

Abstract
Psychological stress-associated immune dysregulation has been shown to disrupt the steady state expression and reactivate latent herpes viruses. One such virus is the Epstein Barr virus (EBV), which is associated with several human malignancies. EBV infects >90% of people living in North America and persists for life in latently infected cells. Although several studies have shown that glucocorticoids (GCs) can directly induce reactivation of the latent virus, the mechanism of stress hormone involvement in the control of EBV gene expression is not well understood. In this study, we tested the hypothesis that GCs can induce the latent EBV genome to lytically replicate through the induction of the EBV immediate early gene BZLF1 which encodes the lytic transactivator protein ZEBRA. We show a dose-dependent upregulation of BZLF1 mRNA expression by hydrocortisone (HC) and dexamethasone (Dex) in Daudi cells, an EBV genome positive Burkitt’s lymphoma cell line, and Dex-induction of the early gene products BLLF3 (encoding for the EBV dUTPase) and BALF5 (encoding for the EBV DNA polymerase). We show that Daudi cells express glucocorticoid receptors (GR) that mediate Dex-dependent upregulation of BZLF1 mRNA levels. This effect was inhibited by both the glucocorticoid receptor antagonist RU486 and by cycloheximide. The results suggest that GCs, in addition to inducing stress-related immune dysregulation, can mediate latent EBV reactivation through the induction of the BZLF1 gene.
Keywords: Epstein Barr virus, glucocorticoids, glucocorticoid receptor, herpesvirus reactivation, psychological stress
Introduction
It is now well established that the central nervous system (CNS), the endocrine system, and the immune system interact with each other and that psychological stress can down-regulate/dysregulate the immune response by affecting the interplay between these systems. These interactions are complex, involving both the hypothalamic–pituitary–adrenal (HPA) and the sympathetic-adrenal medullary (SAM) axes (Glaser and Kiecolt-Glaser, 2005; Rabin, 1999). Dysregulation of these interactions results in the suppression of immune function, poor responses to vaccines and in the reactivation of latent herpes viruses including Epstein Barr virus (EBV).
In a series of studies from our laboratory with medical students, examination stress was associated with changes in the steady-state expression of latent EBV. Higher titers of anti-EBV virus capsid antigen (VCA) IgG were observed at the time of examinations compared to baseline blood samples drawn approximately one month before suggesting that EBV was induced to replicate (Glaser et al., 1994; Glaser et al., 1991; Glaser et al., 1987). In follow-up studies using the same model, we examined the impact of stress on two different aspects of EBV-specific memory T-cell responses. In the first study, we found a significant decrease in the ability of EBV-specific cytotoxic T-cells (from EBV seropositive students) to kill EBV-infected autologous B-lymphocytes associated with examination stress (Glaser et al., 1987). In the second study, peripheral blood leukocytes (PBLs) obtained from medical students at the time of exams compared to the baseline blood sample showed a decrease in proliferation when exposed to several purified EBV polypeptides (Glaser et al., 1993). There is a growing literature showing that different types of psychological stressors can reactivate latent EBV (Esterling et al., 1992; Lutgendorf et al., 1994; Mehta et al., 2000; Payne et al., 1999; Sarid et al., 2001; Stowe et al., 2000).
EBV is a human gamma herpesvirus that has been implicated in the pathogenesis of a variety of human malignancies including African Burkitt’s lymphoma (BL), nasopharyngeal carcinoma (NPC), Hodgkin’s disease and post-transplant lymphoproliferative disorder. It is the etiological agent for infectious mononucleosis and has also been associated with chronic fatigue syndrome (Buchwald et al., 1995; Glaser et al., 2005; Jason et al., 2000; Klimas et al., 1990). EBV latently infects human B-lymphocytes and epithelial cells in the nasopharynx after primary infection (Zhang et al., 1989). Studies have shown that EBV genome positive BL cells, such as Daudi cells, express the glucocorticoid receptor (GR) and that glucocorticoids (GCs) are able to reactivate the latent virus in vitro (Bauer, 1983; Joncas and Leyritz, 1974; Magrath et al., 1979). In an earlier study from our laboratory we explored the possibility that other hormones associated with the HPA axis and that are modulated by psychological stress might have the ability to induce reactivation of the latent EBV in Daudi cells. We confirmed that GCs including hydrocortisone (HC) and dexamethasone (Dex) can induce reactivation of the latent virus using pharmacological and physiological concentrations (Glaser et al., 1995). Other stress hormones including corticotrophin-releasing hormone and adrenocorticotropic hormone did not induce reactivation but did enhance the lytic replication of the HR-1 strain of EBV in super-infected Raji cells (Glaser et al., 1995). The results of these studies suggest that multiple endocrine interactions may be involved in the stress-induced reactivation/replication of latent EBV.
In order to further elucidate the mechanism of stress-associated reactivation of latent EBV, we examined the impact of autonomic activity and GCs on the steady-state expression of latent EBV (Cacioppo et al., 2002). We first investigated whether the expression of latent EBV in vivo differed between women who were high and low sympathetic cardiac reactors determined by stress-induced changes in cardiac pre-ejection period (Cacioppo et al., 2002). Results showed that subjects who were high stress reactors had higher anti-EBV-VCA antibody titers than low stress reactors. High reactors also exhibited greater stress-related increases in cortisol than low reactors. The study suggested that individuals who respond to psychological stressors with large increases in cardiac sympathetic activation are also characterized by having high antibody titers to EBV and therefore greater changes in the steady state expression of the latent virus.
Although several studies have shown that GCs can directly induce reactivation of latent EBV, the mechanism of this effect on EBV gene expression is not well understood. In this study we measured the effects of two GCs, HC and Dex, on the steady state expression of latent EBV. We focused on the mRNA expression of the EBV immediate early gene BZLF1 that encodes for the lytic transactivator protein ZEBRA, and the early genes BLLF3 and BALF5, which encode the EBV dUTPase and EBV DNA polymerase, respectively. Furthermore, we compared the Dex-dependent modulation of expression of the above genes to that resulting from the exposure of Daudi cells to 12-O-tetradecanoylphorbol-13-acetate and sodium butyrate (TPA/NaB), potent inducers of EBV reactivation. The results suggest that GC-mediated EBV reactivation may result from the induction of BZLF1 which plays a central role in controlling the switch from latency to lytic replication.
