

Abstract
Notch signaling is firmly established as a form of cell-to-cell communication that is critical throughout development. Dysregulation of Notch has been linked to cancer and developmental disorders, making it an important target for therapeutic intervention. One aspect of this pathway that sets it apart from others is its apparent reliance on endocytosis by signal-sending and signal-receiving cells. The subtle details of endocytosis-mediated molecular processing within both ligand- and receptor-presenting cells that are required for the Notch signal to maintain fidelity remain unclear. The endosomal system has long been known to play an important role in terminating signal transduction by directing lysosomal trafficking and degradation of cell surface receptors. More recently, endocytic trafficking has also been shown to be critical for activation of signaling. This review highlights four models of endocytic processing in the context of the Notch pathway. In ligand-presenting cells, endocytosis may be required for pre-processing of ligands to make them competent for interaction with Notch receptors and/or for exerting a pulling force on the ligand/Notch complex. In receptor-presenting cells, endocytosis may be a prerequisite for Notch cleavage and thus activation and/or it could be a means of limiting ligand-independent Notch activation. Recent advances in our understanding of how and why endocytosis of Notch receptors and ligands is required for activation and termination of signaling during normal development and in disease states are discussed.
Keywords: Notch, Endocytosis, Signaling, Receptor, DSL ligands
Introduction
In the past decade, endocytic trafficking has been shown to be a critical component of many signaling pathways—including the well-studied Notch signaling pathway, which is essential for a wide range of developmental processes. Many features of the Notch signal transduction cascade have been elucidated, but a key question that remains to be fully answered is why endocytic trafficking is required in both signal sending and receiving cells for the pathway to function. This brief review will cover what is known about the role of endocytosis in Notch signaling and will highlight questions remaining in the field.
Several recent reviews provide an in depth description of the core features of Notch signaling (Kopan and Ilagan, 2009; Tien et al., 2009). Briefly, Notch proteins are single pass transmembrane receptors that transduce signals via a unique mechanism involving receptor proteolysis, resulting in the release of an active intracellular Notch fragment. The extracellular domain of the prototypical Notch receptor contains tandem arrays of Epidermal Growth Factor (EGF1)-like repeats and a conserved negative regulatory region (NRR) that consists of three Lin12/Notch repeats (LNRs) and a heterodimerization (HD) domain (Fig. 1A). The NRR functions to prevent ligand-independent activation of Notch, as illustrated by the fact that mutations within this domain generate a constitutively active receptor, leading to developmental defects and cancers (Greenwald and Seydoux, 1990; Weng et al., 2004). During intracellular receptor maturation, mammalian Notch is cleaved at the S1 site within the HD domain by furin, or a related member of the proprotein convertase family. This generates extracellular and transmembrane subunits that are held together by the HD domain (Fig. 1A). Furin cleavage is not required for mammalian Notch to reach the cell surface (Bush et al., 2001), but it is required for activation of Notch signaling. Curiously, Drosophila Notch lacks a consensus furin cleavage site and only the uncleaved form is detected on the cell surface (Kidd and Lieber, 2002), suggesting that pathway activation differs between vertebrates and invertebrates.
Figure 1. Schematic illustration of Notch structure and pathway activation.
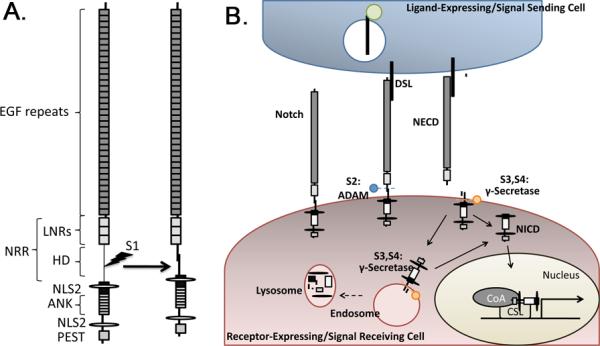
(A) Notch receptors have an extracellular domain composed of reiterated Epidermal Growth Factor (EGF)-like repeats and a conserved negative regulatory region (NRR) consisting of Lin12/Notch repeats (LNRs) and a heterodimerization (HD) domain. The intracellular portion of Notch contains repeated ankyrin (ANK) repeats, nuclear localization signals (NLS) and a PEST domain that controls receptor half life. Vertebrate Notch undergoes S1 cleavage within the secretory pathway to generate the heterodimeric receptor that is found on the cell surface. (B) Notch is activated by binding to ligands of the Delta/Serrate/Lag-2 (DSL) family. The ligands are ubiquitinated (green circle) and internalized into signal sending cells before and/or after receptor activation. Activated Notch undergoes sequential cleavage, initially at the S2 site by members of the ADAM family of metalloproteases (blue ball), and then at the S3 and S4 sites by γ-secretase (orange circle). S2 cleavage occurs at the cell surface and releases the Notch extracellular domain (NECD) from the heterodimer. γ-secretase mediated cleavages take place on the plasma membrane and/or in endosomes. These cleavages release the Notch intracellular domain (NICD), which translocates to the nucleus where it interacts with members of the CBF1/Su(H)/Lag-1 (CSL) family of transcription factors, and recruits co-activators (CoA) to activate transcription of Notch target genes. NICD signaling is terminated by lysosomal degradation.
Ligands of the Delta/Serrate/Lag-2 (DSL) family activate Notch. These ligands are membrane-anchored proteins and thus receptor activation requires cell-cell contact in most circumstances. Ligand binding triggers a sequential cascade of cleavages within Notch, named S2, S3 and S4 (Fig. 1B). The S2 site, which resides within the carboxyl (C)-terminal portion of the HD domain, is cleaved by members of the ADAM/TACE metalloprotease family (Mumm et al., 2000). This cleavage releases the Notch extracellular domain (NECD) from the heterodimer (Figure 1B) (Kopan et al., 1996; Struhl and Adachi, 1998). A recent structural analysis showed that the S2 cleavage site is normally masked by extensive interdomain interactions within the NRR (Gordon et al., 2007). Thus, ligand induced conformational changes in the Notch receptor are presumably required to expose the S2 site for ADAM-mediated cleavage. S2 cleavage is a prerequisite for subsequent intramembranous cleavage of Notch at the S3 and S4 sites by the γ-secretase complex. These cleavages release the Notch intracellular domain (NICD) (Struhl and Adachi, 2000; Fiuza and Arias, 2007). The NICD then translocates to the nucleus, where it interacts with members of the CBF1/Su(H)/Lag-1 (CSL) family of transcription factors, displacing co-repressors and recruiting co-activators to activate transcription of Notch target genes (Fortini and Artavanis-Tsakonas, 1994; Fiuza and Arias, 2007).
Studies in Drosophila have shown that dynamin-dependent endocytosis is essential in both the ligand- and receptor-presenting cells for successful transduction of Notch signals (Seugnet et al., 1997), and several models have since been proposed to explain this requirement (Illustrated in Figure 2). First, endocytosis has been proposed to direct DSL ligands to an intracellular compartment where they undergo essential post-translational modifications prior to recycling to the cell surface for receptor activation (Fig. 2A) (Wang and Struhl, 2004; Wang and Struhl, 2005). Alternatively, or perhaps in addition, endocytosis of DSL ligand bound to the Notch receptor may be necessary to provide a pulling force that dissociates the Notch heterodimer and/or induces a conformational change, thereby exposing the S2 ADAM cleavage site (Fig. 2B) (Parks et al., 2000; Nichols et al., 2007). In the signal-receiving cell, Notch endocytosis may be required for γ-secretase cleavage of Notch, perhaps because the enzyme complex is primarily active in an intracellular compartment (Fig. 2C) (Gupta-Rossi et al., 2004). Finally, Notch receptor endocytosis and lysosomal targeting may be required to prevent “accidental” ligand-independent activation of Notch (Fig. 2D) (Childress et al., 2006; Gallagher and Knoblich, 2006; Jaekel and Klein, 2006). In the following sections, we present evidence for and against each of these models, which are not mutually exclusive.
Figure 2. Models for how endocytosis activates Notch signaling.
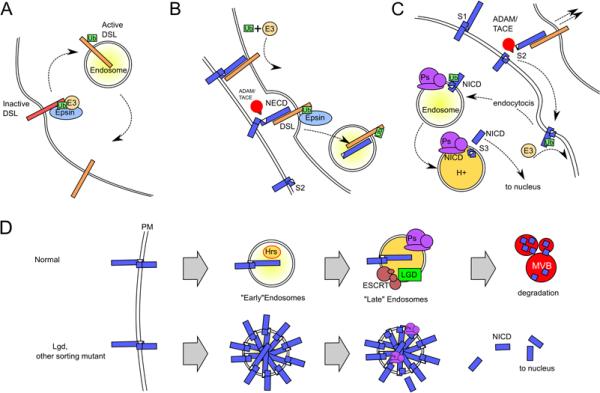
(A) Endocytosis as a means of generating active DSL ligand. In this model, DSL ligand is synthesized, and reaches the plasma membrane in an inactive (red) form. E3 ubiquitin ligases monoubiquitinate (Ub) the cytoplasmic tail of DSL, leading to epsin-dependent endocytosis. The ligand is converted to its active form (orange) in an intracellular compartment and then returned to the plasma membrane. The nature of the postranslational modification that activates the ligand is unknown. (B) Ligand endocytosis as a means of generating mechanical strain. In this model, endocytosis of DSL ligand (orange) bound to the Notch heterodimer (blue) on adjacent cells generates mechanical strain that unmasks the S2 cleavage site on Notch, enabling it to be cleaved by ADAM/TACE metalloproteases, thereby generating the remaining transmembrane fragment that is the substrate for the γ-secretase complex in the signal receiving cell. The completion of endocytosis results in trans-endocytosis of NECD and DSL into the signal sending cell. C. Notch endocytosis is required for S3 cleavage. Following ligand binding and S2 cleavage, ubiquitination of the cytoplasmic tail of the remaining transmembrane fragment of Notch triggers its endocytosis. The more acidic environment of the endosomes is required for the proteolytic activity of presenilin (Ps), the component of the γ-secretase complex (purple) that cleaves the S3 site to generate active NICD. D. Endocytosis as a means of preventing ligand-independent activation of the Notch receptor. A fraction of full length Notch undergoes ligand independent internalization from the plasma membrane and traffics to endocytic and lysosomal vesicles as part of its natural turnover. Mutations in components of the intracellular trafficking machinery, such as lethal giant discs (Lgd), that obstruct endosomal trafficking at a step after the formation of hepatocyte growth factor-regulated tyrosine kinase substrate (Hrs)-positive endosomes but before maturation into lysosomes, enable Notch to either enter or accumulate in an early endosomal compartment that allows for excessive, ligand independent cleavage of the S3 site to generate the active NICD fragment.
Ligand endocytosis: required for ligand activation?
As summarized above, the requirement for endocytosis in signal sending cells might reflect a need to internalize the ligand prior to receptor interaction in order to generate an active ligand and/or a need to internalize the ligand receptor complex in order to activate signaling. There is good evidence supporting both hypotheses and, indeed, endocytosis in the signal-presenting cell may serve multiple functions. The observation that DSL ligand activation requires a specialized endocytic pathway, rather than simple bulk endocytosis, supports the first model. However, the precise effect endocytosis is having on ligand activity remains unresolved. Evidence and proposed mechanisms for this model are discussed in this section.
Endocytosis of DSL ligand is triggered by monoubiquitination of its cytoplasmic tail, by the E3 ubiquitin ligases Neuralized and Mindbomb (Wang and Struhl, 2004). Ubiquitination targets DSL ligands for Epsin-mediated endocytosis, which is essential for signaling. Epsin proteins facilitate membrane curvature in addition to targeting membrane proteins for endocytosis. DSL ligands generated in Epsin mutant cells are efficiently transported to the cell surface, but cannot signal to their neighbors (Wang and Struhl, 2004). Because bulk endocytosis, which does not require Epsin function, is insufficient to facilitate DSL-Notch signaling, these results have been interpreted to suggest that Epsin targets ligands to a special endocytic compartment that they must enter to acquire signaling activity.
Recent studies suggest that ligand recycling may be a prerequisite for receptor binding. An ubiquitination-defective mutant form of the mouse Delta homolog, Dll1, can be internalized, albeit not as efficiently as the wild type species, but is unable to recycle back to the cell surface or to bind and activate Notch in neighboring cells (Heuss et al., 2008). While these studies demonstrate the importance of ligand recycling for receptor binding and activation, the mechanism by which this generates a more potent ligand remains a mystery. One possibility is that DSL ligands undergo post-translational modification of the extracellular domain within a specialized intracellular compartment, and that this enhances receptor binding (Fig. 2A). Consistent with this possibility, a potential DSL cleavage fragment is detected in wild type cells, but not in Epsin mutant cells, which cannot endocytose ligands (Wang and Struhl, 2004). A second possibility is that DSL ligands are initially diffuse over the entire surface of the cell, but are recycled to the outside of the cell in a clustered state, and that this enhances the strength of ligand-receptor interactions. If endocytosis-mediated clustering is occurring, interactions of monomers within endocytic vesicles may promote multimer formation, or monomers could become concentrated and interact upon recycling to localized plasma membrane domains. This model is supported by the finding that soluble forms of Delta are unable to activate Notch unless they are preclustered (Hicks et al., 2002). Finally, ligands may be recycled to specific lipid microdomains that contain essential cofactors, or are otherwise optimized for signal transmission. Lipid rafts are one example of specialized membrane domains that are believed to function as signaling platforms (Lajoie et al., 2009). Mouse Dll1 is partially localized to lipid raft microdomains, whereas an ubiquitin-defective, recycling incompetent form of Dll1 is not (Heuss et al., 2008). The latter ligand is unable to signal, consistent with the idea that localization to particular microdomains is important for signal transmission and is dependent on endocytosis.
It seems likely from these results that at least some DSL ligands must travel through the endocytic pathway in order to become active Notch ligands. This theory does not rule out the potential requirement for endocytosis during interaction with the Notch receptor. It is possible that DSL ligands undergo two rounds of endocytosis, the first to generate an active ligand and the second to generate an active receptor, as discussed below.
Ligand endocytosis: a means of mechanical strain?
A second hypothesis for why endocytosis in the signal-presenting cell is essential is that ligand induced mechanical uncoupling of the extracellular and transmembrane domains of Notch is critical for efficient DSL-Notch signaling. Such a model features the endocytosis of DSL bound to Notch by the ligand-presenting cell, which, in turn generates a pulling force necessary for mechanical uncoupling of the ligand-bound Notch extracellular domain (NECD) from the Notch transmembrane domain (Fig. 2B). This uncoupling subsequently unmasks the S2 ADAM cleavage site and allows Notch receptor processing and, thus, intracellular signaling.
The initial clues for ligand-endocytosis-mediated Notch activation were provided by the observation that endocytosis-deficient clones of cells in Drosophila behave in a manner consistent with an inability to either send or receive Notch signals (Seugnet et al., 1997; Parks et al., 2000). In addition, signaling required the separation of the NECD and NICD into cellular compartments within the signal-sending (called ‘trans-endocytosis”) and signal-receiving cells, respectively, in such clones (Parks et al., 2000). Trans-endocytosis is also seen during Notch activation in mammals (Parks et al., 2000; Nichols et al., 2007): NECD and ligand colocalize in intracellular vesicles of the ligand-sending cell.
A potential structural basis for the mechanical force uncoupling model was recently discovered in the crystal structure of a modified human Notch protein (Gordon et al., 2007). The crystal structure provided clues to the mechanism of auto-inhibition by the extracellular LNR domains that suppress ligand-independent signaling at the plasma membrane (Weinmaster, 1997). The presence of the globular LNR domains in a solvent-inaccessible pocket surrounding the critical S2 cleavage site supports a model in which major conformational changes are necessary to expose the S2 site to allow for metalloprotease cleavage, which is a prerequisite for the subsequent γ-secretase mediated cleavage that generates the active NICD signaling molecule. It is unlikely that minor allosteric effects would be sufficient to disrupt the broad and stable interactions of the S2 cleavage site and LNR domains.
If force generated by the signal-sending cell is required to activate Notch on the receiving cell, then one would predict that endocytosis-deficient DSL ligands would fail to activate Notch. Analysis of endocytosis-incompetent delta mutants is indeed consistent with this model (Parks et al., 2006). Delta molecules lacking their intracellular domain (deltaΔICD) can traffic to the plasma membrane but are unable to activate Notch. Endocytic localization of deltaΔICD is not observed in these situations and it is reasonable to infer that transendocytosis fails to occur. The finding that deltaΔICD expressing cells retain their ability to aggregate with Notch expressing cells suggests that DSL endocytosis is not required for ligand-receptor interactions in vivo, and hence, that these interactions are not sufficient to activate signaling (with some caveats, see discussion below). Further evidence that deltaΔICD can bind endogenous Notch receptors was provided by the demonstration of its ability to act as a dominant negative in several in vivo contexts, including Drosophila, Xenopus and chicken (Chitnis et al., 1995; Sun and Artavanis-Tsakonas, 1996; Henrique et al., 1997). In these systems deltaΔICD induces an overabundance of neuronal tissue—the classic ‘neurogenic’ phenotype that is characteristic of impaired Notch signaling.
These observations appear superficially at odds with earlier cell culture experiments. Trans-endocytosis observed in Drosophila cell culture was shown to involve translocation of full-length Notch into Delta and Serrate expressing cells in the presence of canonical Notch signal activation (Klueg et al., 1998). However, saturation of processing and signaling machinery in the context of overexpression experiments is a potentially serious confound to these experiments. There is, in fact, evidence that some limiting components of the Drosophila delta-notch signaling pathway exist that can be titrated out in overexpression studies (Selkoe and Kopan, 2003). It is therefore possible that trans-endocytosis of the NECD fragment also occurred in the above experiments, (and triggered the signal activation) but was not observed due to abundance of unprocessed/full-length Notch.
In an attempt to address this issue more rigorously, Nichols et al. performed a quantitative analysis of the NECD-to-NICD ratio in the ligand-presenting cells in a mammalian cell culture system expressing wild type proteins (Nichols et al., 2007). Their careful examination of relative amounts of mammalian NECD and NICD that undergo trans-endocytosis in the presence or absence of pharmacological inhibition of S2 or S3 cleavage showed that the majority of transendocytosed Notch consists of NECD independent of NICD. These results are consistent with the finding that separation of the two halves of Notch is sufficient to activate signaling (Rand et al., 2000). Still unresolved, however, is whether trans-endocytosis of S1-cleaved Notch occurs prior to or plays a causal role in S2 cleavage. There is good evidence that S2 cleavage does not occur until after ligand binding (Mumm et al., 2000), but the temporal relationship between dissociation of S1-cleaved heterodimeric forms of Notch and S2 processing has yet to be elucidated. It is possible to have NECD trans-endocytosis in the absence of S2 cleavage, but this may not represent the normal sequence of events. Biochemical experiments that follow the short peptide fragment between the S1 and S2 cleavage sites could shed light on this matter. Trans-endocytosis of that peptide fragment would be strong evidence that S2 cleavage occurs in parallel or prior to any mechanical strain; alternatively, shedding of the small peptide into the extracellular space or internalization into the signal-receiving cell would be consistent with S2 cleavage following NECD trans-endocytosis.
Unlike its mammalian homologue, Drosophila Notch exists on the cell surface predominantly in the uncleaved, full-length form, and appears to lack a consensus motif for S1 cleavage (Kidd and Lieber, 2002). It follows that S2, rather than S1, cleavage must be necessary for NECD trans-endocytosis in flies, unless there is a smaller population of heterodimeric Notch that constitutes the active form of the receptor. There is experimental evidence for the presence of heterodimeric Notch in Drosophila, most notably the finding that divalent cation-chelation leads to the release of NECD even when added in the presence of protease inhibitors (Rand et al., 2000). Because the structural integrity of the LNR domain is dependent on millimolar levels of calcium (Aster et al., 1999), this result is interpreted to mean that calcium chelation disrupts interactions between the LNR and HD domains, leading to release of the S1-cleaved NECD. Importantly, though, it is unclear whether the protease inhibitor cocktail used in these experiments was effective against ADAM metalloproteases and/or γ-secretase. This leaves open the possibility that calcium-chelation merely relieves the structural auto-inhibition by LNR domains and leads to ligand-independent S2 or S3 cleavage (Rand et al., 2000). More experiments are needed to unequivocally determine whether the active form of Drosophila Notch undergoes S1 cleavage and heterodimerization. For example, immunopurification and sequencing of trans-endocytosed NECD would demonstrate whether the peptide fragment ends at a putative furin (S1) or metalloprotease (S2) cleavage site.
One caveat of the mechanical strain model with respect to signaling in Drosophila is borne out of this controversy surrounding Notch S1 pre-processing: if the functional Notch molecule is not expressed as a heterodimer, the pulling force would not separate but instead unfold and expose a cleavage site for processing, but this appears to be insufficient for Notch signaling in other model systems. In mammals, S1 cleavage and heterodimerization must precede S2 cleavage. Mammalian S1 cleavage-incompetent Notch is only observed to transendocytose as full-length protein and no signal activation is detected (implying S2 and S3 processing are absent) (Nichols et al., 2007). If the ligand-expressing cell does pull on the ligand-receptor complex, ‘stretching out’ of the Notch LNR autoinhibitory domains would still be predicted to occur in these mutants, which would expose the S2 cleavage site for processing and then cause NICD liberation. Perhaps this reflects differences between mammalian and Drosophila processing machinery (Kidd and Lieber, 2002). It may be that mammalian Notch is obliged to separate to entirely expose cleavage sites or allow for ADAM/metalloprotease activity whereas the homologous Drosophila S2 cleavage machinery has access to, or can act on the unprocessed, full-length receptor. Expression and analysis of mammalian notch—either wild-type forms in the presence of potent furin inhibitors or S1 cleavage-incompetent mutants—in Drosophila cell culture could address this discrepancy.
The most difficult experimental observation to reconcile with the mechanical strain model is the discovery of some soluble Notch ligands that have signaling capability. Under this model, soluble ligands that are unable to provide traction are predicted to be antagonists of Notch signaling, and some reports bear this prediction out (Sun and Artavanis-Tsakonas, 1997; Qi et al., 1999; Hicks et al., 2002). However, there are also examples of soluble DSL ligands that activate Notch signaling in a myriad of contexts (Fitzgerald and Greenwald, 1995; Varnum-Finney et al., 1998; Han et al., 2000; Hicks et al., 2002; Chen and Greenwald, 2004). Most soluble ligands, including the one known endogenous example (DSL-1 in C. elegans), occur as multimers (DSL-1 is a dimer) or are bound to artificial substrates. This configuration may somehow lead to another form of mechanical strain sufficient for Notch activation. It is equally plausible that these soluble signaling events occur via non-canonical Notch pathways. A recent study of human hematopoietic stem cells suggests soluble and membrane-bound Delta4 have differential effects on cell proliferation via Notch signaling and that soluble ligand signaling can only be partially accounted for by canonical, S3/γ-secretase cleavage (Lahmar et al., 2008).
Receptor endocytosis: required for γ-secretase cleavage of Notch?
Genetic and cell biological studies have identified an important role for the endocytosis of not only the DSL ligands, but also the Notch receptor in generating an active signal. Endocytosis by the signal-receiving cell has been proposed to be required for S3 γ-secretase cleavage of Notch (Fig. 2C), although this remains controversial. Consistent with this possibility, γ-secretase is more enzymatically active at a lower pH characteristic of endosomes and lysosomes, and is found primarily in intracellular compartments, although a small fraction can be detected at the plasma membrane as well (Pasternak et al., 2003). Studies in vertebrates and flies, however, offer conflicting evidence for whether or not γ-secretase cleavage requires endocytosis of Notch. Further, if receptor-endocytosis is necessary, how it influences NICD cleavage and stability is also an area of interest. Evidence against and in favor of a role for endocytosis by the Notch receptor-presenting cell in canonical signal activation is presented in this section.
Genetic studies in Drosophila suggest that S3 γ-secretase cleavage occurs at the plasma membrane, rather than intracellularly, and does not require receptor internalization. One such study showed that whereas γ-secretase dependent cleavage of full length Notch does not occur in shibire mutant embryos, which are defective in endocytosis due to loss of Dynamin activity, γ-secretase dependent cleavage of a truncated form of Notch lacking most of the extracellular domain (and mimicking S2-cleaved Notch) proceeds normally in these embryos (Struhl and Adachi, 1998). This suggests that endocytosis is required for S2 cleavage (perhaps through models 1 and/or 2 discussed above), but not for S3 cleavage. A second study examined embryos mutant for Nicastrin, an essential component of the γ-secretase complex. Nicastrin is required for S3 cleavage of Notch and, thus, in cells defective for this protein Notch processing stalls following S2 ADAM/TACE-mediated ectodomain shedding. If γ-secretase cleavage occurs intracellularly, one would predict that the S2 cleaved form of Notch would undergo endocytosis and accumulate in γ-secretase containing intracellular vesicles in Nicastrin mutant cells, but instead it is detected primarily at the plasma membrane (Lopez-Schier and St Johnston, 2002).
More recent studies in mammalian cell lines support the model that endosomal entry is essential for γ-secretase mediated activation of Notch. In cultured cells, dynamin-dependent endocytosis was shown to be essential for γ-secretase mediated cleavage of a truncated form of murine Notch that mimics the S2-cleaved form (Gupta-Rossi et al., 2004). These same studies showed that Notch is monoubiquitinated on a juxtamembrane lysine residue, by an as yet unidentified ubiquitin-ligase, and that this is critical for generation and nuclear accumulation of the NICD. Careful analysis of endocytic uptake and subcellular localization of full length Notch mutants that cannot be monoubiquitinated revealed that these are retained at the plasma membrane, whereas mutant forms that lack the γ-secretase cleavage motif are internalized, but not cleaved or transported to the nucleus. Collectively, these studies suggest that monoubiquitination takes place at the membrane, and is essential for S2-cleaved Notch to reach internal compartments within the cell where S3 γ-secretase cleavage takes place.
Only recently has a requirement for internalization of endogenous Notch for signaling been examined in detail in Drosophila. Vaccari et al (2008) looked at Notch endocytosis, cleavage and pathway activation in a panel of Drosophila mutant cells that are defective in various stages of endocytosis. This study revealed that there is a sharp increase in γ-secretase cleavage and pathway activation upon entry into endosomes, indicating that endosomal entry of Notch is required for efficient signaling. Curiously, signaling was not completely abolished in mutants with severely restricted endosomal entry, and this residual activity was shown to be due to an alternate, dynamin-independent internalization route. These alternate pathways may be sufficient for activation of Notch when it is highly overexpressed, which could account for the seemingly conflicting data showing that truncated Notch can still signal in flies that are defective for dynamin-dependent endocytosis (Struhl and Adachi, 1998). Alternatively, it is possible that there is a low level of γ-secretase activity at the plasma membrane that can generate enough activated Notch to trigger signaling when the receptor is overexpressed, but not when it is present at endogenous levels.
To complicate matters, recent studies suggest the rate of endocytosis in a given cell type may influence the site of S3 cleavage and potency of the resultant NICD fragment. Biochemical analysis in a mammalian system has revealed that cleaves Notch at more than one site, generating ligands that differ in their stability, and thus signaling potency. The choice of cleavage site varies according to subcellular location (Tagami et al., 2008). At the plasma membrane, is more likely to generate a relatively stable NICD fragment containing valine at the amino (N)-terminus, whereas in endosomes, cleavage is likely to occur at a more C-terminal site that generates a less stable fragment containing a serine or threonine residue at the N-terminus. The serine-ended NICD is not only more easily degraded, but also shows reduced activation of Notch signaling. These findings demonstrate that, at least in cell culture, cleavage occurs both at the cell surface and in intracellular compartments. Since Notch is important in many tissues for a variety of functions, this complex activation scheme may allow for nuanced regulation of Notch for its diverse functions. It remains to be examined, however, whether there are in vivo circumstances in which the rate of endocytosis modulates Notch signaling, much less the mechanism for such a hypothetical shift.
Receptor endocytosis: a mechanism to restrict ligand-independent Notch activation?
Endocytosis has long been described as a means of signal termination for various plasma membrane signaling events. Likewise, Notch endocytosis appears to terminate signaling, and to prevent inappropriate ligand-independent activity. Indeed, HECT-type E3-ubiquitin ligases of the Nedd4 family have been implicated in the targeting of full-length Notch from the plasma membrane to endocytic and then lysosomal vesicles as a part of its natural turnover (Cornell et al., 1999; Qiu et al., 2000; Sakata et al., 2004; Wilkin et al., 2004). However, if endocytosis by the receptor-presenting cell promotes ligand-dependant Notch activation (as discussed in the previous section), could the same set of machinery go awry and lead to ligand-independent Notch activation? This section examines this possibility and discusses research that indicates endocytic function serves a critical role in tamping down spontaneous Notch activation.
Analogous to mutations in the Notch receptor that have shed light on auto-inhibition (Sanchez-Irizarry et al., 2004), examination of proteins involved in endosomal trafficking, sorting, or both have revealed circumstances in which their dysregulation may lead to ‘accidental’ Notch signaling in the absence of ligand-receptor interaction. One protein that has been identified as being necessary for proper endosomal trafficking of Notch is lethal giant disks (Lgd). Lgd is a conserved cytosolic protein containing a lipid-interacting, C2 motif. In lgd mutant cells, Notch accumulates in early endosomal vesicles marked by expression of Hrs (hepatocyte growth factor-regulated tyrosine kinase substrate) and ectopic pathway activation occurs in a dependent but ligand independent fashion (Childress et al., 2006; Gallagher and Knoblich, 2006; Jaekel and Klein, 2006). Interestingly, overexpression of wild-type Lgd leads to the same ligand-independent activation of canonical Notch signaling, suggesting that Lgd is titrating out one or more other requisite trafficking proteins that are required to target Notch for lysosomal degradation (Klein, 2003). A similar accumulation of Notch in early endosomal vesicles is observed in cells mutant for either hrs, which recruits monoubiquitinated plasma membrane proteins to endosomes, or for components of the endosomal sorting complexes required for transport (ESCRT) complex that is necessary for maturation of endosomes into multivesicular bodies. Whereas ligand-independent activation of Notch is observed in cells mutant for ESCRT components, for lgd or for both, it is not observed in hrs or in hrs/lgd double mutant cells. This indicates that Lgd functions downstream of Hrs, and that constitutive activation of Notch in lgd mutant cells requires transit through an early endosomal compartment. It should be noted that although mutations in hrs rescue ligand-independent Notch signaling in lgd mutant cells, Hrs is not required for endogenous ligand-induced signaling. Recently, a thorough study of how blockade of endocytosis at discrete stages affects levels of endogenous Notch signaling was performed. Obstruction of Notch trafficking after formation of Hrs-positive early endosomes, but before maturation into lysosomes, resulted in ectopic Notch activation similar to that observed in lgd mutants (Vaccari et al., 2008). Taken together, these findings suggest that Lgd normally functions to traffic Notch to the lysosome, and that blockade of endocytic maturation at late stages enables Notch to either enter or accumulate in an endosomal compartment that promotes excessive cleavage and signal activation (Fig. 2D).
The most obvious difference between endogenous Notch activation and the ectopic signaling observed in lgd mutants is the dependence of the former, but not the latter, on ligand interaction. This raises the question of whether activation occurs independent of S2 and/or S3 cleavage in lgd mutants. As previously discussed, binding of DSL ligands at the cell surface is believed to relieve autoinhibition mediated by Notch's LNR domains, such that the S2 cleavage event can occur. Given the new appreciation for the structural basis of autoinhibition (Gordon et al., 2007), it is reasonable to hypothesize that the progressively lower pH found in endocytic vesicles distorts H-bonding networks and electrostatic interactions, thus relieving steric hindrance to allow for S2-, and subsequent S3-cleavage in the absence of ligand. In fact, there is evidence that a sequence of cleavages similar or identical to the canonical ligand-induced signaling is required for the endocytic ligand-independent signal in lgd mutants. First, S2 cleavage of Notch has recently been found to depend on post-translational O-linked-glycosylation of the extracellular EGF-repeats of Notch by the O-glucosyltransferase Rumi (Acar et al., 2008). Endogenous Notch signaling is lost in rumi mutant flies, and loss of rumi suppresses ectopic activation of Notch in lgd mutant cells. Thus, Rumi function and, by inference, S2 cleavage of Notch are required for both ligand-dependant and ligand-independent Notch signaling. Analysis of flies mutant for both lgd and kuzbanian (the ADAM protease involved in S2 Notch processing) would clarify the necessity of S2 cleavage in endocytic ligand-independent signaling. The evidence that S3 cleavage is required for endocytic, ligand-independent signaling is more direct since decreasing the dose of presenilin (the enzymatic component of the γ-secretase complex) by half attenuates Notch activation in lgd mutant flies (Jaekel and Klein, 2006). Given that γ-secretase may target different cleavage sites within Notch depending on cellular compartmentalization (Tagami et al., 2008), it would be interesting to compare cleavage site usage in wild type tissue with that in lgd, or other endosomal mutants in which Notch is ectopically activated. To this end, western blot and/or proteomic analysis of Notch fragments may shed light on the nature of endosomal, ligand-independent Notch cleavage.
Conclusions
While Notch signaling is, to date, unique in its requirement for endocytosis of both the ligand and the receptor for full pathway activation, it is becoming increasing apparent that cell signaling and endocytic membrane trafficking are intimately connected for many different signaling pathways. For example, there is evidence that presenilins, the catalytic component of the complex, modulate diverse biologica l processes independent of conventional protease activity--including regulation of protein trafficking (Hass et al., 2009). With this in mind, we cannot rule out the possibility that presenilins participate in directing Notch for internalization via dynamin-independent pathways.
Overlap between Notch signaling components and those of other signaling pathways imply that some of the models described here may have broader influence. For instance, there is a growing list of substrates for which γ-secretase is involved in their regulated intramembrane proteolysis (Beel and Sanders, 2008). One such target for γ-secretase cleavage is amyloid precursor protein (APP), which has been extensively studied for its role in neurodegenerative diseases such as Alzheimer's Disease. It is entirely possible that greater similarities between these two pathways may emerge when the ligand for APP is revealed (Ma et al., 2008). In conclusion, mechanisms for how endocytosis and membrane trafficking activate and restrain the Notch signaling pathway will likely extend beyond the specific proteins we have highlighted in this review.
Acknowledgements
This work was supported in part by a grant from the NIH to JSW, JEM, LC and DH (T32, HD049309) and by grants from the NIH to JLC (RO1 HD37976 and R03 HD058841) and to EBP (5F30DK081305).
Abbreviations used
- EGF
Epidermal Growth Factor
- NRR
negative regulatory region (structural domain of Notch that shields S2 cleavage site in absence of ligand binding and activation)
- LNR
Lin12/Notch repeat
- HD
heterodimerization domain
- NECD
Notch extracellular domain
- NICD
Notch intracellular domain (the portion of Notch that traffics and signals to the nucleus)
- CSL
CBF/Su(H)/Lag-1
- DSL
Delta/Serrate/Lag-2 (family of ligands that activate Notch)
- Lgd
lethal giant disks
- Hrs
hepatocyte growth factor-regulated tyrosine kinase substrate
- ESCRT
endosomal sorting complexes required for transport
- S1 cleavage
(furin-mediated cleavage resulting in heterodimerization of Notch)
- S2 cleavage
(ADAM/TACE cleavage resulting in release of NECD fragment)
- S3 and S4 cleavage
(γ-secretase cleavages resulting in release of NICD fragment)
References
- Acar M, Jafar-Nejad H, Takeuchi H, Rajan A, Ibrani D, Rana NA, Pan H, Haltiwanger RS, Bellen HJ. Rumi is a CAP10 domain glycosyltransferase that modifies Notch and is required for Notch signaling. Cell. 2008;132:247–58. doi: 10.1016/j.cell.2007.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aster JC, Simms WB, Zavala-Ruiz Z, Patriub V, North CL, Blacklow SC. The folding and structural integrity of the first LIN-12 module of human Notch1 are calcium-dependent. Biochemistry. 1999;38:4736–42. doi: 10.1021/bi982713o. [DOI] [PubMed] [Google Scholar]
- Beel AJ, Sanders CR. Substrate specificity of gamma-secretase and other intramembrane proteases. Cell Mol Life Sci. 2008;65:1311–34. doi: 10.1007/s00018-008-7462-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bush G, diSibio G, Miyamoto A, Denault JB, Leduc R, Weinmaster G. Ligand-induced signaling in the absence of furin processing of Notch1. Dev Biol. 2001;229:494–502. doi: 10.1006/dbio.2000.9992. [DOI] [PubMed] [Google Scholar]
- Chen N, Greenwald I. The lateral signal for LIN-12/Notch in C. elegans vulval development comprises redundant secreted and transmembrane DSL proteins. Dev Cell. 2004;6:183–92. doi: 10.1016/s1534-5807(04)00021-8. [DOI] [PubMed] [Google Scholar]
- Childress JL, Acar M, Tao C, Halder G. Lethal giant discs, a novel C2-domain protein, restricts notch activation during endocytosis. Curr Biol. 2006;16:2228–33. doi: 10.1016/j.cub.2006.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chitnis A, Henrique D, Lewis J, Ish-Horowicz D, Kintner C. Primary neurogenesis in Xenopus embryos regulated by a homologue of the Drosophila neurogenic gene Delta. Nature. 1995;375:761–6. doi: 10.1038/375761a0. [DOI] [PubMed] [Google Scholar]
- Cornell M, Evans DA, Mann R, Fostier M, Flasza M, Monthatong M, Artavanis-Tsakonas S, Baron M. The Drosophila melanogaster Suppressor of deltex gene, a regulator of the Notch receptor signaling pathway, is an E3 class ubiquitin ligase. Genetics. 1999;152:567–76. doi: 10.1093/genetics/152.2.567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzgerald K, Greenwald I. Interchangeability of Caenorhabditis elegans DSL proteins and intrinsic signalling activity of their extracellular domains in vivo. Development. 1995;121:4275–82. doi: 10.1242/dev.121.12.4275. [DOI] [PubMed] [Google Scholar]
- Fiuza UM, Arias AM. Cell and molecular biology of Notch. J Endocrinol. 2007;194:459–74. doi: 10.1677/JOE-07-0242. [DOI] [PubMed] [Google Scholar]
- Fortini ME, Artavanis-Tsakonas S. The suppressor of hairless protein participates in notch receptor signaling. Cell. 1994;79:273–82. doi: 10.1016/0092-8674(94)90196-1. [DOI] [PubMed] [Google Scholar]
- Gallagher CM, Knoblich JA. The conserved c2 domain protein lethal (2) giant discs regulates protein trafficking in Drosophila. Dev Cell. 2006;11:641–53. doi: 10.1016/j.devcel.2006.09.014. [DOI] [PubMed] [Google Scholar]
- Gordon WR, Vardar-Ulu D, Histen G, Sanchez-Irizarry C, Aster JC, Blacklow SC. Structural basis for autoinhibition of Notch. Nat Struct Mol Biol. 2007;14:295–300. doi: 10.1038/nsmb1227. [DOI] [PubMed] [Google Scholar]
- Greenwald I, Seydoux G. Analysis of gain-of-function mutations of the lin-12 gene of Caenorhabditis elegans. Nature. 1990;346:197–9. doi: 10.1038/346197a0. [DOI] [PubMed] [Google Scholar]
- Gupta-Rossi N, Six E, LeBail O, Logeat F, Chastagner P, Olry A, Israel A, Brou C. Monoubiquitination and endocytosis direct gamma-secretase cleavage of activated Notch receptor. J Cell Biol. 2004;166:73–83. doi: 10.1083/jcb.200310098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han W, Ye Q, Moore MA. A soluble form of human Delta-like-1 inhibits differentiation of hematopoietic progenitor cells. Blood. 2000;95:1616–25. [PubMed] [Google Scholar]
- Hass MR, Sato C, Kopan R, Zhao G. Presenilin: RIP and beyond. Semin Cell Dev Biol. 2009;20:201–10. doi: 10.1016/j.semcdb.2008.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henrique D, Hirsinger E, Adam J, Le Roux I, Pourquie O, Ish-Horowicz D, Lewis J. Maintenance of neuroepithelial progenitor cells by Delta-Notch signalling in the embryonic chick retina. Curr Biol. 1997;7:661–70. doi: 10.1016/s0960-9822(06)00293-4. [DOI] [PubMed] [Google Scholar]
- Heuss SF, Ndiaye-Lobry D, Six EM, Israel A, Logeat F. The intracellular region of Notch ligands Dll1 and Dll3 regulates their trafficking and signaling activity. Proc Natl Acad Sci U S A. 2008;105:11212–7. doi: 10.1073/pnas.0800695105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hicks C, Ladi E, Lindsell C, Hsieh JJ, Hayward SD, Collazo A, Weinmaster G. A secreted Delta1-Fc fusion protein functions both as an activator and inhibitor of Notch1 signaling. J Neurosci Res. 2002;68:655–67. doi: 10.1002/jnr.10263. [DOI] [PubMed] [Google Scholar]
- Jaekel R, Klein T. The Drosophila Notch inhibitor and tumor suppressor gene lethal (2) giant discs encodes a conserved regulator of endosomal trafficking. Dev Cell. 2006;11:655–69. doi: 10.1016/j.devcel.2006.09.019. [DOI] [PubMed] [Google Scholar]
- Kidd S, Lieber T. Furin cleavage is not a requirement for Drosophila Notch function. Mech Dev. 2002;115:41–51. doi: 10.1016/s0925-4773(02)00120-x. [DOI] [PubMed] [Google Scholar]
- Klein T. The tumour suppressor gene l(2)giant discs is required to restrict the activity of Notch to the dorsoventral boundary during Drosophila wing development. Dev Biol. 2003;255:313–33. doi: 10.1016/s0012-1606(02)00052-0. [DOI] [PubMed] [Google Scholar]
- Klueg KM, Parody TR, Muskavitch MA. Complex proteolytic processing acts on Delta, a transmembrane ligand for Notch, during Drosophila development. Mol Biol Cell. 1998;9:1709–23. doi: 10.1091/mbc.9.7.1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopan R, Ilagan MX. The canonical Notch signaling pathway: unfolding the activation mechanism. Cell. 2009;137:216–33. doi: 10.1016/j.cell.2009.03.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopan R, Schroeter EH, Weintraub H, Nye JS. Signal transduction by activated mNotch: importance of proteolytic processing and its regulation by the extracellular domain. Proc Natl Acad Sci U S A. 1996;93:1683–8. doi: 10.1073/pnas.93.4.1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahmar M, Catelain C, Poirault S, Dorsch M, Villeval JL, Vainchenker W, Albagli O, Lauret E. Distinct effects of the soluble versus membrane-bound forms of the notch ligand delta-4 on human CD34+CD38low cell expansion and differentiation. Stem Cells. 2008;26:621–9. doi: 10.1634/stemcells.2007-0428. [DOI] [PubMed] [Google Scholar]
- Lajoie P, Goetz JG, Dennis JW, Nabi IR. Lattices, rafts, and scaffolds: domain regulation of receptor signaling at the plasma membrane. J Cell Biol. 2009;185:381–5. doi: 10.1083/jcb.200811059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Schier H, St Johnston D. Drosophila nicastrin is essential for the intramembranous cleavage of notch. Dev Cell. 2002;2:79–89. doi: 10.1016/s1534-5807(01)00109-5. [DOI] [PubMed] [Google Scholar]
- Ma QH, Futagawa T, Yang WL, Jiang XD, Zeng L, Takeda Y, Xu RX, Bagnard D, Schachner M, Furley AJ, Karagogeos D, Watanabe K, Dawe GS, Xiao ZC. A TAG1-APP signalling pathway through Fe65 negatively modulates neurogenesis. Nat Cell Biol. 2008;10:283–94. doi: 10.1038/ncb1690. [DOI] [PubMed] [Google Scholar]
- Mumm JS, Schroeter EH, Saxena MT, Griesemer A, Tian X, Pan DJ, Ray WJ, Kopan R. A ligand-induced extracellular cleavage regulates gamma-secretase-like proteolytic activation of Notch1. Mol Cell. 2000;5:197–206. doi: 10.1016/s1097-2765(00)80416-5. [DOI] [PubMed] [Google Scholar]
- Nichols JT, Miyamoto A, Olsen SL, D'Souza B, Yao C, Weinmaster G. DSL ligand endocytosis physically dissociates Notch1 heterodimers before activating proteolysis can occur. J Cell Biol. 2007;176:445–58. doi: 10.1083/jcb.200609014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parks AL, Klueg KM, Stout JR, Muskavitch MA. Ligand endocytosis drives receptor dissociation and activation in the Notch pathway. Development. 2000;127:1373–85. doi: 10.1242/dev.127.7.1373. [DOI] [PubMed] [Google Scholar]
- Parks AL, Stout JR, Shepard SB, Klueg KM, Dos AA, Santos TR Parody, Vaskova M, Muskavitch MA. Structure-function analysis of delta trafficking, receptor binding and signaling in Drosophila. Genetics. 2006;174:1947–61. doi: 10.1534/genetics.106.061630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasternak SH, Bagshaw RD, Guiral M, Zhang S, Ackerley CA, Pak BJ, Callahan JW, Mahuran DJ. Presenilin-1, nicastrin, amyloid precursor protein, and gamma-secretase activity are co-localized in the lysosomal membrane. J Biol Chem. 2003;278:26687–94. doi: 10.1074/jbc.m304009200. [DOI] [PubMed] [Google Scholar]
- Qi H, Rand MD, Wu X, Sestan N, Wang W, Rakic P, Xu T, Artavanis-Tsakonas S. Processing of the notch ligand delta by the metalloprotease Kuzbanian. Science. 1999;283:91–4. doi: 10.1126/science.283.5398.91. [DOI] [PubMed] [Google Scholar]
- Qiu L, Joazeiro C, Fang N, Wang HY, Elly C, Altman Y, Fang D, Hunter T, Liu YC. Recognition and ubiquitination of Notch by Itch, a hect-type E3 ubiquitin ligase. J Biol Chem. 2000;275:35734–7. doi: 10.1074/jbc.M007300200. [DOI] [PubMed] [Google Scholar]
- Rand MD, Grimm LM, Artavanis-Tsakonas S, Patriub V, Blacklow SC, Sklar J, Aster JC. Calcium depletion dissociates and activates heterodimeric notch receptors. Mol Cell Biol. 2000;20:1825–35. doi: 10.1128/mcb.20.5.1825-1835.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakata T, Sakaguchi H, Tsuda L, Higashitani A, Aigaki T, Matsuno K, Hayashi S. Drosophila Nedd4 regulates endocytosis of notch and suppresses its ligand-independent activation. Curr Biol. 2004;14:2228–36. doi: 10.1016/j.cub.2004.12.028. [DOI] [PubMed] [Google Scholar]
- Sanchez-Irizarry C, Carpenter AC, Weng AP, Pear WS, Aster JC, Blacklow SC. Notch subunit heterodimerization and prevention of ligand-independent proteolytic activation depend, respectively, on a novel domain and the LNR repeats. Mol Cell Biol. 2004;24:9265–73. doi: 10.1128/MCB.24.21.9265-9273.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkoe D, Kopan R. Notch and Presenilin: regulated intramembrane proteolysis links development and degeneration. Annu Rev Neurosci. 2003;26:565–97. doi: 10.1146/annurev.neuro.26.041002.131334. [DOI] [PubMed] [Google Scholar]
- Seugnet L, Simpson P, Haenlin M. Requirement for dynamin during Notch signaling in Drosophila neurogenesis. Dev Biol. 1997;192:585–98. doi: 10.1006/dbio.1997.8723. [DOI] [PubMed] [Google Scholar]
- Struhl G, Adachi A. Nuclear access and action of notch in vivo. Cell. 1998;93:649–60. doi: 10.1016/s0092-8674(00)81193-9. [DOI] [PubMed] [Google Scholar]
- Struhl G, Adachi A. Requirements for presenilin-dependent cleavage of notch and other transmembrane proteins. Mol Cell. 2000;6:625–36. doi: 10.1016/s1097-2765(00)00061-7. [DOI] [PubMed] [Google Scholar]
- Sun X, Artavanis-Tsakonas S. The intracellular deletions of Delta and Serrate define dominant negative forms of the Drosophila Notch ligands. Development. 1996;122:2465–74. doi: 10.1242/dev.122.8.2465. [DOI] [PubMed] [Google Scholar]
- Sun X, Artavanis-Tsakonas S. Secreted forms of DELTA and SERRATE define antagonists of Notch signaling in Drosophila. Development. 1997;124:3439–48. doi: 10.1242/dev.124.17.3439. [DOI] [PubMed] [Google Scholar]
- Tagami S, Okochi M, Yanagida K, Ikuta A, Fukumori A, Matsumoto N, Ishizuka-Katsura Y, Nakayama T, Itoh N, Jiang J, Nishitomi K, Kamino K, Morihara T, Hashimoto R, Tanaka T, Kudo T, Chiba S, Takeda M. Regulation of Notch signaling by dynamic changes in the precision of S3 cleavage of Notch-1. Mol Cell Biol. 2008;28:165–76. doi: 10.1128/MCB.00863-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tien AC, Rajan A, Bellen HJ. A Notch updated. J Cell Biol. 2009;184:621–9. doi: 10.1083/jcb.200811141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaccari T, Lu H, Kanwar R, Fortini ME, Bilder D. Endosomal entry regulates Notch receptor activation in Drosophila melanogaster. J Cell Biol. 2008;180:755–62. doi: 10.1083/jcb.200708127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varnum-Finney B, Purton LE, Yu M, Brashem-Stein C, Flowers D, Staats S, Moore KA, Le Roux I, Mann R, Gray G, Artavanis-Tsakonas S, Bernstein ID. The Notch ligand, Jagged-1, influences the development of primitive hematopoietic precursor cells. Blood. 1998;91:4084–91. [PubMed] [Google Scholar]
- Wang W, Struhl G. Drosophila Epsin mediates a select endocytic pathway that DSL ligands must enter to activate Notch. Development. 2004;131:5367–80. doi: 10.1242/dev.01413. [DOI] [PubMed] [Google Scholar]
- Wang W, Struhl G. Distinct roles for Mind bomb, Neuralized and Epsin in mediating DSL endocytosis and signaling in Drosophila. Development. 2005;132:2883–94. doi: 10.1242/dev.01860. [DOI] [PubMed] [Google Scholar]
- Weinmaster G. The ins and outs of notch signaling. Mol Cell Neurosci. 1997;9:91–102. doi: 10.1006/mcne.1997.0612. [DOI] [PubMed] [Google Scholar]
- Weng AP, Ferrando AA, Lee W, Morris JPt, Silverman LB, Sanchez-Irizarry C, Blacklow SC, Look AT, Aster JC. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science. 2004;306:269–71. doi: 10.1126/science.1102160. [DOI] [PubMed] [Google Scholar]
- Wilkin MB, Carbery AM, Fostier M, Aslam H, Mazaleyrat SL, Higgs J, Myat A, Evans DA, Cornell M, Baron M. Regulation of notch endosomal sorting and signaling by Drosophila Nedd4 family proteins. Curr Biol. 2004;14:2237–44. doi: 10.1016/j.cub.2004.11.030. [DOI] [PubMed] [Google Scholar]