacting markers. Local restructuring was described by changes in node degree centrality, a measure of direct connectivity, and eigenvector centrality, a combined measure of direct and indirect connectivity. Results presented in Figure 2 indicate that nodes representing IL-1b, 2, 4, IFNγ and TNFα concentrations became better integrated into the core network of CFS, both in terms of their association with direct and remote neighbors. Despite maintaining similar eigenvector centrality in both networks, the strength of direct connections from neighboring nodes to IL-10 substantially increased (degree centrality) in CFS. In addition, IL-10 shifted from having a weak association to a core node (IFNγ) in HC to having stronger associations to an opposite group of nodes in CFS (IL-6, 13, 17, 23) (Figures 2, 3). Markers that were much less strongly connected in CFS were IL-5, 6, 12, 13, and 17 (Table S1). By the same token cytokines IL-8, 15, and 23 remained unchanged in their degree of overall integration in the CFS and HC networks.
Fig. 2.
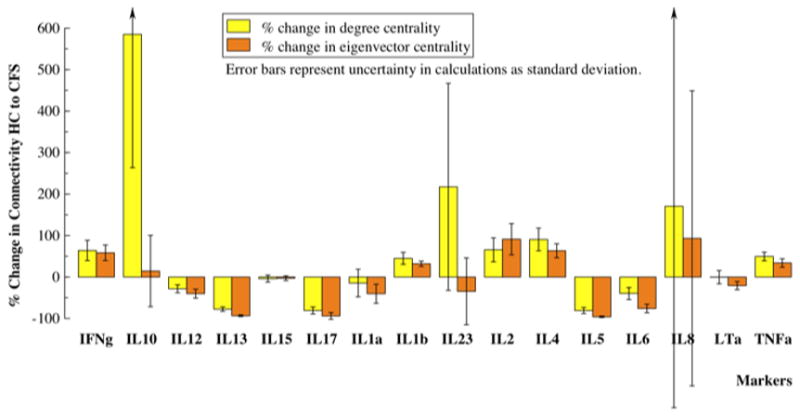
Most cytokines significantly modified their connectivity in the CFS state. Theses network alterations were revealed by the relative change in the total weight of edges connected at each node (node degree centrality) as well as edges acquired through first neighbors (normalized eigenvector centrality). Interleukins (IL), 2, 4, and 1b, interferon-gamma (IFNγ), and tumor necrosis factor-alpha (TNFα) became much better integrated into the core network in CFS, while interleukins, 5, 6, 12, 13, and 17 became more weakly associated.
Fig. 3.
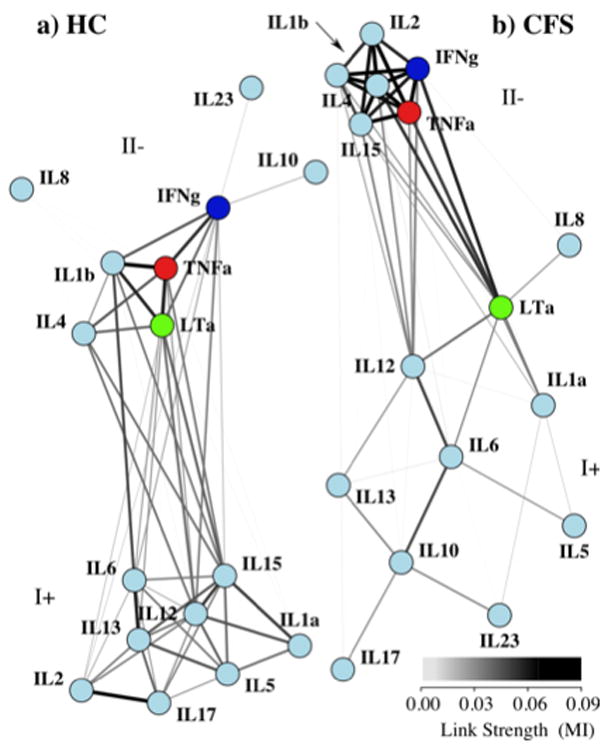
Both HC and CFS networks are composed of two distinct communities. Visually “relaxing” the links between identified communities of nodes and allowing them to drift apart emphasizes community structure in both networks. Overall modularity was maximized when each network was separated into two communities with differing compositions, labeled I+ at the top and II−. Each community represents a clustering of nodes with a greater internal linkage than would be expected compared to a random sampling of similar nodes.
3.3. Mid-scale shifts in network structure
The distribution of connections in each network among sets of nodes suggested that both the HC and CFS networks were made up of sub-networks. To analyze the extent of community structure within each network we iteratively divided the set of cytokine nodes into subsets and calculated increase in overall network modularity. Results indicated that the extent of community structure in the HC and CFS networks was about the same with maximal modularity values of 0.398 and 0.394 respectively. These values were achieved when the networks were broken down into two component modules, labeled I+ and II− (Table 1, Figure 3). Separation into additional modules either lead to a decrease in modularity, or did not significantly increase the modularity index at p < 0.05 confidence.
Results in Table 1 show that although both HC and CFS networks were made up of two mid-scale communities; these constituent modules possessed important differences in internal structure. Cluster I+ became less densely linked in among CFS patients as measured by a significant decline in number and strength of internal node associations. In cluster I+ of the CFS network the mean number of links per node fell from 5.3 to 2.6 (p<0.01) and the mean node degree fell from 0.217 to 0.088 (p<0.01). In addition cluster I+ became structurally more hub-like in CFS with an increase in centrality index to 0.609 from 0.187 in HC (p<0.001). Conversely cluster II− became structurally less focused in CFS dropping in centrality index from 0.562 to 0.112 (p<0.001). More evenly connected, cluster II− was also more densely linked in CFS patients with significant increases in the number and strength of internal node associations. The mean number of links per node rose from 2.8 to 5.0 (p< 0.01) in cluster II−, and the mean node degree increased from 0.121 to 0.332 (p<0.01) in the CFS network.
In addition to changes in structure we also observed changes in the composition of modules. The membership of an individual node to its respective module was measured by its centrality within the modularity matrix. This shifted significantly in CFS as a result of changing pair-wise associations (Table 2). In CFS the markers IL-10, IL-23 and LT-α shifted from cluster II− to cluster I+. While IL-6 strengthened its position in cluster I+ of the CFS network it shed the direct and strong association it held with IL-1b in HC. Conversely the markers, IL-2 and IL-15 moved in the opposite direction, significantly shifting centrality away from cluster I+ and towards cluster II− in CFS. These changes in centrality were significant at p<0.001. In contrast IL-8 maintained marginal association with either of these node communities in both CFS and HC.
Table 2. Cytokines change community membership in CFS.
Membership score with standard error ( ) to either of the two modules, I+ or II− for each cytokine node in HC and CFS networks. The magnitude of the membership score indicates the strength with which nodes are associated to the module they belong. Change in modularity membership score tracks differences in community association for each marker.
| Module Membership | Change in | |||
|---|---|---|---|---|
| Marker | HC | CFS | Membership | P-value |
| IL1a | 0.17(0.09) | 0.04 (0.06) | −0.13 (0.15) | 0.004 |
| IL1b | −0.34 (0.04) | −0.28 (0.04) | 0.06 (0.07) | 0.002 |
| IL2 | 0.24 (0.06) | −0.36 (0.02) | −0.60 (0.07) | 0.000 |
| IL4 | −0.24 (0.03) | −0.29 (0.04) | −0.05 (0.07) | 0.010 |
| IL5 | 0.33 (0.04) | 0.13(0.03) | −0.19 (0.07) | 0.000 |
| IL6 | 0.11 (0.09) | 0.41 (0.03) | 0.30 (0.11) | 0.000 |
| IL8 | −0.07 (0.09) | 0.02 (0.01) | 0.10 (0.10) | 0.176 |
| IL10 | −0.04 (0.01) | 0.41 (0.04) | 0.46 (0.05) | 0.000 |
| IL12 | 0.13(0.06) | 0.17 (0.05) | 0.04 (0.11) | 0.199 |
| IL13 | 0.28 (0.03) | 0.23 (0.05) | −0.05 (0.09) | 0.028 |
| IL15 | 0.05 (0.04) | −0.24 (0.03) | −0.30 (0.07) | 0.000 |
| IL17 | 0.36 (0.03) | 0.12(0.03) | −0.24 (0.06) | 0.000 |
| IL23 | −0.03 (0.04) | 0.15(0.03) | 0.17 (0.06) | 0.000 |
| IFNg | −0.29 (0.03) | −0.26 (0.03) | 0.03 (0.07) | 0.082 |
| LTa | −0.30 (0.04) | 0.03 (0.04) | 0.33 (0.08) | 0.000 |
| TNFa | −0.42 (0.02) | −0.29 (0.01) | 0.13 (0.03) | 0.000 |
4. Discussion
In order to explore changes in the patterns of immune activity in CFS we constructed two distinct association networks linking the expression of 16 cytokines measured in plasma for 40 female patients and 59 case-matched healthy controls (HC). Quantitative analysis of these two networks indicated that their topologies differed far beyond what would be expected by chance alone. Indeed variation separating the patterns of cytokine-cytokine association from each subject group was 10 times greater than the variability found within each group. Interestingly the average cytokine node in either network supported the same overall exchange of mutual information. This being said a typical CFS network node relied on one less connection to do so. This is an important point as it suggests that despite differences in cytokine expression between groups both networks were equally coherent overall (p=0.689, Table 1). Even at the basal levels of cytokine expression found in the HC group the correlation linking cytokines into a network was not only significant (all edges p<0.001) but it was virtually equivalent to the overall strength of association supporting the CFS network. Instead the difference between CFS and HC networks arose from a redistribution in the routing of mutual information with the CFS network relying more strongly on a minority of highly connected hubs. Driving these changes in structure we found that cytokines IL-1b, 2, 4, IFNγ, TNFα became much better integrated into the core CFS network, so much so that these formed a distinct subnetwork. Direct connections to anti-inflammatory cytokine IL-10 also increased substantially in CFS while the reverse was true of IL-13, 17 as well as IL-5 and 6. Despite this local restructuring these very different cytokine networks still shared a similar overall granularity. Using a novel measure of modularity we dissected these cytokine networks and found that two mid-scale communities could be isolated in both the CFS and HC group: clusters I+ and II−. However a closer look at the internal structure of these communities revealed diametrically opposite designs across illness groups. In CFS cytokine nodes in cluster I+ were more sparsely connected and adopted a more hub-like architecture whereas cytokine nodes in cluster II− were more strongly and more uniformly interconnected. The exact opposite is true of these same clusters in the control network. Differences such as these reinforce the notion that CFS manifests not only as a difference in the expression level of individual cytokines but also as an important shift in the patterns of association linking these cytokines.
The emergence of a tight-knit cluster dominated by Th1 cytokines was perhaps the most significant and most visible feature of the CFS network. Consisting of cytokine nodes IL-1b, IL-4, IFN-γ and TNF-α cluster II− also saw the recruitment of cytokines IL-2 and IL-15 from their position in cluster I+ of the HC network. This group became much more tightly associated in CFS and less centered about any individual cytokine. Interestingly IL-2, 4 and 15 belong to a family of cytokines that also includes IL-7, IL-9 and IL-21. Members of this family share a receptor complex consisting of IL-2 specific IL-2 receptor alpha (CD25), IL-2 receptor beta (CD122) and a common gamma chain (γc). It is not surprising therefore to observe a strong association between these network nodes upon immune activation. IL-2 and IL-4 are both T cell growth factors though the latter is a much more effective promoter of B cell proliferation (Burke et al., 1997). In these data, the IL-4 median concentration was increased 3-fold in CFS while IL-2, IFNγ and TNFα concentrations remained unchanged. This would support the presence of an active Th2 component in CFS and an antagonistic role for IL-4 towards Th1 cytokines such as IFNγ within cluster II−. Additionally new recruits, IL-2 and IL-15, both contribute to NK cell proliferation. Though NK cell response was not assessed directly in this work, the lower levels of IL-15 and unchanged levels of IL-2 observed here appear consistent with reports of deficient NK cell response in CFS (Maher et al., 2005).
Contrary to cluster II−, cluster I+ was dominated by cytokines typically associated with innate immunity and/or Th2 adaptive response namely IL-5, 6, 10, 12 and 13. For the most part associations between cytokine nodes in cluster I+ were fewer in number and visibly weaker than those linking their counterparts in cluster II−. Despite having weaker ties the circulating levels of IL-5, IL- 6 and IL-1a were significantly elevated suggesting an established Th-2 inflammatory environment. Indeed in CFS the mean node degree within cluster I+ was 4-fold lower than that of cluster II− (Table 1) and the centrality index 6-fold higher suggesting a much sparser and more centrally directed pattern of interaction. Especially recognizable in CFS cluster I+ is the relatively strong association of pro-inflammatory cytokine IL-6 with anti-inflammatory counterpart IL-10. Recall that IL-10, though not differentially expressed, shifted from having a weak association with cluster II− in the HC network to this much more central role in cluster I+ opposite IL-6 in CFS. This altered role would have gone unnoticed in a more conventional analysis. Also recognizable are elements of the IL-23/Th17/IL-17 response (Boniface et al., 2008; Aggarwal et al., 2003; McGeachy et al., 2007). The direct antagonism of IL-17 response by IL-2 (Laurence et al., 2007) observed in the HC network was absent in CFS. Instead an alternative response emerged whereby IL-17, IL-23 and IL-6 were all separated by IL-10. IL-6 typically enhances IL-1b–driven IL-17 production (Louten et al., 2009; Perona-Wright et al., 2009) while IL-10 is known to effectively down-regulate Th17 cytokine expression in macrophages and T cells (Gu et al., 2008). In these data median concentrations of IL-17 and 23 were unchanged despite elevated levels of IL-1b and IL-6. Though Th17 activation was not measured directly these observations suggest that responsiveness of this subset, like that of NK cells, may be altered in CFS.
Another key feature of the CFS network is the central role that the hub nodes LTα and IL-12 (Figure 3b) play in linking cytokine clusters I+ and II−. In contrast this role is almost evenly shared between IL-6, IL-15 and IL-2 in the HC network. No longer a member of cluster II− in CFS, the LTα hub nonetheless maintains strong associations to IL-1b, TNFα and IFNγ. Primarily a product of activated T and B-lymphocytes, LTα shares a strong homology with TNFα and IL-1b and is a powerful inducer of both these cytokines (Kasid et al., 1990). Moreover IFNγ has been shown to increase the number of receptors for TNFα and LTα further promoting their action (Aggarwal et al., 1985). In opposition to this, IL-4 will inhibit IL-2 triggered production of TNFα and LTα in mixed PBMC populations (Kasid et al., 1990). The network links identified here indicate that these known responses of IL-1b and TNFα to LTα, and to a lesser extent IFNγ, remained consistently expressed in the data. However, while the expression of IL-1b increased 2-fold in CFS, that of TNFα remained unchanged despite an almost 4-fold increase in LTα. This attenuated TNFα response in CFS could in principle be linked with the absence of IFNγ engagement and the elevated levels of IL-4 (> 3-fold) observed in these patients. In comparison to LTα, the association of IL-12 with the nodes of cluster II− is much weaker. Typically released by macrophages and dendritic cells, IL-12 is known to stimulate the production of IFNγ and TNFα from NK and T cells. This effect is enhanced by IL-2 (Wang et al., 2000) and to a lesser extent by IL-4 (Bream et al., 2003), a cytokine normally suppressive of IFNγ production. Though elevated 2-fold in this CFS cohort, the absence of a concordant IFNγ response further supports a dampened sensitivity of NK cells to IL-12 signaling in CFS. This may be due at least partially to inadequate IL-2 priming of IL-12 receptor expression (Wang et al., 2000) since IL-2 concentrations remained unchanged.
Viral triggers such as EBV and human cytomegalovirus (HCMV) have long been suspected of involvement in the onset and persistence of CFS. Recent evidence of xenotropic murine leukemia virus-related virus (XMRV) involvement in CFS (Lombardi et al., 2009) further supports this hypothesis. While other causes may underlie the cytokine expression patterns observed in this work many of these are at least consistent with some of the disruptive effects of chronic viral infection. In one potential model, infection with one or several viral agents may trigger or exploit deficient responsiveness of NK cells to IL-12 and LTα, both of which are actively produced by EBV-immortalized B cells (Airoldi et al., 2002; Thompson et al., 2003), leading to impaired IFNγ production and Th1 activation. In this scenario increased IL-6, also produced by EBV-infected B cells, together with depressed levels of IL-15 may interfere with LT-α and IL-12 activation of NK cells and the resulting IFN-γ production (Wilson et al., 2001; Saghafian-Hedengren et al., 2009). It is important to note however that while many of the patterns found here aligned with known EBV processes others did not; for example the lack of elevated IL-10 (Samanta et al., 2008) and IL-13 (Tsai et al., 2009). As very distinct illnesses arise from the expression of specific subsets of the 12 known EBV induced genes (Tsuge et al., 2001) the notion that CFS may involve a form of restricted viral latency may be worthy of consideration. Finally from a methodological perspective we observed that several significant shifts in network structure involved cytokines that were not differentially expressed across subject groups. This underscores the significance of co-expression analysis in understanding complex illnesses such as CFS. In particular such an analysis makes it possible to detect low-grade immune processes that may operate consistently with relatively modest changes in marker expression.
Supplementary Material
Acknowledgments
Special thanks to Dr. Andrea Califano and the members of his laboratory at Columbia University for many helpful discussions and their assistance in deploying ARACNe. This analysis was funded by grants from the US National Institute of Health, including R21AA016635 (PI MA Fletcher) and R01AI065723 (PI MA Fletcher); the CFIDS Association of America to G Broderick and N Klimas; the US Department of Veterans Affairs, Merit Awards to N Klimas. Ms Kreitz was funded through the generous support of the Patient Alliance for Neuroendocrine-immune Disorders Organization for Research and Advocacy (PANDORA).
Footnotes
Authors’ contributions
Conceived and designed the experiments: MAF NGK GB. Performed the experiments: MAF NGK. Analyzed the data: JF AK GB. Contributed reagents/materials/analysis tools: MAF AK GB JF. Wrote the paper: GB MAF JF AK SDV NGK.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Gordon Broderick, Email: gordon.broderick@ualberta.ca.
Jim Fuite, Email: jfuite@phys.ualberta.ca.
Andrea Kreitz, Email: akreitz@ualberta.ca.
Suzanne D Vernon, Email: sdvernon@cfids.org.
Nancy Klimas, Email: Nancy.Klimas@va.gov.
Mary Ann Fletcher, Email: MFletche@med.miami.edu.
References
- 1.Aggarwal BB, Eessalu TE, Hass PE. Characterization of receptors for human tumour necrosis factor and their regulation by gamma-interferon. Nature. 1985;318(6047):665–667. doi: 10.1038/318665a0. [DOI] [PubMed] [Google Scholar]
- 2.Aggarwal S, Ghilardi N, Xie MH, de Sauvage FJ, Gurney AL. Interleukin-23 promotes a distinct CD4Tcell activation state characterized by the production of interleukin-17. J Biol Chem. 2003;278:1910–1914. doi: 10.1074/jbc.M207577200. [DOI] [PubMed] [Google Scholar]
- 3.Airoldi I, Guglielmino R, Carra G, Corcione A, Gerosa F, Taborelli G, Trinchieri G, Pistoia V. The interleukin-12 and interleukin-12 receptor system in normal and transformed human B lymphocytes. Haematologica. 2002;87(4):434–442. Review. [PubMed] [Google Scholar]
- 4.Aspler AL, Bolshin C, Vernon SD, Broderick G. Evidence of Inflammatory Immune Signaling in Chronic Fatigue Syndrome: A Pilot Study of Gene Expression in Peripheral Blood. Behav Brain Funct. 2008;4:44. doi: 10.1186/1744-9081-4-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barabási AL. Network medicine - from obesity to the “diseasome”. N Engl J Med. 2007;357(4):404–407. doi: 10.1056/NEJMe078114. [DOI] [PubMed] [Google Scholar]
- 6.Beirlant J, Dudewicz E, Gyorfi L, van der Meulen E. Nonparametric entropy estimation: An overview. Int J Math Stat Sci. 1997;6(1):17–39. [Google Scholar]
- 7.Boniface K, Blom B, Liu YJ, de Waal Malefyt R. From interleukin-23 to T-helper 17 cells: human T-helper cell differentiation revisited. Immunol Rev. 2008;226:132–146. doi: 10.1111/j.1600-065X.2008.00714.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bream JH, Curiel RE, Yu CR, Egwuagu CE, Grusby MJ, Aune TM, Young HA. IL-4 synergistically enhances both IL-2- and IL-12-induced IFN-gamma expression in murine NK cells. Blood. 2003;102(1):207–214. doi: 10.1182/blood-2002-08-2602. [DOI] [PubMed] [Google Scholar]
- 9.Bunke H. Graph matching: Theoretical foundations, algorithms, and applications. Proc. Vision Interface 2000; Montreal. May 14–17; 2000. pp. 82–88. [Google Scholar]
- 10.Burke MA, Morel BF, Oriss TB, Bray J, McCarthy SA, Morel PA. Modeling the proliferative response of T cells to IL-2 and IL-4. Cell Immunol. 1997;178(1):42–52. doi: 10.1006/cimm.1997.1125. [DOI] [PubMed] [Google Scholar]
- 11.Butte AJ, Kohane IS. Mutual information relevance networks: functional genomic clustering using pairwise entropy measurements. Proc Pac Symp Biocomput. 2000:418–429. doi: 10.1142/9789814447331_0040. [DOI] [PubMed] [Google Scholar]
- 12.Carlo-Stella N, Badulli C, De Silvestri A, Bazzichi L, Martinetti M, Lorusso L, Bombardieri S, Salvaneschi L, Cuccia M. A first study of cytokine genomic polymorphisms in CFS: Positive association of TNF-857 and IFNgamma 874 rare alleles. Clin Exp Rheumatol. 2006;24(2):179–182. [PubMed] [Google Scholar]
- 13.Chandler HK, Ciccone D, MacBride RJ, Natelson B. Medically unexplained illness in short- and long-term disability applicants: prevalence and cost of salary reimbursement. Disabil Rehabil. 2008;30(16):1185–1191. doi: 10.1080/09638280701500109. [DOI] [PubMed] [Google Scholar]
- 14.Cover TM, Thomas JA. Elements of Information Theory. John Wiley & Sons; New York: 2006. p. 776. [Google Scholar]
- 15.Craddock RC, Taylor R, Broderick G, Whistler T, Klimas N, Unger ER. Exploration of statistical dependence between illness parameters using the Entropy Correlation Coefficient. Pharmacogenomics. 2006;7(3):421–428. doi: 10.2217/14622416.7.3.421. [DOI] [PubMed] [Google Scholar]
- 16.Dantzer R, O’Connor JC, Freund GG, Johnson RW, Kelley KW. From inflammation to sickness and depression: when the immune system subjugates the brain. Nat Rev Neurosci. 2008;9(1):46–56. doi: 10.1038/nrn2297. Review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dickinson P, Bunke H, Dadej A, Kraetzl M. Matching Graphs with Unique Node Labels. Pattern Analysis and Applications. 2004;7:243–254. [Google Scholar]
- 18.Emmert-Streib F. The chronic fatigue syndrome: a comparative pathway analysis. J Comput Biol. 2007;14(7):961–972. doi: 10.1089/cmb.2007.0041. [DOI] [PubMed] [Google Scholar]
- 19.Fletcher MA, Zeng XR, Barnes Z, Levis S, Klimas NG. Plasma cytokines in women with chronic fatigue syndrome. J Transl Med. 2009;7:96. doi: 10.1186/1479-5876-7-96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fuite J, Vernon SD, Broderick G. Neuroendocrine and immune network re-modeling in chronic fatigue syndrome: an exploratory analysis. Genomics. 2008;92(6):393–399. doi: 10.1016/j.ygeno.2008.08.008. [DOI] [PubMed] [Google Scholar]
- 21.Fukuda K, Straus SE, Hickie I, Sharpe MC, Dobbins JG, Komaroff A. The chronic fatigue syndrome: a comprehensive approach to its definition and study. International Chronic Fatigue Syndrome Study Group. Ann Intern Med. 1994;12:953–959. doi: 10.7326/0003-4819-121-12-199412150-00009. [DOI] [PubMed] [Google Scholar]
- 22.Gaab J, Rohleder N, Heitz V, Engert V, Schad T, Schürmeyer TH, Ehlert U. Stress-induced changes in LPS-induced proinflammatory cytokine production in chronic fatigue syndrome. Psychoneuroendocrinology. 2005;30:188–198. doi: 10.1016/j.psyneuen.2004.06.008. [DOI] [PubMed] [Google Scholar]
- 23.Gerrity TR, Bates J, Bell DS, Chrousos G, Furst G, Hedrick T, Hurwitz B, Kula RW, Levine SM, Moore RC, Schondorf R. Chronic fatigue syndrome: what role does the autonomic nervous system play in the pathophysiology of this complex illness? Neuroimmunomodulation. 2003;10:134–141. doi: 10.1159/000067176. [DOI] [PubMed] [Google Scholar]
- 24.Girvan M, Newman MEJ. Community Structure in Social and Biological Networks. Proc Natl Acad Sci USA. 2002;99(12):7821–7826. doi: 10.1073/pnas.122653799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gu Y, Yang J, Ouyang X, Liu W, Li H, Yang J, Bromberg J, Chen SH, Mayer L, Unkeless JC, Xiong H. Interleukin 10 suppresses Th17 cytokines secreted by macrophages and T cells. Eur J Immunol. 2008;38(7):1807–1813. doi: 10.1002/eji.200838331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gupta S, Aggarwal S, See D, Starr A. Cytokine production by adherent and non-adherent mononuclear cells in chronic fatigue syndrome. J Psychiatr Res. 1997;31(1):149–156. doi: 10.1016/s0022-3956(96)00063-5. [DOI] [PubMed] [Google Scholar]
- 27.Harper G, Bravi GS, Pickett SD, Hussain J, Green DV. The reduced graph descriptor in virtual screening and data-driven clustering of high-throughput screening data. J Chem Inf Comput Sci. 2004;44(6):2145–2156. doi: 10.1021/ci049860f. [DOI] [PubMed] [Google Scholar]
- 28.Hilgers A, Frank J. Chronic fatigue syndrome: immune dysfunction, role of pathogens and toxic agents and neurologic and cardial changes. Weiner Medizinische Wochenschrift. 1994;144(16):399–406. [PubMed] [Google Scholar]
- 29.Jason LA, Benton MC, Valentine L, Johnson A, Torres-Harding S. The Economic impact of ME/CFS: Individual and societal costs. Dyn Med. 2008;7:6. doi: 10.1186/1476-5918-7-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kasid A, Director EP, Stovroff MC, Lotze MT, Rosenberg SA. Cytokine regulation of tumor necrosis factor-alpha and -beta (lymphotoxin)-messenger RNA expression in human peripheral blood mononuclear cells. Cancer Res. 1990;50(16):5072–5076. [PubMed] [Google Scholar]
- 31.Keller RH, Lane JL, Klimas N, Reiter WM, Fletcher MA, van Riel F, Morgan R. Association between HLA class II antigens and the chronic fatigue immune dysfunction syndrome. Clin Infect Dis. 1994;18(Suppl 1):S154–156. doi: 10.1093/clinids/18.supplement_1.s154. [DOI] [PubMed] [Google Scholar]
- 32.Kerr JR, Petty R, Burke B, Gough J, Fear D, Sinclair LI, Mattey DL, Richards SC, Montgomery J, Baldwin DA, Kellam P, Harrison TJ, Griffin GE, Main J, Enlander D, Nutt DJ, Holgate ST. Gene expression subtypes in patients with chronic fatigue syndrome/myalgic encephalomyelitis. J Infect Dis. 2008a;197(8):1171–1184. doi: 10.1086/533453. [DOI] [PubMed] [Google Scholar]
- 33.Kerr JR, Burke B, Petty R, Gough J, Fear D, Mattey DL, Axford JS, Dalgleish AG, Nutt DJ. Seven genomic subtypes of chronic fatigue syndrome/myalgic encephalomyelitis: a detailed analysis of gene networks and clinical phenotypes. J Clin Patho. 2008b;61(6):730–739. doi: 10.1136/jcp.2007.053553. [DOI] [PubMed] [Google Scholar]
- 34.Klemm K, Bornholdt S. Topology of biological networks and reliability of information processing. Proc Natl Acad Sci USA. 2005;102(51):18414–18419. doi: 10.1073/pnas.0509132102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kleinberg JM. Authoritative Sources in a Hyperlinked Environment. J Assoc Comp Mach. 1999;46(5):604–632. [Google Scholar]
- 36.Klimas N, Salvato F, Morgan R, Fletcher MA. Immunologic abnormalities in chronic fatigue syndrome. J Clin Microbiol. 1990;28(6):1403–1410. doi: 10.1128/jcm.28.6.1403-1410.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Laurence A, Tato CM, Davidson TS, Kanno Y, Chen Z, Yao Z, Blank RB, Meylan F, Siegel R, Hennighausen L, Shevach EM, O’shea JJ. Interleukin-2 signaling via STAT5 constrains T helper 17 cell generation. Immunity. 2007;26(3):371–81. doi: 10.1016/j.immuni.2007.02.009. [DOI] [PubMed] [Google Scholar]
- 38.Lombardi VC, Ruscetti FW, Das Gupta J, Pfost MA, Hagen KS, Peterson DL, Ruscetti SK, Bagni RK, Petrow-Sadowski C, Gold B, Dean M, Silverman RH, Mikovits JA. Detection of an infectious retrovirus, XMRV, in blood cells of patients with chronic fatigue syndrome. Science. 2009;326(5952):585–589. doi: 10.1126/science.1179052. [DOI] [PubMed] [Google Scholar]
- 39.Louten J, Boniface K, de Waal Malefyt R. Development and function of TH17 cells in health and disease. J Allergy Clin Immunol. 2009;123(5):1004–1011. doi: 10.1016/j.jaci.2009.04.003. [DOI] [PubMed] [Google Scholar]
- 40.Maher K, Klimas NG, Fletcher MA. Immunology. In: Jason LA, Fennell PA, Taylor RR, editors. Handbook of Chronic Fatigues. John Wiley & Sons; Hoboken: 2003. pp. 124–151. [Google Scholar]
- 41.Maher KJ, Klimas NG, Fletcher MA. Chronic fatigue syndrome is associated with diminished intracellular perforin. Clin Exp Immunol. 2005;142(3):505–511. doi: 10.1111/j.1365-2249.2005.02935.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Margolin AA, Wang K, Lim WK, Kustagi M, Nemenman I, Califano A. Reverse engineering cellular networks. Nat Protoc. 2006a;1(2):662–71. doi: 10.1038/nprot.2006.106. [DOI] [PubMed] [Google Scholar]
- 43.Margolin AA, Nemenman I, Basso K, Wiggins C, Stolovitzky G, Dalla Favera R, Califano A. ARACNE: an algorithm for the reconstruction of gene regulatory networks in a mammalian cellular context. BMC Bioinformatics. 2006b;7(Suppl 1):S7. doi: 10.1186/1471-2105-7-S1-S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McGeachy MJ, Bak-Jensen KS, Chen Y, Tato CM, Blumenschein W, McClanahan T, Cua DJ. TGF-β and IL-6 drive the production of IL-17 and IL-10 by T cells and restrain TH-17 cell-mediated pathology. Nat Immunol. 2007;8:1390–1397. doi: 10.1038/ni1539. [DOI] [PubMed] [Google Scholar]
- 45.Milo R, Kashtan N, Itzkowitz S, Newman MEJ, Alon U. On the uniform generation of random graphs with prescribed degree sequences. 2004 http://arxiv.org/pdf/cond-mat/0312028.
- 46.Newman MEJ. Detecting Community Structure in Networks. Eur Phys J B - Condensed Matter and Complex Systems. 2004a;38(2):321–330. [Google Scholar]
- 47.Newman MEJ. Analysis of weighted graphs. Phys Rev E. 2004b;70(5):56131-1–56131-9. [Google Scholar]
- 48.Newman MEJ, Girvan M. Finding and Evaluating Community Structure in Networks. Phys Rev E. 2004;69(2):026113(15). doi: 10.1103/PhysRevE.69.026113. [DOI] [PubMed] [Google Scholar]
- 49.Newman MEJ. Modularity and Community Structure in Networks. Proc Natl Acad Sci USA. 2006a;103(23):8577–8582. doi: 10.1073/pnas.0601602103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Newman MEJ. Finding Community Structure in Networks Using the Eigenvectors of Matrices. Phys Rev E. 2006b;74(3):036104(19). doi: 10.1103/PhysRevE.74.036104. [DOI] [PubMed] [Google Scholar]
- 51.Orosz CG. An introduction to immuno-ecology and immuno-informatics. In: Segel LA, Cohen IR, editors. Design principles for the Immune System and Other Distributed Autonomous Systems. Oxford Univ. Press; New York: 2001. pp. 125–149. [Google Scholar]
- 52.Patarca R, Fletcher MA, Podack E. Cytolytic cell functions. In: Rose N, deMacario E, Folds J, Lane HC, Nakamura R, editors. Manual of Clinical Laboratory Immunology. ASM Press; Washington: 1997. pp. 296–303. [Google Scholar]
- 53.Patarca-Montero R, Antoni M, Fletcher MA, Klimas NG. Cytokine and other immunologic markers in chronic fatigue syndrome and their relation to neuropsychological factors. Appl Neuropsychol. 2001;8(1):51–64. doi: 10.1207/S15324826AN0801_7. [DOI] [PubMed] [Google Scholar]
- 54.Pemmaraju S, Skiena S. Computational Discrete Mathematics: Combinatorics and Graph Theory with Mathematica®. Cambridge University Press; Cambridge & New York: 2003. pp. 220–223. [Google Scholar]
- 55.Perona-Wright G, Jenkins SJ, O’Connor RA, Zienkiewicz D, McSorley HJ, Maizels RM, Anderton SM, MacDonald AS. A pivotal role for CD40-mediated IL-6 production by dendritic cells during IL-17 induction in vivo. J Immunol. 2009;182(5):2808–2815. doi: 10.4049/jimmunol.0803553. [DOI] [PubMed] [Google Scholar]
- 56.Prins J, van der Meer J, Bleijenberg G. Chronic Fatigue Syndrome. Lancet. 2006;28:367(9507):346–55. doi: 10.1016/S0140-6736(06)68073-2. [DOI] [PubMed] [Google Scholar]
- 57.Reeves WC, Lloyd A, Vernon SD, Klimas N, Jason LA, Bleijenberg G, Evengard B, White PD, Nisenbaum R, Unger ER International Chronic Fatigue Syndrome Study Group. Identification of ambiguities in the 1994 chronic fatigue syndrome research case definition and recommendations for resolution. BMC Health Serv Res. 2003;3(1):25. doi: 10.1186/1472-6963-3-25. Review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Reeves WC, Jones JF, Maloney E, Heim C, Hoaglin DC, Boneva RS, Morrissey M, Devlin R. Prevalence of chronic fatigue syndrome in metropolitan, urban, and rural Georgia. Popul Health Metr. 2007;5:5. doi: 10.1186/1478-7954-5-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Reynolds KJ, Vernon SD, Bouchery E, Reeves WC. The economic impact of chronic fatigue syndrome. Cost Eff Resour Alloc. 2004;2(1):4. doi: 10.1186/1478-7547-2-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ruhnau B. Eigenvector-centrality – a Node Centrality? Social Networks. 2000;22:357–365. [Google Scholar]
- 61.Saghafian-Hedengren S, Sundström Y, Sohlberg E, Nilsson C, Linde A, Troye-Blomberg M, Berg L, Sverremark-Ekström E. Herpesvirus seropositivity in childhood associates with decreased monocyte-induced NK cell IFN-gamma production. J Immunol. 2009;182(4):2511–2517. doi: 10.4049/jimmunol.0801699. [DOI] [PubMed] [Google Scholar]
- 62.Samanta M, Iwakiri D, Takada K. Epstein-Barr virus-encoded small RNA induces IL-10 through RIG-I-mediated IRF-3 signaling. Oncogene. 2008;27(30):4150–4160. doi: 10.1038/onc.2008.75. [DOI] [PubMed] [Google Scholar]
- 63.Samanta M, Takada K. Modulation of innate immunity system by Epstein-Barr virus-encoded non-coding RNA and oncogenesis. Cancer Sci. 2009 Sep 29; doi: 10.1111/j.1349-7006.2009.01377.x. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Siegel SD, Antoni MH, Fletcher MA, Maher K, Segota MC, Klimas N. Impaired natural immunity, cognitive dysfunction, and physical symptoms in patients with chronic fatigue syndrome: preliminary evidence for a subgroup? J Psychosom Res. 2006;60:559–566. doi: 10.1016/j.jpsychores.2006.03.001. [DOI] [PubMed] [Google Scholar]
- 65.Skowera A, Cleare A, Blair D, Bevis L, Wessely SC, Peakman M. High levels of type 2 cytokine-producing cells in chronic fatigue syndrome. Clin Exp Immunol. 2004;135(2):294–302. doi: 10.1111/j.1365-2249.2004.02354.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Straus SE, Fritz S, Dale JK, Gould B, Strober W. Lymphocyte phenotyping and function in chronic fatigue syndrome. J Clin Immunol. 1993;13(1):30–40. doi: 10.1007/BF00920633. [DOI] [PubMed] [Google Scholar]
- 67.Szymańska E, Markuszewski MJ, Capron X, van Nederkassel AM, Heyden YV, Markuszewski M, Krajka K, Kaliszan R. Increasing conclusiveness of metabonomic studies by chem-informatic preprocessing of capillary electrophoretic data on urinary nucleoside profiles. J Pharm Biomed Anal. 2007;43(2):413–420. doi: 10.1016/j.jpba.2006.08.015. [DOI] [PubMed] [Google Scholar]
- 68.ter Wolbeek M, van Doornen LJ, Kavelaars A, van de Putte EM, Schedlowski M, Heijnen CJ. Longitudinal analysis of pro- and anti-inflammatory cytokine production in severely fatigued adolescents. Brain Behav Immun. 2007;21(8):1063–1074. doi: 10.1016/j.bbi.2007.04.007. [DOI] [PubMed] [Google Scholar]
- 69.Thompson MP, Aggarwal BB, Shishodia S, Estrov Z, Kurzrock R. Autocrine lymphotoxin production in Epstein-Barr virus-immortalized B cells: induction via NF-kappaB activation mediated by EBV-derived latent membrane protein 1. Leukemia. 2003;17(11):2196–2201. doi: 10.1038/sj.leu.2403130. [DOI] [PubMed] [Google Scholar]
- 70.Tirelli U, Bernardi D, Improta S, Pinto A. Immunologic abnormalities in chronic fatigue syndrome. J Chronic Fatigue Syndrome. 1996;2(1):85–96. [Google Scholar]
- 71.Tsai SC, Lin SJ, Chen PW, Luo WY, Yeh TH, Wang HW, Chen CJ, Tsai CH. EBV Zta protein induces the expression of interleukin-13, promoting the proliferation of EBV-infected B cells and lymphoblastoid cell lines. Blood. 2009;114(1):109–118. doi: 10.1182/blood-2008-12-193375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tsuge I, Morishima T, Kimura H, Kuzushima K, Matsuoka H. Impaired cytotoxic T lymphocyte response to Epstein-Barr virus-infected NK cells in patients with severe chronic active EBV infection. J Med Virol. 2001;64(2):141–148. doi: 10.1002/jmv.1029. [DOI] [PubMed] [Google Scholar]
- 73.Vollmer-Conna U, Aslakson E, White PD. An empirical delineation of the heterogeneity of chronic unexplained fatigue in women. Pharmacogenomics. 2006;7(3):355–364. doi: 10.2217/14622416.7.3.355. [DOI] [PubMed] [Google Scholar]
- 74.Wang KS, Frank DA, Ritz J. Interleukin-2 enhances the response of natural killer cells to interleukin-12 through up-regulation of the interleukin-12 receptor and STAT4. Blood. 2000;95(10):3183–3190. [PubMed] [Google Scholar]
- 75.Wilson AD, Hopkins JC, Morgan AJ. In vitro cytokine production and growth inhibition of lymphoblastoid cell lines by CD41 T cells from Epstein-Barr virus (EBV) seropositive donors. Clin Exp Immunol. 2001;126:101–110. doi: 10.1046/j.1365-2249.2001.01641.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.World Health Organization. Composite Interview Diagnostic Interview (CIDI core), version 2.1. 1997. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.