Methods
Cell Lines and Culture Conditions
The EBV genome positive BL cell line Daudi, the EBV positive marmoset B-lymphoblastoid cell line B95-8, the EBV positive epithelial cell lines C666-1 (an NPC cell line) (Cheung et al., 1999) and D98/HR-1 (a hybrid cell line) (Glaser and O’Neill, 1972) were utilized in these studies. The Daudi, B95-8 and C666-1 cells were maintained in RPMI-1640 medium supplemented with 10% fetal bovine serum (FBS), L-glutamine, and 1X antibiotic-antimycotic (Invitrogen). D98/HR-1 cells were maintained in complete MEM with 10% FBS, L-glutamine, 1X antibiotic-antimycotic and hypoxanthine, aminopterine and thymidine (HAT). All cells were maintained at 37°C in a humidified atmosphere with 5% CO2.
Cell Treatments
Daudi and B95-8 cells were seeded (2 × 106 cells/well) in 6-well plates in media containing 10% charcoal-stripped serum and treated with HC or Dex (Sigma-Aldrich) for 6, 12, 24, 48 or 72 hr. Cells were homogenized in TRIzol reagent and stored at −80°C until assayed by real-time PCR as described below. In order to show the role of GR, Daudi cells were either left untreated, or treated with 100 nM Dex, 100 nM of the GR antagonist RU486, or 100 nM Dex and 100 nM RU486 for 6, 12, or 24 hr and prepared for real-time PCR.
In order to compare the kinetics of induction of BZLF1, BLLF3, and BALF5 expression, Daudi cells were treated with 100 nM HC, 100 nM Dex, or 40 ng/ml TPA and 3 mM NaB (TPA/NaB) for 3, 6, 12, 24, 48 or 72 hr and samples were collected for real-time PCR and western blot analyses.
Finally, we examined whether the influence of Dex and TPA/NaB on BZLF1 gene expression in Daudi cells is prevented by cyclohexamide (CHX), an inhibitor of protein synthesis. Daudi cells were either untreated or treated with 100 nM Dex, 33 μg/ml CHX plus 100 nM Dex, TPA/NaB, or 33 μg/ml CHX plus TPA/NaB for 6, 12, and 24 hr and samples were collected for real-time PCR.
Real-time PCR Assessment of BZLF1, BLLF3, and BALF5 Gene Expression
Total RNA from cultured cells was isolated using TRIzol following the manufacturer’s instructions (Invitrogen). First strand cDNAs were synthesized using random primers and MultiScribe Reverse Transcriptase (Applied Biosystems). Levels of BZLF1, BLLF3, and BALF5 mRNA were analyzed using Custom Taqman Gene Expression Assays (Applied Biosystems). Sequences of primers and probes were designed by using Primer Express for Mac v1.5 (BZLF1 and BLLF3; Applied Biosystems) or derived from previous publications (BALF5) (Kimura et al., 1999) (Table 1). The housekeeping gene GAPDH was analyzed using the Taqman Human GAPDH Endogenous Control Reagent (Assay ID: 4326317E, Applied Biosystems) and was used as an internal positive control. Levels of each target mRNA were measured with TaqMan fluorogenic probes listed in Table 1, and amplified using the 7300 Real Time PCR system (Applied Biosystems). Reactions were carried out in a 20 μl volume, and each sample was run in duplicate. The PCR thermal cycle conditions used were according to the manufacturer’s recommendations. The levels of expression of BZLF1, BLLF3, and BALF5 mRNA in each sample were normalized to GAPDH mRNA levels. For the assessment of BZLF1 mRNA levels in B95-8 cells, we utilized the Human ACTB (beta actin) Endogenous Control Reagent (Assay ID; 4333762F; Applied Biosystems) since it was determined that GAPDH mRNA levels were modulated by TPA/NaB treatment in these cells. The relative expression of mRNA species was calculated using the comparative CT method as described by the manufacturer (see User bulletin #2 Applied Biosystems, P/N 4303859, 1997) (Livak and Schmittgen, 2001).
Table 1.
Real-time PCR primers and probes
| Target Gene | Upstream primer (5′–3′) | Downstream primer (5′–3′) | FAM-Probe (5′–3′) |
|---|---|---|---|
| BZLF1 | AATTTAAGAGATCCTCGTGTAAAACATCT | GCGCCTCCTGTTGAAGCA | AATGGAGTCAACATCCAG |
| BLLF3 | GGACGTCAGCCTGACCAACT | CAGAAGCGCCGGTACTTGTT | CCAGACCGTGTTCCT |
| BALF5 | CGGAAGCCCTCTGGACTTC | CCCTGTTTATCCGATGGAATG | TGTACACGCACGAGAATGCGCC |
Western Blot Analysis of Glucocorticoid Receptor Expression
Proteins were extracted from cell pellets using M-PER (Thermo Fisher) according to the manufacturer’s instructions. The proteins were denatured in NuPage LDS Sample Buffer (Invitrogen) containing 2.5% β-mercaptoethanol at 95°C for 5 min. Proteins (10–20 μg) were loaded onto a Nupage Novex 4–12% bis-Tris acrylamide gel (Invitrogen) and run at 150 V for 1 hr. Proteins were transferred to nitrocellulose membrane and blocked with 5% non-fat dry milk in TBST for 1 hr at room temperature and then incubated for 1 hr at room temperature with the primary antibody in 5% non-fat dry milk in TBST. The following primary antibodies were used; anti-GR (PA1-511A; 0.5 mg/ml; Thermo Fisher), or anti-GR (sc-8992; Santa Cruz; 1:1000). Following incubation with the primary antibody the membrane was washed in TBST and then incubated for 1 hr at room temperature in Zymax goat anti-rabbit IgG HRP conjugate (Invitrogen, 1:2000) and signal detected with ECL plus (GE Healthcare).
Western Blot Analysis of EBV ZEBRA, and Early Antigen (EA) Protein Expression
Cell lysates were prepared from 5 × 106 Daudi cells in 200 μl of buffer containing 100 mM Tris-HCl, pH 7.6; 0.5% Triton X-100, 2 mM PMSF, 1 μl of Protease Inhibitor Cocktail (Sigma-Aldrich). Proteins were separated on 10% polyacrylamide gels, transferred to Hybond-ECL membranes (GE Healthcare), and probed for ZEBRA using 1.0 μg/ml of the mouse anti-ZEBRA antibody (HHV-4 ZEBRA (BZ-1); sc-53904; Santa Cruz) overnight at 4°C. Blots were then incubated in 1:1000 dilution of goat anti-mouse IgG Peroxidase Conjugate (A0168; Sigma -Aldrich) for 1 hr at room temperature. To detect total EBV EA expression, blots were incubated with an EBV seropositive human serum followed by incubation with a 1:10,000 dilution of goat anti-human IgG Peroxidase Conjugate (sc-2907; Santa Cruz). Positive reactivities were visualized using the Phototope-HRP Western Detection System (Cell Signaling).
Transient Transfection of hGR
C666-1 and D98/HR-1 cells were transfected with various amounts of the hGR expression plasmid (Spanjaard and Chin, 1993) using Lipofectamine LTX (Invitrogen) according to the manufacturer’s instructions. Successful transfection was determined by western blotting as described above. Cells were subsequently treated with 100 nM Dex for 24 hr, cells were homogenized in TRIzol reagent and stored at −80°C until assayed by real-time PCR.
Statistical Analysis
Experiments were performed in duplicate and performed at least four times. Results are expressed as mean ± SD. Differences in expression levels were evaluated with independent t test or one-way analysis of variance (ANOVA) with an LSD post-hoc test using PASW Statistics 17.0.2 for Macintosh (SPSS). Results were considered statistically significant for p < 0.05.
Results
GCs induce EBV immediate early BZLF1 gene expression in Daudi cells
To test the effect of GCs on the expression of the EBV immediate early BZLF1 gene, Daudi cells were treated with increasing concentrations of HC and Dex. Induction of BZLF1 mRNA (normalized to GAPDH mRNA levels) by HC was measured after 6, 12, 24, 48, and 72 hrs (Fig. 1A). Overall, expression of BZLF1 mRNA increased in response to HC at all time points (6 hr: F(3, 12) = 6.799, p = 0.006; 12 hr: F(3, 12) = 3.074, p = 0.067; 24 hr: F(3, 12) = 8.002, p = 0.003; 48 hr: F(3, 12) = 21.04, p < 0.001; 72 hr: F(3, 12) = 8.845, p = 0.002). Treatment of Daudi cells with physiological concentrations of HC (1 nM) did not result in a significant increase in BZLF1 expression above that of untreated controls at any time point. However, cells treated with stress levels (100 nM) and pharmacological levels (10 μM) of HC induced BZLF1 gene expression, reaching a maximum increase of 3.07 ± 1.28 fold above control levels (p = 0.001) after treatment with 10 μM HC for 72 hrs.
Figure 1.

The GC-dependent upregulation of EBV BZLF1 mRNA levels in Daudi cells is mediated by GR. Real-time PCR quantification of EBV BZLF1 mRNA levels in Daudi cells after treatment with HC (A) or Dex (B). Cells were collected after 6, 12, 24, 48, and 72 hrs. Total RNA was isolated and BZLF1 mRNA levels (normalized to GAPDH mRNA levels) were measured using real-time PCR. (C) Western blot showing the expression of GR in Daudi cells. (D) Real-time PCR assay measuring EBV BZLF1 mRNA levels in Daudi cells after treatment with Dex, RU486, or Dex plus RU486 for 6, 12, and 24 hrs to assess the role of GR in the Dex-dependent modulation of BZLF1 gene expression.
Values are presented as fold change from untreated control levels. Error bars represent the means ± SD, n = 4; *, p ≤ 0.05; #, p ≤ 0.001.
Daudi cells were then treated with Dex to examine the effect of this synthetic and more potent glucocorticoid on BZLF1 gene expression. Daudi cells were treated with 1 nM, 100 nM, or 10 μM Dex and collected after 6, 12, 24, 48, and 72 hrs of treatment for analysis of EBV BZLF1 gene expression (Fig. 1B). BZLF1 gene expression in Daudi cells increased in response to Dex treatment with the greatest increase observed after 72 hrs of treatment with 100 nM Dex (7.31 ± 2.59 fold above untreated control levels p < 0.001).
Western blot analysis showed that Daudi cells express GR (Fig. 1C). In order to assess the role of GR binding in mediating the observed effect described above, Daudi cells were treated with either 100 nM Dex, 100 nM of the GR antagonist RU486, or co-treated with 100 nM Dex and 100 nM RU486 for 6, 12, and 24 hrs. RU486 efficiently blocked the Dex-induced upregulation of BZLF1 gene expression in Daudi cells. For example, treatment of cells with 100 nM Dex for 12 hrs resulted in a significant increase (2.72 ± 0.23 fold above untreated control levels (p < 0.001)) in BZLF1 expression. Co-treatment with 100 nM Dex and 100 nM RU486 resulted in the abrogation of the Dex-induced increase in BZLF1 mRNA levels but was still significantly higher (1.67 ± 0.44 fold; p = 0.016) than untreated control levels (Fig. 1D). Treatment with RU486 alone did not significantly change BZLF1 gene expression from that observed in untreated control levels (p=0.099).
Kinetics of GC-mediated EBV reactivation in vitro is different from chemical induction with TPA/NaB in Daudi and B95-8 cells
Co-treatment with TPA/NaB has been shown to effectively induce reactivation of latent EBV in Daudi cells through demethylation of the BZLF1 gene (Falk and Ernberg, 1999). We therefore examined the expression of genes associated with lytic replication after exposure of Daudi cells to TPA/NaB compared with treatment with GCs. The expression of BZLF1, BLLF3, and BALF5 mRNA (Fig. 2A, B, and D) was measured at 3, 6, 12, 24, 48 and 72 hrs after treatment with Dex (100 nM), HC (100 nM), or the combined treatment of 40 ng/ml TPA and 3 mM NaB.
Figure 2.

The GCs HC and Dex modulate BZLF1, BLLF3, and BALF5 mRNA expression in Daudi and B98-5 cells with different kinetics from chemical induction with TPA/NaB. Real-time PCR quantification of EBV BZLF1 (A), BLLF3 (C), and BALF5 (D) mRNA in untreated Daudi cells and after treatment with HC, Dex, and TPA/NaB for 3, 6, 12, 24, 48, and 72 hrs. Modulation of BZLF1 gene expression in Daudi cells was compared to that observed in B95-8 cells after treatment with HC, Dex, and TPA/NaB (B). Values are presented as fold change from untreated control levels. Error bars represent the means ± SD, n = 4; *, p ≤ 0.05; #, p ≤ 0.001. (E) Western blot analysis showing the protein expression of ZEBRA, and EAs in untreated Daudi cells and after treatment with HC, Dex, and TPA/NaB for 3, 6, 12, 24, 48, and 72 hrs.
A significant increase in BZLF1 mRNA expression was apparent by 6 hrs and remained upregulated thereafter in GC treated cells. A greater induction of BZLF1 gene expression resulted from Dex treatment compared to HC that is consistent with the former’s greater potency. Compared to the effect of treatment with GCs, BZLF1 expression induced by TPA/NaB occurred later at the 24 hr time point (Fig. 2A). The highest level of BZLF1 mRNA expression (77.36 ± 29.52 fold higher than control levels; p < 0.001) was observed after 72 hrs treatment with TPA/NaB in Daudi cells.
To test whether this effect on BZLF1 gene expression is unique to Daudi cells we compared the effects of HC, Dex, and TPA/NaB on the EBV positive marmoset B-lymphoblastoid cell line B95-8. Unlike Daudi cells, GCs did not induce BZLF1 gene expression in B95-8 cells. Similar to Daudi cells, TPA/NaB treatment resulted in increased BZLF1 mRNA levels in B95-8 cells. Significant upregulation of BZLF1 gene expression was observed as early as 6 hrs of treatment with TPA/NaB resulting in levels 1.91 ± 0.24 fold higher than in untreated B95-8 cells (p = 0.002). Continued treatment for 72 hrs resulted in further increases in BZLF1 mRNA levels which reached 148.82 ± 14.01 fold higher expression levels than untreated controls (p < 0.001). This induction was greater than that observed in Daudi cells.
We then tested the effect of GCs on mRNA expression of the EBV early genes downstream of BZLF1, i.e., BLLF3 and BALF5, which encode for the EBV dUTPase and DNA polymerase, respectively in Daudi cells (Fig. 2C and D). The greatest significant induction of BLLF3 gene expression by HC was observed after 6 hrs of treatment (1.64 ± 0.34 fold; p = 0.001) while peak induction of BALF5 gene expression was observed at the 12 hr time point (1.44 ± 0.20 fold, p = 0.003). As expected, lesser induction of BLLF3 and BALF5 was observed after treatment with HC compared to cells treated with Dex and TPA/NaB. Treatment with Dex resulted in significant increase in both BLLF3 and BALF5 levels at the 12 hr time point (2.66 ± 0.23 fold above control levels; p = 0.002 and 2.32 ± 0.27 fold above control levels; p < 0.001, respectively). As was observed for BZLF1 mRNA levels, TPA/NaB-dependent upregulation of BLLF3 and BALF5 mRNA was observed later (48 hr for BLLF3 and after 24 hr for BALF5) compared to HC and Dex. A decrease in BLLF3 and BALF5 mRNA levels compared to untreated controls was initially observed. The highest TPA/NaB-induced expression was observed after 72 hrs of treatment for BLLF3 mRNA (44.83 ± 11.63 fold increase, p < 0.001) and after 48 hrs for BALF5 mRNA (9.25 ± 1.09, p < 0.001). This difference in the patterns of expression following the treatments reflects the difference in magnitude of the stimuli presented by these inducers of EBV gene expression. Alternatively, these patterns of expression may suggest that the mechanisms by which the GCs and TPA/NaB modulate BZLF1 gene expression involve different pathways.
GC-induced ZEBRA expression mediates the differential expression of EBV EAs
We subsequently performed western blot analysis for the kinetic evaluation of the expression of the BZLF1 gene product ZEBRA at the same time points to compare transcriptional with translational changes after each treatment (Fig. 2E). Expression of ZEBRA in Daudi cells was elevated after 6 hrs of treatment with both GCs and remained upregulated thereafter. On the other hand, increased levels of ZEBRA in Daudi cells was not observed until after 24 hrs of treatment with TPA/NaB (Fig. 2E).
We detected the expression of EBV EAs by western blotting using a previously characterized EBV seropositive human serum that detects total EBV EAs. In HC treated cells, a band at 75 kDa was the only detectable EA (Fig. 2E). This band was detectable at all time points examined except after 72 hrs of treatment. Dex-treated cells also expressed this 75 kDa band with low levels after 6 hr of treatment and increasing thereafter. Other EAs migrating at 15 kDa, and 35 kDa, were also expressed after 24 hrs of Dex treatment with low levels of a 250 kDa band faintly apparent at 48 and 72 hrs (Fig 2E). EBV EA bands migrating at 15 kDa, 35 kDa, 75 kDa and 250 kDa were detectable in TPA/NaB treated cells. The 15 kDa band is present after 3 hrs and decreased thereafter while the larger EAs were detected at lower levels and upregulated at later time points (e.g., high levels of the 75 kDa band are present at 24 and 48 hrs while the 250 kDa band is abundant at 48 and 74 hrs; Fig 2E). These data show that GCs induce the expression of EBV proteins with a different kinetic profile than TPA/NaB. The significance of these differences and the mechanism involved in the modulation of these protein expressions is not fully understood.
BZLF1 induction in latent EBV infected Daudi cells is prevented by cyclohexamide
Modulation of BZLF1 gene expression in EBV latently infected lymphoblastoid cell lines by various chemical inducers of EBV reactivation has been shown to depend on de novo protein synthesis (Laux et al., 1988; Ye et al., 2007). To test whether this is also true for Daudi cells, we co-treated cells with 33 μg/ml cycloheximide (CHX) and with Dex or TPA/NaB. Treatment with CHX prevented the Dex- and TPA/NaB-dependent induction of BZLF1 mRNA expression after 12 and 24 hrs (Fig. 3). Preliminary experiments of Daudi cells treated with CHX alone showed no significant effect on BZLF1 mRNA expression (data not shown).
Figure 3.
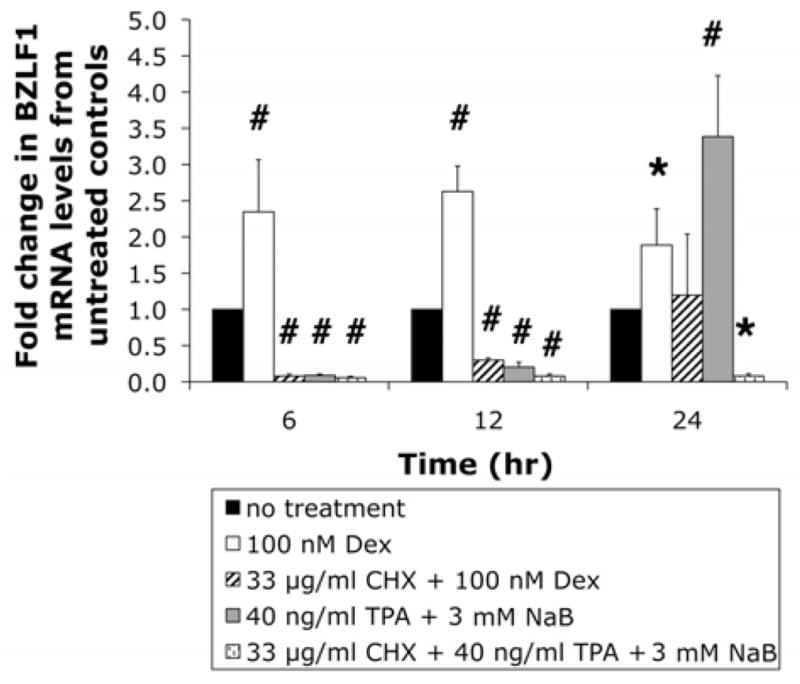
Dex- and TPA/NaB-dependent upregulation of BZLF1 gene expression is inhibited by cycloheximide. Real-time PCR quantification of EBV BZLF1 mRNA in untreated Daudi cells and after treatment with Dex, Dex plus CHX, TPA/NaB, and TPA/NaB plus CHX for 6, 12, and 24 hrs. Values are presented as fold change from untreated control levels. Error bars represent the means ± SD, n = 4; *, p ≤ 0.05; #, p ≤ 0.001.
Increased GR expression in EBV positive epithelial cells stimulates GC mediated BZLF1 mRNA expression
Because of our long-standing interest in the role of EBV in NPC and the impact of stress on this disease (Glaser et al., 1989), we further assessed the association between GR expression and the GC-dependent induction of BZLF1 gene expression in the NPC cell line C666-1 and the epithelial hybrid cell line D98/HR-1. To this end, we transfected cells with various amounts of a GR expression plasmid and then measured Dex mediated induction of BZLF1 gene expression (Fig. 4A & B). Western blotting confirmed increased expression of GR in both these cell lines. When transfected with increasing amounts of a GR expression plasmid, a significant increase in BZLF1 mRNA expression was only observed in C666-1 cells transfected with 3 μg hGR (1.62 ± 0.19 fold increase above control levels, p = 0.002; Fig. 4A). In D98/HR-1 cells, BZLF1 gene expression significantly increased with the transfection of 1.5 μg of the hGR plasmid (2.49 ± 1.13 fold increase above control levels, p = 0.021; Fig. 4B). Compared to cells with 1.5 μg hGR, cells transfected with 3 μg hGR exhibited less of an increase above untreated control levels (1.75 ± 0.58 fold increase above control levels, p = 0.024).
Figure 4.
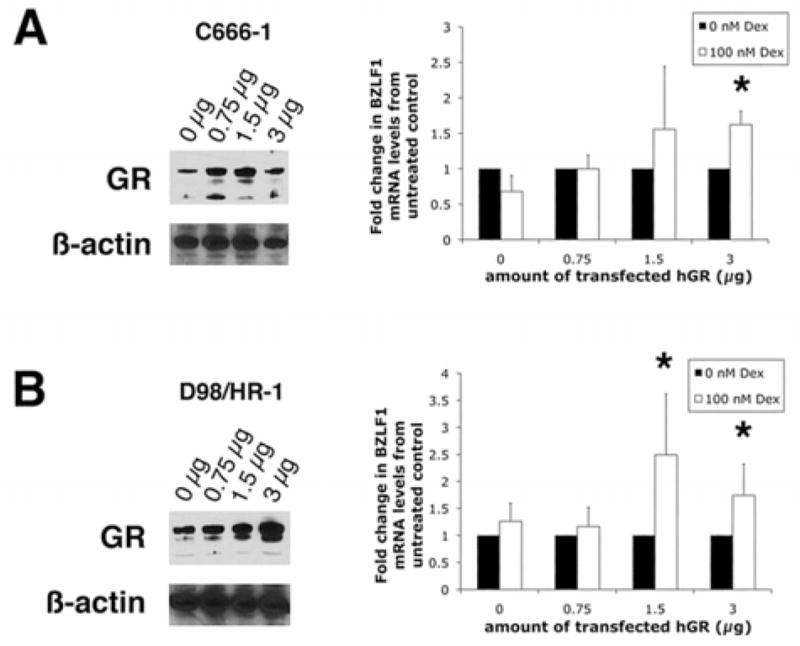
Increased GR expression in EBV positive epithelial cells stimulates Dex-mediated BZLF1 mRNA expression. Western blot showing expression of GR after transfection of 0.75, 1.5, or 3 μg of a GR expression plasmid and real-time PCR quantification of EBV BZLF1 in C666-1 (A) and D98/HR-1 (B) cells after growth for 24 hr in the presence or absence of 100 nM Dex. Values are presented as fold change from untreated control levels. Error bars represent the means ± SD, (A) n = 5, (B) n = 6; *, p ≤ 0.05; #, p ≤ 0.001.
Discussion
Our lab and others have shown that the hormonal/physiological changes associated with psychological stress can induce the reactivation of latent herpesviruses such as EBV and herpes simplex virus (Esterling et al., 1992; Lutgendorf et al., 1994; Mehta et al., 2000; Payne et al., 1999; Sarid et al., 2001; Stowe et al., 2000). Studies have shown that cortisol is an important player in this process (Bauer, 1983; Glaser et al., 1995; Joncas and Leyritz, 1974; Magrath et al., 1979). In this study, we have explored how GCs can reactivate latent EBV. We found that the treatment of Daudi cells with HC or Dex resulted in a dose-dependent upregulation of BZLF1 mRNA expression. BZLF1 has been shown to play a pivotal role in the switch from the latent condition to virus replication. We further show that co-treatment of Daudi cells with Dex and the GR antagonist RU486 effectively abrogated the Dex-dependent upregulation of BZLF1 gene expression supporting the hypothesis that GR mediates the activation of BZLF1 gene expression. The upregulated BZLF1 gene expression results in the synthesis of the BZLF1 gene product ZEBRA and the expression of downstream genes BLLF3 (dUTPase) and BALF5 (DNA polymerase). Finally, our results show that the GC and TPA/NaB mediated upregulation of BZLF1 mRNA expression is abrogated by CHX. However, our current study does not distinguish whether the effect of CHX on the GC-and TPA/NaB-dependent induction of BZLF1 gene expression is due to inhibition of de novo protein synthesis or prevention of degradation of labile proteins. Further studies are needed to distinguish between these possibilities.
To our knowledge this is the first description of the GC-dependent stimulation of the expression of the EBV BZLF1 gene product ZEBRA, a transcription factor that acts as a transactivator of viral genes and cellular genes and thus is a key mediator of reactivation from latency to the viral productive cycle (Speck et al., 1997). Interestingly, we did not observe a GC-dependent upregulation of BZLF1 gene expression in the marmoset derived B98-5 cells. This is consistent with the previous observation of GC resistance in New World primates (Chrousos et al., 1984; Chrousos et al., 1982).
The observations described here provide an explanation for previous studies by our lab and others showing stress-associated changes in the steady-state expression of latent EBV in stressed subjects (Cacioppo et al., 2002; Glaser et al., 1995; Mehta et al., 2007; Mehta et al., 2000; Pierson et al., 2005). In vitro studies have shown that HC and Dex can induce reactivation of latent EBV in Daudi cells and enhance the number of EBV antigen positive Raji cells superinfected with the HR-1 strain of EBV (Glaser et al., 1995). It has been further shown that HC can induce the replication of latent EBV in lymphoblastoid cells (Bauer, 1983; Joncas et al., 1973; Prachova and Roubal, 1981). Here we further confirm that Dex and HC can reactivate latent EBV and that Dex can more efficiently induce reactivation by virtue of its greater potency compared to HC.
We have previously proposed at least three ways that hormones and neuropeptides could influence EBV latency/lytic replication; 1) the hormone(s) directly reactivate the EBV genome in latently infected target cells in peripheral blood or in tissue; 2) the hormone(s) enhance EBV replication after reactivation has taken place, and 3) reactivation/replication takes place as a result of a down-regulated virus specific memory immune T-cell response (Glaser et al., 1995). Here we provide evidence to support the first possibility by showing that exposure to Dex or HC can reactivate latent EBV by enhancing the expression of BZLF1 mRNA. That this enhanced expression of BZLF1 can lead to full viral replication is supported by our observation that Dex treatment also stimulates the expression of the downstream genes (expressed after the reactivation of the latent virus genome) BLLF3 and BALF5, as well as the protein expression of other EBV EAs. We further showed that both the GR antagonist RU486 and cycloheximide prevent the Dex-dependent upregulation of BZLF1 gene expression. Further work is needed to determine if GR directly regulates the BZLF1 gene or if intermediary proteins are involved that play a role in the overall process of EBV reactivation.
Western blot analysis confirmed that Daudi cells express GR which when bound to its ligand results in modulation of EBV gene expression. Ligand binding results in the nuclear translocation of GR where it binds as a homodimer to glucocorticoid response elements (GREs) in the regulatory regions of genes. This binding leads to the enhancement and/or repression of gene transcription depending on the promoter sequence (Duma et al., 2006). A functional GRE has been described in the B95-8 strain of EBV (Kupfer and Summers, 1990; Schuster et al., 1991). Our results showing that RU486, a GR antagonist, blocked the Dex-dependent upregulation of BZLF1 gene expression lends support to the role of this receptor in mediating this effect. However, direct binding of GR to this promoter remains to be shown. The role of GR in the Dex-related stimulation of BZLF1 mRNA expression is further supported by our observation that transfection of C666-1 and D98/HR-1 cells with increasing amounts of a construct containing the hGR resulted in an enhanced BZLF1 gene expression. The fact that BZLF1 gene expression peaked with transfection of 0.75 μg hGR in D98/HR-1 cells and no further increase associated with the addition of 1.5 μg hGR suggests that other co-regulators may become limiting in these cells.
Comparison of the BZLF1, BLLF3, and BALF5 gene expression in Daudi cells treated with HC, Dex or with the chemical inducer, TPA/NaB, demonstrated differences in the timing of expression of these mRNAs indicating differences in the magnitude of the stimuli when Daudi cells are exposed to these inducers of EBV gene expression. The late induction of BZLF1 by TPA/NaB is not unique to Daudi cells as a similar, although slightly earlier, induction of BZLF1 by TPA/NaB was seen in B98-5 cells. The different patterns of expression observed above indicate that expression of EBV EAs is differentially regulated by various inducers of the EBV lytic cycle. The significance of the difference in expression profiles of EA antigens observed in western blots under these three conditions is not fully understood and warrants further investigation. These results further suggest that while the lower BZLF1 mRNA expression induced by GCs results in limited EA expression (and partial or incomplete reactivation of EBV) the greater BZLF1 mRNA expression induced by TPA/NaB can result in the expression of a larger set of EBV EAs that may lead to the complete reactivation of EBV (Glaser et al., 2005).
The results described here contribute to our understanding of how psychological stressors can affect the steady-state expression of latent EBV. We, and others, have shown that stress can induce the HPA and SAM axes resulting in the production of cortisol, which we have shown in this study (in the form of HC) to induce the latent EBV genome to be activated. Further studies aimed at elucidating the mechanism(s) involved in the stress-associated reactivation of latent EBV will enhance our ability to counter the adverse effects of psychological stressors on EBV latency and EBV-associated diseases.
Acknowledgments
The authors thank Drs. Marshall Williams and William B. Malarkey for their helpful suggestions, and Amy C. Gross and Carly Safier for valuable technical assistance. This work was supported by The Gilbert and Kathryn Mitchell Endowment to RG, the Ohio State University Comprehensive Cancer Center CA16058 (NCI), and NCI grant CA100243.
Footnotes
Conflict of Interest Statement: All authors declare that there are no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bauer G. Induction of Epstein-Barr virus early antigens by corticosteroids: Inhibition by TPA and retinoic acid. International Journal of Cancer. 1983;31:291–295. doi: 10.1002/ijc.2910310307. [DOI] [PubMed] [Google Scholar]
- Buchwald D, Umali P, Umali J, Kith P, Pearlman T, Komaroff AL. Chronic fatigue and the chronic fatigue syndrome: prevalence in a Pacific Northwest health care system. Annals of Internal Medicine. 1995;123:81–88. doi: 10.7326/0003-4819-123-2-199507150-00001. [DOI] [PubMed] [Google Scholar]
- Cacioppo J, Kiecolt-Glaser J, Malarkey W, Laskowski B, Rozlog L, Poehlmann K, Burleson M, Glaser R. Autonomic and glucocorticoid associations with the steady-state expression of latent Epstein-Barr virus. Hormones and Behavior. 2002;42:32–41. doi: 10.1006/hbeh.2002.1801. [DOI] [PubMed] [Google Scholar]
- Cheung ST, Huang DP, Hui AB, Lo KW, Ko CW, Tsang YS, Wong N, Whitney BM, Lee JC. Nasopharyngeal carcinoma cell line (C666-1) consistently harbouring Epstein-Barr virus. International Journal of Cancer. 1999;83:121–126. doi: 10.1002/(sici)1097-0215(19990924)83:1<121::aid-ijc21>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- Chrousos GP, Loriaux DL, Brandon D, Shull J, Renquist D, Hogan W, Tomita M, Lipsett MB. Adaptation of the mineralocorticoid target tissues to the high circulating cortisol and progesterone plasma levels in the squirrel monkey. Endocrinology. 1984;115:25–32. doi: 10.1210/endo-115-1-25. [DOI] [PubMed] [Google Scholar]
- Chrousos GP, Renquist D, Brandon D, Eil C, Pugeat M, Vigersky R, Cutler GB, Jr, Loriaux DL, Lipsett MB. Glucocorticoid hormone resistance during primate evolution: receptor-mediated mechanisms. Proceedings of the National Academy of Sciences. 1982;79:2036–2040. doi: 10.1073/pnas.79.6.2036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duma D, Jewell CM, Cidlowski JA. Multiple glucocorticoid receptor isoforms and mechanisms of post-translational modification. The Journal of Steroid Biochemistry and Molecular Biology. 2006;102:11–21. doi: 10.1016/j.jsbmb.2006.09.009. [DOI] [PubMed] [Google Scholar]
- Esterling BA, Antoni MH, Schneiderman N, Carver CS, LaPerriere A, Ironson G, Klimas NG, Fletcher MA. Psychosocial modulation of antibody to Epstein-Barr viral capsid antigen and human herpesvirus type-6 in HIV-1-infected and at-risk gay men. Psychosomatic Medicine. 1992;54:354–371. doi: 10.1097/00006842-199205000-00011. [DOI] [PubMed] [Google Scholar]
- Falk KI, Ernberg I. Demethylation of the Epstein-barr virus origin of lytic replication and of the immediate early gene BZLF1 is DNA replication independent. Brief report. Archives Of Virology. 1999;144:2219–2227. doi: 10.1007/s007050050636. [DOI] [PubMed] [Google Scholar]
- Glaser R, Kiecolt-Glaser JK. Stress-induced immune dysfunction: implications for health. Nature Reviews. Immunology. 2005;5:243–251. doi: 10.1038/nri1571. [DOI] [PubMed] [Google Scholar]
- Glaser R, Kutz LA, MacCallum RC, Malarkey WB. Hormonal modulation of Epstein-Barr virus replication. Neuroendocrinology. 1995;62:356–361. doi: 10.1159/000127025. [DOI] [PubMed] [Google Scholar]
- Glaser R, O’Neill FJ. Hybridization of Burkitt lymphoblastoid cells. Science. 1972;176:1245–1247. doi: 10.1126/science.176.4040.1245. [DOI] [PubMed] [Google Scholar]
- Glaser R, Padgett DA, Litsky ML, Baiocchi RA, Yang EV, Chen M, Yeh PE, Klimas NG, Marshall GD, Whiteside T, Herberman R, Kiecolt-Glaser J, Williams MV. Stress-associated changes in the steady-state expression of latent Epstein-Barr virus: Implications for chronic fatigue syndrome and cancer. Brain, Behavior, and Immunity. 2005;19:91–103. doi: 10.1016/j.bbi.2004.09.001. [DOI] [PubMed] [Google Scholar]
- Glaser R, Pearl DK, Kiecolt-Glaser JK, Malarkey WB. Plasma cortisol levels and reactivation of latent Epstein-Barr virus in response to examination stress. Psychoneuroendocrinology. 1994;19:765–772. doi: 10.1016/0306-4530(94)90023-x. [DOI] [PubMed] [Google Scholar]
- Glaser R, Pearson GR, Bonneau RH, Esterling BA, Atkinson C, Kiecolt-Glaser JK. Stress and the memory T-cell response to the Epstein-Barr virus in healthy medical students. Health Psychology: official journal of the Division of Health Psychology American Psychological Association. 1993;12:435–442. doi: 10.1037//0278-6133.12.6.435. [DOI] [PubMed] [Google Scholar]
- Glaser R, Pearson GR, Jones JF, Hillhouse J, Kennedy S, Mao HY, Kiecolt-Glaser JK. Stress-related activation of Epstein-Barr virus. Brain, Behavior, and Immunity. 1991;5:219–232. doi: 10.1016/0889-1591(91)90018-6. [DOI] [PubMed] [Google Scholar]
- Glaser R, Rice J, Sheridan J, Fertel R, Stout J, Speicher CE, Pinsky D, Kotur M, Post A, Beck M, Kiecolt-Glaser JK. Stress-related immune suppression: Health implications. Brain, Behavior, and Immunity. 1987;1:7–20. doi: 10.1016/0889-1591(87)90002-x. [DOI] [PubMed] [Google Scholar]
- Glaser R, Zhang HY, Yao KT, Zhu HC, Wang FX, Li GY, Wen DS, Li YP. Two epithelial tumor cell lines (HNE-1 and HONE-1) latently infected with Epstein-Barr virus that were derived from nasopharyngeal carcinomas. Proceedings of the National Academy of Sciences of America. 1989;86:9524–9528. doi: 10.1073/pnas.86.23.9524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jason LA, Taylor RR, Kennedy CL, Jordan K, Song S, Johnson DE, Torres SR. Chronic fatigue syndrome: sociodemographic subtypes in a community-based sample. Evaluation and the Health Professions. 2000;23:243–263. doi: 10.1177/01632780022034598. [DOI] [PubMed] [Google Scholar]
- Joncas J, Boucher J, Boudreault A, Granger-Julien M. Effect of hydrocortisone on cell viability, Epstein-Barr virus genome expression and interferon synthesis in human lymphoblastoid cell lines. Cancer Research. 1973;33:2142–2148. [PubMed] [Google Scholar]
- Joncas J, Leyritz M. The effect of hydrocortisone and bromodeoxyuridine (BUdR) on the Epstein-Barr herpes virus in human lymphoblastoid cell lines. Rev Can Biol. 1974;33:135–147. [PubMed] [Google Scholar]
- Kimura H, Morita M, Yabuta Y, Kuzushima K, Kato K, Kojima S, Matsuyama T, Morishima T. Quantitative analysis of Epstein-Barr virus load by using a real-time PCR assay. Journal of Clinical Microbiology. 1999;37:132–136. doi: 10.1128/jcm.37.1.132-136.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klimas NG, Salvato FR, Morgan R, Fletcher MA. Immunologic abnormalities in chronic fatigue syndrome. Journal of Clinical Microbiology. 1990;28:1403–1410. doi: 10.1128/jcm.28.6.1403-1410.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kupfer SR, Summers WC. Identification of a glucocorticoid-responsive element in Epstein-Barr virus. J Virol. 1990;64:1984–1990. doi: 10.1128/jvi.64.5.1984-1990.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laux G, Freese UK, Fischer R, Polack A, Kofler E, Bornkamm GW. TPA-inducible Epstein-Barr virus genes in Raji cells and their regulation. Virology. 1988;162:503–507. doi: 10.1016/0042-6822(88)90496-5. [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Lutgendorf SK, Antoni MH, Kumar M, Schneiderman N. Changes in cognitive coping strategies predict EVA-antibody titre change following a stressor disclosure induction. Journal of Psychosomatic Research. 1994;38:63–78. doi: 10.1016/0022-3999(94)90009-4. [DOI] [PubMed] [Google Scholar]
- Magrath IT, Pizzo PA, Novikova L, Levine AS. Enhancement of Epstein-Barr virus replication in producer cell lines by a combination of low temperature and corticosteroids. Virology. 1979;97:477–481. doi: 10.1016/0042-6822(79)90360-x. [DOI] [PubMed] [Google Scholar]
- Mehta SK, Crucian B, Pierson DL, Sams C, Stowe RP. Monitoring immune system function and reactivation of latent viruses in the Artificial Gravity Pilot Study. Journal of gravitational physiology: a journal of the International Society for Gravitational Physiology. 2007;14:21. [PubMed] [Google Scholar]
- Mehta SK, Pierson DL, Cooley H, Dubow R, Lugg D. Epstein-Barr virus reactivation associated with diminished cell-mediated immunity in Antarctic expeditioners. Journal of Medical Virology. 2000;61:235–240. doi: 10.1002/(sici)1096-9071(200006)61:2<235::aid-jmv10>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- Payne DA, Mehta SK, Tyring SK, