

Abstract
Inflammatory cell infiltration and resident microglial activation within the central nervous system (CNS) are pathological events in multiple sclerosis (MS) and experimental autoimmune encephalomyelitis (EAE). While MS therapies target the peripheral immune system, no treatment is currently known to also modulate microglia. FMS-like tyrosine-3 (FLT-3) is expressed on hematopoietic and dendritic cells. We reported that FLT-3 inhibition ameliorates early actively induced EAE by predominantly modulating dendritic cell function as compared to microglia. We demonstrate in this report that FLT-3 is expressed in perivascular cuffs, brain parenchyma and in non-lesioned gray and white matter within MS brain but not in these regions within control brain. Furthermore, we demonstrate that FLT-3 is expressed on two populations of cells within MS brain; one which expresses the dendritic cell marker CD209, and the other which does not, suggesting that FLT-3 within MS brain is expressed on infiltrating dendritic cells and a non-dendritic cell such as microglia. Additionally, we report that FLT-3 inhibition in murine microglia blocks, in a dose dependent manner, IFN-γ-induced expression of MHC class II and CD86, and LPS-induced secretion of IL-6. These data suggest that FLT-3 is involved in microglial cell’s capacity to respond to environmental cues to function as antigen presenting cells and mediate CNS inflammation. Furthermore these data suggest that FLT-3 may be a therapeutic target on microglia to mitigate CNS inflammation.
INTRODUCTION
The pathology of both multiple sclerosis (MS) and experimental autoimmune encephalomyelitis (EAE), an animal model for MS, are characterized by central nervous system (CNS) inflammation, demyelination, glial activation, and axonal damage (Bjartmar et al., 1999). Evidence from in vitro and in vivo studies suggests an important role for activated microglia in propagating CNS inflammation and subsequent neural injury (Block and Hong, 2005; Heppner et al., 2005; Jack et al., 2005; Kreutzberg, 1996). Present MS immunomodulatory therapies affect peripheral immune cells but currently there are no therapies that also modulate microglial cells.
Although microglia cells are resident immune cells within the brain, they possess some dendritic cell-like characteristics. Both microglia and dendritic cells are derived from hematopoietic precursors, express common surface markers, and can function as antigen presenting cells (Alliot et al., 1999; Aloisi et al., 1999; Santambrogio et al., 2001; Town et al., 2005). Dendritic cells are more efficient at presenting antigen than microglia cells(Aloisi et al., 1999). However, during the course of EAE, microglia increase expression of major histocompatibility complex class II (MHCII) and CD86, and thereby increase their capability to function as antigen presenting cells (Juedes and Ruddle, 2001). In humans, HLA-Class II is necessary to activate pathogenic CD4+ T cells and has been found to be elevated in MS brain(Steinman, 2008). Furthermore, HLA-Class II is up-regulated on activated microglia in pre-active lesions of MS brain (van der Valk and Amor, 2009). In addition to an increased capacity to present antigen during EAE and MS, activated microglia have been found to secrete toxic (cytokines) molecules such as IL-6 (Gottschall et al., 1995). IL-6 promotes the differentiation of pathogenic Th-17 cells, is up-regulated during MS, and contributes to EAE development (McGeachy et al., 2007; Mendel et al., 1998; Samoilova et al., 1998; Serada et al., 2008; Steinman, 2008). The important role that microglia cells play in the development of EAE is further illustrated by data showing that inhibiting microglia function blocks EAE initiation(Heppner et al., 2005).
We previously demonstrated that EAE could be ameliorated by treating mice with a selective FMS-like tyrosine-3 (FLT-3) inhibitor, CEP-701, which was found to modulate the maturation of dendritic cells (DCs) but had no direct effect on T cells (Whartenby et al., 2005). FLT-3 is a receptor found on hematopoietic cells and DCs which, upon binding to FLT-3 ligand (FL), promotes cell differentiation, proliferation and survival (Markovic et al., 2005). The importance of FLT-3 in contributing to EAE is also evident by data showing that the onset of EAE is delayed in mice lacking FL (Skarica et al., 2009). Although the mechanism by which FLT-3 inhibition ameliorates EAE has been attributed partially to a decrease in the number of dendritic cells within the brain, it has also been reported that both the number of microglia cells and the percentage of CD86hi microglia cells decrease within the brain of mice with EAE after treatment with the FLT-3 inhibitor, CEP-701(Skarica et al., 2009). To expand upon these findings, the aim of this study was to determine whether FLT-3 is a relevant target within MS brain and to further probe the potential role that FLT-3 plays in modulating microglia function.
Dendritic cells participate in immune surveillance of the CNS and are found in normal brain in choroid plexus and brain meninges but not within normal brain parenchyma(Matyszak and Perry, 1996; McMenamin, 1999). In MS brain however, dendritic cells have been detected within perivascular cuffs of acute and chronic active lesions and in non-lesioned gray matter (Cudrici et al., 2007; Serafini et al., 2000). We therefore used immunohistochemistry to assess the localization of FLT-3 and CD209, a marker for immature dendritic cells, in MS lesions as compared to normal brain and to determine the pattern of FLT-3 expression relative to dendritic cells. We show in this report that FLT-3 is elevated in MS lesions compared to normal control brain tissue and furthermore that two populations of FLT-3 expressing cells are evident. We find that in MS plaques there exists a population of cells expressing both FLT-3 and CD209 in perivascular regions, while another population of cells expressing only FLT-3 is present in these regions and in the brain parenchyma. Taken together, these data suggest FLT-3 is expressed on dendritic cells in perivascular regions of the MS brain, but FLT-3 is also expressed on a non-dendritic cell type such as the microglia in these regions and within the brain parenchyma. Furthermore, we find that treatment of activated microglia with FLT-3 inhibitor, CEP-701, results in a dose-dependent decrease of surface expression of MHCII and CD86 and secretion of IL-6. These findings suggest that FLT-3 is a relevant target within MS brain and furthermore, that FLT-3 modulates microglia function.
MATERIALS AND METHODS
Animals
Female C57 BL/6 mice were purchased from NCI (Frederick, MD) and housed in microisolater cages to maintain a pathogen-free environment. Animals were provided with autoclaved food and water ad libitum, and used between 8–14 weeks of age. All procedures were performed in accordance with NIH guidelines and approved by Johns Hopkins Animal Care and Use Committee.
Immunohistochemistry
Frozen human brain tissue was obtained at autopsy from patients with clinically diagnosed MS from Human Brain and Spinal Fluid Resource Center, Veterans Affairs West Los Angeles Health Care Center, Los Angeles, CA. Active MS lesions, normal appearing gray matter (NAGM) and normal adjacent white matter (NAWM) were identified as described previously(Cudrici et al., 2007). Control brain tissue was obtained from Cooperative Human Tissue Network, Charlottesville, VA. Immunohistochemistry was performed as described previously(Cudrici et al., 2007). Briefly, 4–6 μM cryrostat sections were fixed in acetone with 0.3% of H2O2 for 10 minutes. Sections were incubated with rabbit polyclonal antibody against FLT-3 (clone M-20) diluted 1:50, (Santa Cruz, San Diego) and incubated for 2 hours at room temperature. After washing, sections were incubated with goat anti-rabbit horseradish peroxidase-conjugated IgG (Jackson Immunoresearch Laboratories, West Grove, PA). NovaRED (Vector Labs, Burlingame, CA) was used to develop specific reaction. For double staining, sections were prepared as described above for the FLT3 staining then deperoxidated in 0.3% H2O2 to remove excess peroxidase. Cryosections were then incubated with mouse monoclonal antibody against CD209 (clone 120507) diluted 1:50 (R&D systems; Minneapolis, MN) for two hours at room temperature. After washing, sections were reacted with horseradish-peroxidase conjugated goat anti-mouse antibody (Jackson Immunoresearch Laboratories) and then diaminobenzidine (Pierce, Rockford, IL). Double stained cells were quantified by light microscopy using a 40X objective. Control sections were prepared by immunostaining without the primary antibody, or using control isotype IgG instead of the primary antibody.
Confocal methods
Brain tissues for confocal microcopy were obtained at autopsy and fixed in either 10% buffered formalin or 4% paraformaldehyde and fixed overnight. Tissues were cryoprotected consequtively with 10%-, 20% and 30%-sucrose solutions, frozen at −70°C until use. Immediately before use for immunostaining, brain tissues were sectioned with a sliding microtome for obtaining 40 μm thick sections. Following cutting, the sections were blocked and permeabilized with 5% normal goat serum and 0.4% Triton X-100 for one hour and subsequently incubated with the primary antibodies (rabbit anti FLT-3, clone M-20 and mouse anti-CD209) for 48 hours at 4°C. Following the incubation with primary antibodies the sections were rinsed with PBS and incubated with the secondary fluorophore-tagged antibodies (Cy3 or Alexa) for 3 hours. The sections were rinsed, mounted and coverslipped with Mowoil. Brain sections were examined in a LSM510 Zeiss confocal microscope. Z-sectons of areas of interest were then reconstructed for 3D reconstruction.
Quantification method used to identify extent of FLT-3 staining
The staining intensity of the FLT-3 deposits was evaluated independently by two investigators in a blinded fashion. The intensity of staining was graded as follows: negative (−), slightly positive (+), positive (++), or highly positive (+++).
Microglia isolation and culture conditions
Microglia were isolated as previously described (Carson et al., 1998) with slight modifications. Briefly, mice were perfused with chilled PBS. Brains were removed from skulls. Meninges, choroid plexus, and cerebellum were removed from each brain. Brains were minced using a razor blade and pressed through a pre-wet 100uM nylon filter (BD Falcon, San Jose, CA). Cells were incubated in HBSS containing calcium and magnesium with 60 ug/mL of Liberase Blendzyme (Roche Applied Science, Indianapolis, IN) and 28.5 U/mL of DNASE 1 (Sigma) for 20 minutes at 37°C with gentle agitation. After quenching the enzymatic reaction with HBSS containing 10% fetal calf serum, cells were washed. Microglia were collected at the interface of a 37% and 70% Percoll (GE Healthcare, Piscataway, NJ) gradient after centrifuging at 950×G for 20 minutes at room temperature. After washing, microglia cells were plated at 1 × 105 cells/mL in 24 well plates (Becton Dickinson, Franklin Lakes, NJ) in microglia media: low glucose DMEM (Invitrogen, Carlsbad, CA), 50μM 2-B–mercaptoethanol (Sigma), 2mM L-glutamine (Gibco, Carlsbad, CA), 50μg/mL gentamicin (quality biological, Gaithersburg, MD), 10% FCS (Mediatech, Manassas, VA) and 10ng/mL MCSF (R&D Systems, Minneapolis, MN) to maintain microglia in the resting state, while allowing for proliferation and activation in response to stimulating conditions as previously described (Ponomarev et al., 2005a). Cells were incubated at 37°C in 5% CO2.
For functional assays, microglia cells were seeded in 24 well plates (Becton Dickinson) in microglia media (described above). Confluent cultures were stimulated with either 100 ng/mL of LPS (Sigma) or 10ng/mL of IFN-γ (PeproTech, Rocky Hill, NJ) and treated with specified concentrations of FLT-3 inhibitor, CEP-701 (Lestaurtinib; LC Laboratories, Woburn, MA) or vehicle control, Dimethyl Sulfoxide (DMSO) (Fischer Scientific, Hanover Park, IL), for 24 hours in serum free media at 37°C in 5% CO2. We found no effect of DMSO at the concentrations used. (Data not shown.)
Immunocytochemistry of mouse microglia cells in culture
Cultured primary microglia cells were seeded in Lab Tek© permonox or glass chamber slides (Nalge Nunc Intl., Rochester, NY) at 37°C with 5% CO2 for 24 hours. Immunostaining was performed as previously described (Potter et al., 2007) with slight modifications. Briefly, cells were washed in PBS and fixed with 4% paraformaldehyde for 15 minutes at room temperature. After washing with PBS cells were permeablized with 0.4% Triton X 100 (Sigma, St. Louis) for 15 minutes, and blocked with 10% donkey serum + 0.1% Triton X 100 for 1 hour at room temperature. Cells were then incubated at 4°C overnight with either rabbit-anti ionized calcium binding adaptor molecule 1 (Iba-1) (Wako, Richmond, VA) or anti-glial fibrillary acidic protein (GFAP) (Zymed, Carlsbad, CA) at a dilution of 1:200 in blocking solution (5% donkey serum+ 0.1% Triton X 100). After washing, cells were incubated with Alexa Fluor® 488 donkey anti-rabbit antibody (Molecular Probes, Carlsbad, CA) for 50 minutes at room temperature. After washing, Fluor-gel (Electron Microscopy Sciences, Hatfield, PA) containing 0.5 ug/mL DAPI stain was used to adhere cover slips to slides. Cells were visualized using an epi-fluorescence microscope (Olympus; Center Valley, PA).
Enzyme-Linked Immunosorbent Assay (ELISA)
IL-6 protein concentration of supernatants collected from microglia cultures was determined using an IL-6 ELISA kit, BD OPT EIA (BD Biosciences, San Diego, CA) according to manufacturer instructions. Data was collected on a Biorad microplate reader model 680 (Bio-Rad Laboratories, Hercules, CA) and analyzed using Microplate Manager 5.2 (Bio-Rad Laboratories).
FACS analysis
Fc receptor and non-specific antibody interactions were blocked with CD16/32 (clone 2.4G2) (BD Pharmingen; San Diego, CA) in 5% bovine serum albumin in PBS and stained for flow cytometric analysis using the following antibodies, stains, and isotype controls: FITC CD45 (clone Ly-5); PE CD86 (clone GL1); PE CD11b (clone M1/70), PE IgG2b, and FITC rIgG2b, purchased from eBioscience (San Diego, CA) and FITC I-Ab (clone AF6-120.1), 7AAD, FITC mIgG2a and PE rIgG2a, purchased from (BD Pharmingen; San Diego, CA). Data were collected using a BD FACSCalibur (BD Biosciences Immunocytometry, San Jose, CA) and analyzed with CellQuest software (BD Biosciences). For analyses, cells were gated on live cells using forward vs. side scatter and a viability stain (7AAD) and CD11b or CD45 positive cells.
Statistical analysis
Data were normalized to expression detected for the condition in which cells were stimulated with IFN-γ or LPS and treated with vehicle (DMSO) alone. Before analyzing data, we tested for outliers and they were excluded using standard statistical methods. PRISM Graphpad™ software was used to perform statistical analyses. A one-way ANOVA was performed. To determine whether significant differences exist between stimulated microglia cells treated with vehicle (DMSO) vs. stimulated microglia cells treated with FLT-3 inhibitor CEP 701, Bonferroni’s multiple comparison post-hoc test was applied.
RESULTS
Two populations of FLT-3 expressing cells, with and without CD209 colocalization are found in parenchymal and perivascular regions of MS brain
We first investigated whether FLT-3 expression is elevated within brains obtained from MS cases relative to control brains obtained from subjects without clinical evidence of MS. We found increased expression of FLT-3 in plaques in NAGM and NAWM in brains from MS subjects compared to a lack of FLT-3 immunostaining in gray and white matter in normal control brains (see Table 1).
Table 1.
FLT-3 expression in MS brain
| Case | Sex | Age | Lesion types | No of lesion | Lesional activity | Parenchymal staining | Perivascular staining |
|---|---|---|---|---|---|---|---|
| 1 | F | 38 | Frontal Plaque | 3 | Acute | + | ++ |
| NAWM | + | + | |||||
| NAGM | ++ | ++ | |||||
| 2 | M | 62 | Parietal plaque | 3 | Chronic active | ++ | ++ |
| NAWM | ++ | ++ | |||||
| NAGM | ++ | ++ | |||||
| 3 | F | 51 | Frontal Plaque | 2 | Acute | + | + |
| NAWM | + | ++ | |||||
| 4 | F | 51 | Parietal plaque | 1 | NAWM | + | + |
| 5 | F | 61 | Occipital plaque | 3 | Chronic active | + | ++ |
| NAWM | + | ++ | |||||
| NAGM | +++ | +++ | |||||
| 6–9 | Normal | 10 | Control WM | − | − | ||
| Control GM | − | − | |||||
| Meninges | − | + |
F, female; M, male; −, negative; +, slightly positive; ++, positive; +++, highly positive; NAWM, normal adjacent white matter, NAGM, normal adjacent gray matter.
Since it is known that dendritic cells express FLT-3(Markovic et al., 2005) and dendritic cells have been detected within perivascular cuffs of acute and chronic active lesions in MS brain but not within normal brain parenchyma (Matyszak and Perry, 1996; McMenamin, 1999; Serafini et al., 2000; Serafini et al., 2006), we sought to determine if all cells expressing FLT-3 within MS brain tissue also express CD209, a marker for immature dendritic cells. Dendritic cells have been reported to reside in normal brain in choroid plexus and brain meninges for immune surveillance(Matyszak and Perry, 1996; McMenamin, 1999). We therefore used choroid plexus as a positive control and found FLT-3 protein expression in the choroid plexus in normal brain (Figure 1B) as expected and in perivascular regions of MS plaque (Figure 1A). We found FLT-3 (red) and CD209 (black) colocalization in perivascular regions of plaques in MS brain (Figure 1C) and in normal gray matter from MS brain (Figure 1E), and found that some of these FLT-3 expressing cells did not colocalize with CD209. We found no expression of FLT-3 in MS brain tissue when isotype antibody was used as a control (Figure 1F). Figure 1D illustrates FLT-3 expression in parenchymal regions of plaques in MS brain, but in this region, FLT-3 staining did not colocalize with CD209. Taken together, these data suggest that the population of cells that express FLT-3 and CD209 in the perivascular regions of the brain are dendritic cells, but that a population of cells expressing FLT-3, which do not express CD209, interspersed in perivascular regions of MS plaques and extensively within the brain parenchyma, may be a non-dendritic cell type. Based on the morphology these FLT-3+ DC209− cells most probably are microglia. Since some CD209+ cells also express CD68(Cudrici et al., 2007), it is possible that some of these FLT-3+ cells are perivascular macrophages.
Figure 1.

FLT-3 (red) immunostaining in human brain. FLT-3 protein expression (arrow heads) in perivascular regions of an MS plaque (A) and in the choroid plexus in normal brain (B). FLT-3 (red) expression is colocalized (arrows) with a dendritic cell marker CD209 (black) in regions of plaques in MS brain in perivascular regions (C), and in normal non-lesional gray matter from MS brain (E). FLT-3 expression alone is evident in parenchymal regions of plaques in MS brain (D) while no FLT-3 expression is apparent in controls of immunoperoxidase reaction when the isotype control antibody was used (F). (Scale bars indicate 20 μM. Original Magnification: x400)
We next used double immunostaining and confocal microscopy to determine if a subset of FLT-3 expressing cells colocalizes with CD209 in MS plaques. Again, we found two populations of cells expressing FLT-3. One population of cells expresses both FLT-3 and CD209, while the other population expresses FLT-3 alone (Figure 2).
Figure 2.

Fluorescent immunostaining for FLT-3 (green) and CD209 (red) in a human MS plaque. (Scale bars indicate 10 μM. Original Magnification: 40x)
FLT-3 inhibition with CEP-701 induces a dose dependent decrease in MHCII and CD86 protein expression in activated microglia in vitro
To determine if FLT-3 affects microglia function, microglia were first isolated from mouse brains using a method previously described to achieve high purity and placed in culture to induce proliferation to increase yield(Carson et al., 1998; Ponomarev et al., 2005a). Fluorescent immunostaining with microglia marker, ionized calcium binding adaptor molecule 1 (Iba-1), or astrocytic marker, glial fibrillary acidic protein, and DAPI nuclear stain was performed to confirm that cells were microglia. The data show that most cells express Iba-1, while no cells express GFAP, indicating a high purity of microglia. See Figure 3. These data therefore confirm that the cells used in the in-vitro culture express Iba-1, as would be expected for isolated microglia or perivascular macrophages.
Figure 3.
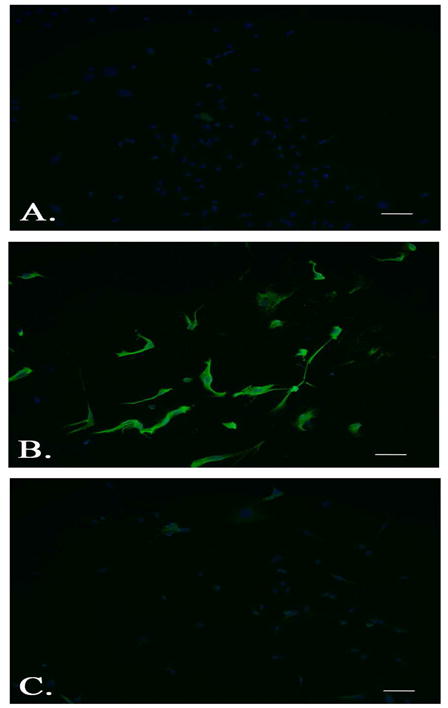
Ionized calcium binding adaptor molecule 1 (Iba-1) expression in murine microglia. Primary cultured microglia immunostained with rabbit monoclonal antibodies against Iba-1 (B.), or glial fibrillary acidic protein (C.) and nuclear DAPI staining compared to no primary control (A.) (Scale bars indicate 50 μM; Original magnification 20x).
We next investigated if FLT-3 inhibition modulates microglia activation. To address this, microglia were isolated from mouse brains, placed in culture to induce proliferation, stimulated with IFN–γ, and treated with a FLT-3 inhibitor, CEP-701, or vehicle control, DMSO. Prior to IFN–γ stimulation, microglia expressed essentially no MHCII but expressed robust MHCII after IFN–γ stimulation as shown in Figure 4, (Ponomarev et al., 2005a). CEP-701 has been reported to be potent (IC50 = 56nM for hematopoietic cells) and a selective inhibitor of FLT-3 where the most closely related receptor tyrosine kinases (PDGFR-B, FMS, and KIT) are not affected by CEP-701 until greater than 500nM (Levis et al., 2002; Weisel et al., 2007). We found no effect of CEP-701 on microglia cell death at the concentrations used in this report (data not shown). We find a statistically significant dose dependent decrease of MHCII and CD86 protein expression in activated microglia cells treated with CEP-701 (P<.05). See Figure 4. Activated microglia cells treated with 10, 25, 50, or 100 nM of CEP-701 for 24 hours have significantly less protein expression of MHCII relative to expression in activated microglia cells treated only with vehicle control, DMSO (P<.05). See Figure 4A. Similarly, activated microglia cells treated with 5, 10, 25, 50, or 100 nM of CEP-701 for 24 hours have significantly less protein expression of CD86 relative to expression found in activated microglia cells treated only with vehicle control, DMSO (P<.05). See figure 4B.
Figure 4.

Expression of (A.) MHCII or (B.) CD86 in un-stimulated (−) microglia cells or microglia cells stimulated with IFN-γ (+) and treated with 0, 5, 10, 25, 50, or 100 nM of CEP-701 for 24 hours. Data are an average of 6 separate experiments and are presented as percent change relative to expression in microglia cells stimulated with IFN-g and treated with vehicle control, DMSO only. Error bars represent standard deviation. * Denotes significant differences (P<.05) compared to expression in cells stimulated with IFN-γ and treated with vehicle control, DMSO only.
FLT-3 inhibition with CEP-701 induces a dose dependent decrease in IL-6 secretion from activated microglia in-vitro
We next examined whether FLT-3 inhibition also suppresses activated microglia cell secretion of proinflammatory cytokine IL-6. Our data corroborate other findings that stimulation with LPS increases microglia cell secretion of IL-6 (Gottschall et al., 1995). Our data further show that LPS-stimulated microglia cells treated with 10, 25, and 50 nM of CEP-701 secrete significantly less IL-6 compared to activated microglia cells treated with only vehicle control, DMSO, in a dose dependent manner (P<.05). See figure 5.
Figure 5.

Expression of secreted IL-6 (pg/mL) from un-stimulated (−) microglia cells or microglia cells stimulated with LPS (+) and treated with 0, 5, 10, 25, or 50 nM of CEP-701 for 24 hours. Data are an average of 4 separate experiments and presented as percent change relative to IL-6 expression in microglia cells stimulated with LPS and treated with vehicle control, DMSO only. Error bars represent standard deviation. * Denotes significant differences (P<.05) compared to IL-6 expression in cells stimulated with LPS and treated with vehicle control, DMSO only.
DISCUSSION
In this study, we report that FLT-3 staining is observed in acute and chronic active lesions as well as in NAWM and NAGM from MS brains but not in normal white or gray matter in control brain. Newly emerging magnetic resonance imaging technology such as diffusion tensor imaging has allowed detection of pathological progression of MS previously undetected in NAWM and NAGM while metabolite changes in these areas have been associated with disability (Guo et al., 2002; Phuttharak et al., 2007; Sastre-Garriga et al., 2005; Vrenken and Geurts, 2007). In addition to axonal damage and neuronal loss, microglia activation has been found in both NAWM and NAGM (Gray et al., 2008; Kutzelnigg et al., 2005; Lassmann, 2008). Therefore, our finding that FLT-3 is expressed in MS brain is not only novel, but provides further illustration of the changes that occur in the NAWM and NAGM in MS brain.
While FLT-3 is known to be expressed on dendritic cells, this report is the first to show FLT-3 protein expression in MS brains. Dendritic cells expressing CD209 have been found in perivascular regions around lymphocyte infiltrate in early active lesions, at the borders of chronic active lesions and within non-lesioned gray matter within MS brain(Cudrici et al., 2007; Serafini et al., 2006). Furthermore, dendritic cells were found to lie in close proximity to lymphocytes(Cudrici et al., 2007). We show in this report colocalization of FLT-3 and CD209 in perivascular regions of plaques in MS brain and in some regions of NAGM from MS brain indicating that these FLT-3+ CD209+ cells are infiltrating dendritic cells. A subset of CD209 cells have been found to express CD68 (Cudrici et al., 2007), which suggests the possibility of macrophages as a subpopulation of FLT-3+ CD209+ cells. Interestingly, we also found that some FLT-3 expressing cells did not colocalize with CD209, suggesting that cells such as microglia may express FLT-3 in these areas. Due to the limitations of specific surface molecules that would distinguish between macrophage and microglia, we cannot conclusively distinguish between infiltrating macrophage and resident microglia in these experiments.
Two key pathological events within the CNS—T cell retention and the development of lesions—are dependent upon the presence of cells within the brain that are capable of presenting antigen to infiltrating T cells(Jack et al., 2005). In addition to dendritic cells, other cell types present antigen to infiltrating T cells and are involved in propagating the inflammatory process in MS and EAE (Greter et al., 2005; Hickey and Kimura, 1988). For example, dendritic cells in rat EAE were found mostly in perivascular cuffs, but comprised only 2% of MHCII expressing cells, suggesting other cells are also participating in antigen presentation during CNS inflammation(Matyszak and Perry, 1996). These findings suggest that other, non-dendritic cells may also be available to present antigen to infiltrating T cells. In addition to infiltrating dendritic cells, microglia, resident macrophage within the brain, respond to environmental cues and after becoming activated are also capable of antigen presentation in MS and EAE(Aloisi et al., 1999; Jack et al., 2005). Microglia contribute to MS pathogenesis by playing a role in initiating the inflammatory process within the CNS, removing debris, and promoting tissue damage(Jack et al., 2005). Microglia activation has been detected in pre-active lesions which consist of perivascular and intravascular accumulation of lymphocytes without demyelination as well as active and chronic demyelinating MS lesions (Barnett and Prineas, 2004; De Groot et al., 2001; Jack et al., 2005). In this report we demonstrate that in addition to some perivascular regions in MS lesions, FLT-3 staining is detected without colocalization with dendritic cell marker CD209 within MS brain parenchyma. This again suggests that FLT-3 is also expressed within MS brain on a non-dendritic cell type such as the microglia. Our finding that FLT-3 is expressed on cells within MS brain, which may have the capacity to present antigen, has implications in therapeutically targeting two cell populations within MS brain to control CNS inflammation.
Once we established that FLT-3 is expressed in MS brain in non-dendritic cells we went on to investigate the role that FLT-3 plays in modulating microglia function. Microglia, as well as other cells, can upregulate HLA-Class II and CD86—surface molecules necessary for antigen presentation during MS and EAE, respectively—and have been shown to be capable of presenting antigen and reactivating infiltrating peripheral lymphocytes(Heppner et al., 2005; Hickey and Kimura, 1988; Juedes and Ruddle, 2001; van der Valk and Amor, 2009). Kinetic studies of microglia activation after induction of EAE have shown that the timing of microglia activation in which MHCII and CD86 are up-regulated coincides with lymphocyte trafficking into the brain, (Ponomarev et al., 2005b). The importance of antigen presenting cells within the CNS in MS is further illustrated by data indicating that whether T cells have high or low pathogenicity is dependent on the extent of T cells reactivation within the CNS (Kawakami et al., 2004). Furthermore, impairing microglia function blocks the initiation of EAE(Heppner et al., 2005).
We show in this report that when microglia are stimulated with IFN-γ in the presence of the FLT-3 inhibitor, CEP-701, expression of surface molecules, MHC II and CD86, is significantly diminished compared to IFN-γ-stimulated-microglia not treated with FLT-3 inhibitor. These data indicate that FLT-3 inhibition impairs microglia capacity to respond to environmental cues to alter cell phenotype to function as an antigen presenting cell. Furthermore, these data extend the findings that FLT-3 inhibition in EAE ameliorates pathogenesis by inhibiting dendritic cell function, (Skarica et al., 2009) by providing new evidence that FLT-3 inhibition also diminishes the ability of microglia to respond to activation signals to express MHCII and CD86. These data provide additional evidence that FLT-3 inhibition of microglia may also be a potential mechanism by which FLT-3 inhibition may ameliorate CNS inflammation in EAE and MS.
Microglia and dendritic cells have both been found to function as antigen presenting cells in the CNS and contribute to the pathogenesis of EAE and MS(Greter et al., 2005; Heppner et al., 2005; Jack et al., 2005; Serafini et al., 2006). While it has been shown that dendritic cells are sufficient to facilitate the entry of previously activated T lymphocytes into the CNS and the subsequent development of EAE it has also been demonstrated in-vivo that microglia are essential for maintenance of the effector phase of EAE (Becher et al., 2003; Greter et al., 2005; Heppner et al., 2005). These data suggest that microglia and dendritic cells may be implicated in promoting CNS inflammation at different times within the course of disease. Similarly, FLT-3 inhibition may mediate CNS inflammation by affecting different cell populations throughout the course of CNS-inflammatory disease. While it has been shown that FLT-3 inhibition ameliorates early stages of EAE primarily by inhibiting DCs, with only a minimal effect on microglia(Skarica et al., 2009), FLT-3 inhibition of microglia may play a more significant role in modulating CNS inflammation at later stages of disease. We show in this report that FLT-3 is expressed in two cell populations within different regions of MS brain and furthermore, we report that FLT-3 modulates the capacity of microglia to function as an antigen presenting cell. Therefore, FLT-3 inhibition may be a therapeutic strategy to hinder the function of both dendritic cells that have entered the CNS from the periphery and CNS-resident microglia, as a means to inhibit the pathogenesis of MS both at the initiation and propagation phases.
Activated microglia, in addition to functioning as an antigen presenting cell, secrete pro-inflammatory cytokines such as IL-6(Gottschall et al., 1995). We report here that inhibiting FLT-3 in LPS-stimulated-microglia reduces microglia secretion of IL-6 compared to stimulated microglia not treated with FLT-3 inhibitor. The ability to therapeutically inhibit activation induced secretion of IL-6 in the CNS by microglia cells have implications in controlling CNS inflammation because of the role that IL-6 has in orchestrating the inflammatory process within the CNS. IL-6 is one of the factors involved in the differentiation of the recently described Th17 lymphocyte which has been implicated in contributing to inflammation in the CNS in EAE and MS (Bettelli et al., 2006; Langrish et al., 2005). Furthermore, additional reports show that IL-6 is necessary for EAE development specifically because of its role in promoting the activation and differentiation of T cells, notably, Th17 cell differentiation(Samoilova et al., 1998; Serada et al., 2008) (Mendel et al., 1998). While it has been clearly demonstrated that IL-6 plays a role in pathogenic T cell differentiation within the periphery, IL-6 production within the CNS has also been found to play a significant role in CNS inflammation. CD14+ monocytes have been suggested to cross the BBB to acquire CD209 dendritic cell phenotype, secrete IL-6, and promote proliferation of infiltrating Th17 lymphocytes(Ifergan et al., 2008). In addition, CSF levels of IL-6 correlated with nitric oxide production, neuronal injury, and predicted disability in patients with inflammatory spinal cord injury from transverse myelitis (TM)(Kaplin et al., 2005). These findings suggest that IL-6 within the CNS is critical for T cell pathogenesis in CNS inflammation. This is further suggested by reports describing IL-6 and IL-17 gene transcripts within MS lesions(Lock et al., 2002). Additionally, IL-6 from non-dendritic cells was found to be important in determining the site of CNS inflammation in a study in which transgenic GFAP-IL-6 mice were used to restrict IL-6 to astrocytes within the cerebellum. In this study, CNS inflammation was targeted specifically to the CNS region in which IL-6 was present(Quintana et al., 2009). Taken together, these data suggest that FLT-3 inhibition within the CNS may be a means to ameliorate CNS inflammation by inhibiting IL-6 secretion in both microglia and dendritic cells within the CNS, and thereby inhibit differentiation and accumulation of pathogenic T cells within the CNS.
In this report we demonstrate two populations of FLT-3 expressing cells in MS brain, one population being dendritic cells and we suggest the other population may be microglia. We go on to show that FLT-3 inhibition modulates microglia cell function by inhibiting activation induced secretion of IL-6 and expression of surface molecules necessary for antigen presentation. Because of the well demonstrated implications that IL-6 production and antigen presentation has in the differentiation and reactivation of pathogenic T cells within the CNS, the data we present in this report suggest that FLT-3 inhibition is a therapeutic strategy to impede CNS inflammation by modulating function of both microglia and dendritic cells.
Acknowledgments
Grant Support: NMSS TR-3760-A-3, FG 1695-A-1, and US Public Health Grant RO1NS 42011
MS brain tissue was received from Human Brain and Spinal Fluid Resource Center, Veterans Affairs West Los Angeles Health Care Center, Los Angeles, CA.
Footnotes
Conflict of Interest Statement
The authors declare that there are no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alliot F, Godin I, Pessac B. Microglia derive from progenitors, originating from the yolk sac, and which proliferate in the brain. Brain Res Dev Brain Res. 1999;117:145–52. doi: 10.1016/s0165-3806(99)00113-3. [DOI] [PubMed] [Google Scholar]
- Aloisi F, Ria F, Columba-Cabezas S, Hess H, Penna G, Adorini L. Relative efficiency of microglia, astrocytes, dendritic cells and B cells in naive CD4+ T cell priming and Th1/Th2 cell restimulation. Eur J Immunol. 1999;29:2705–14. doi: 10.1002/(SICI)1521-4141(199909)29:09<2705::AID-IMMU2705>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- Barnett MH, Prineas JW. Relapsing and remitting multiple sclerosis: pathology of the newly forming lesion. Ann Neurol. 2004;55:458–68. doi: 10.1002/ana.20016. [DOI] [PubMed] [Google Scholar]
- Becher B, Durell BG, Noelle RJ. IL-23 produced by CNS-resident cells controls T cell encephalitogenicity during the effector phase of experimental autoimmune encephalomyelitis. J Clin Invest. 2003;112:1186–91. doi: 10.1172/JCI19079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, Weiner HL, Kuchroo VK. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235–8. doi: 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]
- Bjartmar C, Yin X, Trapp BD. Axonal pathology in myelin disorders. J Neurocytol. 1999;28:383–95. doi: 10.1023/a:1007010205037. [DOI] [PubMed] [Google Scholar]
- Block ML, Hong JS. Microglia and inflammation-mediated neurodegeneration: multiple triggers with a common mechanism. Prog Neurobiol. 2005;76:77–98. doi: 10.1016/j.pneurobio.2005.06.004. [DOI] [PubMed] [Google Scholar]
- Carson MJ, Reilly CR, Sutcliffe JG, Lo D. Mature microglia resemble immature antigen-presenting cells. Glia. 1998;22:72–85. doi: 10.1002/(sici)1098-1136(199801)22:1<72::aid-glia7>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- Cudrici C, Ito T, Zafranskaia E, Niculescu F, Mullen KM, Vlaicu S, Judge SI, Calabresi PA, Rus H. Dendritic cells are abundant in non-lesional gray matter in multiple sclerosis. Exp Mol Pathol. 2007;83:198–206. doi: 10.1016/j.yexmp.2007.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Groot CJ, Bergers E, Kamphorst W, Ravid R, Polman CH, Barkhof F, van der Valk P. Post-mortem MRI-guided sampling of multiple sclerosis brain lesions: increased yield of active demyelinating and (p)reactive lesions. Brain. 2001;124:1635–45. doi: 10.1093/brain/124.8.1635. [DOI] [PubMed] [Google Scholar]
- Gottschall PE, Yu X, Bing B. Increased production of gelatinase B (matrix metalloproteinase-9) and interleukin-6 by activated rat microglia in culture. J Neurosci Res. 1995;42:335–42. doi: 10.1002/jnr.490420307. [DOI] [PubMed] [Google Scholar]
- Gray E, Thomas TL, Betmouni S, Scolding N, Love S. Elevated activity and microglial expression of myeloperoxidase in demyelinated cerebral cortex in multiple sclerosis. Brain Pathol. 2008;18:86–95. doi: 10.1111/j.1750-3639.2007.00110.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greter M, Heppner FL, Lemos MP, Odermatt BM, Goebels N, Laufer T, Noelle RJ, Becher B. Dendritic cells permit immune invasion of the CNS in an animal model of multiple sclerosis. Nat Med. 2005;11:328–34. doi: 10.1038/nm1197. [DOI] [PubMed] [Google Scholar]
- Guo AC, MacFall JR, Provenzale JM. Multiple sclerosis: diffusion tensor MR imaging for evaluation of normal-appearing white matter. Radiology. 2002;222:729–36. doi: 10.1148/radiol.2223010311. [DOI] [PubMed] [Google Scholar]
- Heppner FL, Greter M, Marino D, Falsig J, Raivich G, Hovelmeyer N, Waisman A, Rulicke T, Prinz M, Priller J, Becher B, Aguzzi A. Experimental autoimmune encephalomyelitis repressed by microglial paralysis. Nat Med. 2005;11:146–52. doi: 10.1038/nm1177. [DOI] [PubMed] [Google Scholar]
- Hickey WF, Kimura H. Perivascular microglial cells of the CNS are bone marrow-derived and present antigen in vivo. Science. 1988;239:290–2. doi: 10.1126/science.3276004. [DOI] [PubMed] [Google Scholar]
- Ifergan I, Kebir H, Bernard M, Wosik K, Dodelet-Devillers A, Cayrol R, Arbour N, Prat A. The blood-brain barrier induces differentiation of migrating monocytes into Th17-polarizing dendritic cells. Brain. 2008;131:785–99. doi: 10.1093/brain/awm295. [DOI] [PubMed] [Google Scholar]
- Jack C, Ruffini F, Bar-Or A, Antel JP. Microglia and multiple sclerosis. J Neurosci Res. 2005;81:363–73. doi: 10.1002/jnr.20482. [DOI] [PubMed] [Google Scholar]
- Juedes AE, Ruddle NH. Resident and infiltrating central nervous system APCs regulate the emergence and resolution of experimental autoimmune encephalomyelitis. J Immunol. 2001;166:5168–75. doi: 10.4049/jimmunol.166.8.5168. [DOI] [PubMed] [Google Scholar]
- Kaplin AI, Deshpande DM, Scott E, Krishnan C, Carmen JS, Shats I, Martinez T, Drummond J, Dike S, Pletnikov M, Keswani SC, Moran TH, Pardo CA, Calabresi PA, Kerr DA. IL-6 induces regionally selective spinal cord injury in patients with the neuroinflammatory disorder transverse myelitis. J Clin Invest. 2005;115:2731–41. doi: 10.1172/JCI25141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawakami N, Lassmann S, Li Z, Odoardi F, Ritter T, Ziemssen T, Klinkert WE, Ellwart JW, Bradl M, Krivacic K, Lassmann H, Ransohoff RM, Volk HD, Wekerle H, Linington C, Flugel A. The activation status of neuroantigen-specific T cells in the target organ determines the clinical outcome of autoimmune encephalomyelitis. J Exp Med. 2004;199:185–97. doi: 10.1084/jem.20031064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreutzberg GW. Microglia: a sensor for pathological events in the CNS. Trends Neurosci. 1996;19:312–8. doi: 10.1016/0166-2236(96)10049-7. [DOI] [PubMed] [Google Scholar]
- Kutzelnigg A, Lucchinetti CF, Stadelmann C, Bruck W, Rauschka H, Bergmann M, Schmidbauer M, Parisi JE, Lassmann H. Cortical demyelination and diffuse white matter injury in multiple sclerosis. Brain. 2005;128:2705–12. doi: 10.1093/brain/awh641. [DOI] [PubMed] [Google Scholar]
- Langrish CL, Chen Y, Blumenschein WM, Mattson J, Basham B, Sedgwick JD, McClanahan T, Kastelein RA, Cua DJ. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med. 2005;201:233–40. doi: 10.1084/jem.20041257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lassmann H. The pathologic substrate of magnetic resonance alterations in multiple sclerosis. Neuroimaging Clin N Am. 2008;18:563–76. ix. doi: 10.1016/j.nic.2008.06.005. [DOI] [PubMed] [Google Scholar]
- Levis M, Allebach J, Tse KF, Zheng R, Baldwin BR, Smith BD, Jones-Bolin S, Ruggeri B, Dionne C, Small D. A FLT3-targeted tyrosine kinase inhibitor is cytotoxic to leukemia cells in vitro and in vivo. Blood. 2002;99:3885–91. doi: 10.1182/blood.v99.11.3885. [DOI] [PubMed] [Google Scholar]
- Lock C, Hermans G, Pedotti R, Brendolan A, Schadt E, Garren H, Langer-Gould A, Strober S, Cannella B, Allard J, Klonowski P, Austin A, Lad N, Kaminski N, Galli SJ, Oksenberg JR, Raine CS, Heller R, Steinman L. Gene-microarray analysis of multiple sclerosis lesions yields new targets validated in autoimmune encephalomyelitis. Nat Med. 2002;8:500–8. doi: 10.1038/nm0502-500. [DOI] [PubMed] [Google Scholar]
- Markovic A, MacKenzie KL, Lock RB. FLT-3: a new focus in the understanding of acute leukemia. Int J Biochem Cell Biol. 2005;37:1168–72. doi: 10.1016/j.biocel.2004.12.005. [DOI] [PubMed] [Google Scholar]
- Matyszak MK, Perry VH. The potential role of dendritic cells in immune-mediated inflammatory diseases in the central nervous system. Neuroscience. 1996;74:599–608. doi: 10.1016/0306-4522(96)00160-1. [DOI] [PubMed] [Google Scholar]
- McGeachy MJ, Bak-Jensen KS, Chen Y, Tato CM, Blumenschein W, McClanahan T, Cua DJ. TGF-beta and IL-6 drive the production of IL-17 and IL-10 by T cells and restrain T(H)-17 cell-mediated pathology. Nat Immunol. 2007;8:1390–7. doi: 10.1038/ni1539. [DOI] [PubMed] [Google Scholar]
- McMenamin PG. Distribution and phenotype of dendritic cells and resident tissue macrophages in the dura mater, leptomeninges, and choroid plexus of the rat brain as demonstrated in wholemount preparations. J Comp Neurol. 1999;405:553–62. [PubMed] [Google Scholar]
- Mendel I, Katz A, Kozak N, Ben-Nun A, Revel M. Interleukin-6 functions in autoimmune encephalomyelitis: a study in gene-targeted mice. Eur J Immunol. 1998;28:1727–37. doi: 10.1002/(SICI)1521-4141(199805)28:05<1727::AID-IMMU1727>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- Phuttharak W, Galassi W, Laopaiboon V, Laopaiboon M, Hesselink JR. Abnormal diffusivity of normal appearing brain tissue in multiple sclerosis: a diffusion-weighted MR imaging study. J Med Assoc Thai. 2007;90:2689–94. [PubMed] [Google Scholar]
- Ponomarev ED, Novikova M, Maresz K, Shriver LP, Dittel BN. Development of a culture system that supports adult microglial cell proliferation and maintenance in the resting state. J Immunol Methods. 2005a;300:32–46. doi: 10.1016/j.jim.2005.02.011. [DOI] [PubMed] [Google Scholar]
- Ponomarev ED, Shriver LP, Maresz K, Dittel BN. Microglial cell activation and proliferation precedes the onset of CNS autoimmunity. J Neurosci Res. 2005b;81:374–89. doi: 10.1002/jnr.20488. [DOI] [PubMed] [Google Scholar]
- Potter EG, Cheng Y, Knight JB, Gordish-Dressman H, Natale JE. Basic science; metallothionein I and II attenuate the thalamic microglial response following traumatic axotomy in the immature brain. J Neurotrauma. 2007;24:28–42. doi: 10.1089/neu.2006.0056.R1. [DOI] [PubMed] [Google Scholar]
- Quintana A, Muller M, Frausto RF, Ramos R, Getts DR, Sanz E, Hofer MJ, Krauthausen M, King NJ, Hidalgo J, Campbell IL. Site-specific production of IL-6 in the central nervous system retargets and enhances the inflammatory response in experimental autoimmune encephalomyelitis. J Immunol. 2009;183:2079–88. doi: 10.4049/jimmunol.0900242. [DOI] [PubMed] [Google Scholar]
- Samoilova EB, Horton JL, Hilliard B, Liu TS, Chen Y. IL-6-deficient mice are resistant to experimental autoimmune encephalomyelitis: roles of IL-6 in the activation and differentiation of autoreactive T cells. J Immunol. 1998;161:6480–6. [PubMed] [Google Scholar]
- Santambrogio L, Belyanskaya SL, Fischer FR, Cipriani B, Brosnan CF, Ricciardi-Castagnoli P, Stern LJ, Strominger JL, Riese R. Developmental plasticity of CNS microglia. Proc Natl Acad Sci U S A. 2001;98:6295–300. doi: 10.1073/pnas.111152498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sastre-Garriga J, Ingle GT, Chard DT, Ramio-Torrenta L, McLean MA, Miller DH, Thompson AJ. Metabolite changes in normal-appearing gray and white matter are linked with disability in early primary progressive multiple sclerosis. Arch Neurol. 2005;62:569–73. doi: 10.1001/archneur.62.4.569. [DOI] [PubMed] [Google Scholar]
- Serada S, Fujimoto M, Mihara M, Koike N, Ohsugi Y, Nomura S, Yoshida H, Nishikawa T, Terabe F, Ohkawara T, Takahashi T, Ripley B, Kimura A, Kishimoto T, Naka T. IL-6 blockade inhibits the induction of myelin antigen-specific Th17 cells and Th1 cells in experimental autoimmune encephalomyelitis. Proc Natl Acad Sci U S A. 2008;105:9041–6. doi: 10.1073/pnas.0802218105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serafini B, Columba-Cabezas S, Di Rosa F, Aloisi F. Intracerebral recruitment and maturation of dendritic cells in the onset and progression of experimental autoimmune encephalomyelitis. Am J Pathol. 2000;157:1991–2002. doi: 10.1016/S0002-9440(10)64838-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serafini B, Rosicarelli B, Magliozzi R, Stigliano E, Capello E, Mancardi GL, Aloisi F. Dendritic cells in multiple sclerosis lesions: maturation stage, myelin uptake, and interaction with proliferating T cells. J Neuropathol Exp Neurol. 2006;65:124–41. doi: 10.1097/01.jnen.0000199572.96472.1c. [DOI] [PubMed] [Google Scholar]
- Skarica M, Wang T, McCadden E, Kardian D, Calabresi PA, Small D, Whartenby KA. Signal transduction inhibition of APCs diminishes th17 and Th1 responses in experimental autoimmune encephalomyelitis. J Immunol. 2009;182:4192–9. doi: 10.4049/jimmunol.0803631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinman L. Nuanced roles of cytokines in three major human brain disorders. J Clin Invest. 2008;118:3557–63. doi: 10.1172/JCI36532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Town T, Nikolic V, Tan J. The microglial “activation” continuum: from innate to adaptive responses. J Neuroinflammation. 2005;2:24. doi: 10.1186/1742-2094-2-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Valk P, Amor S. Preactive lesions in multiple sclerosis. Curr Opin Neurol. 2009;22:207–13. doi: 10.1097/WCO.0b013e32832b4c76. [DOI] [PubMed] [Google Scholar]
- Vrenken H, Geurts JJ. Gray and normal-appearing white matter in multiple sclerosis: an MRI perspective. Expert Rev Neurother. 2007;7:271–9. doi: 10.1586/14737175.7.3.271. [DOI] [PubMed] [Google Scholar]
- Weisel KC, Yildirim S, Schweikle E, Kanz L, Mohle R. Effect of FLT3 inhibition on normal hematopoietic progenitor cells. Ann N Y Acad Sci. 2007;1106:190–6. doi: 10.1196/annals.1392.020. [DOI] [PubMed] [Google Scholar]
- Whartenby KA, Calabresi PA, McCadden E, Nguyen B, Kardian D, Wang T, Mosse C, Pardoll DM, Small D. Inhibition of FLT3 signaling targets DCs to ameliorate autoimmune disease. Proc Natl Acad Sci U S A. 2005;102:16741–6. doi: 10.1073/pnas.0506088102. [DOI] [PMC free article] [PubMed] [Google Scholar]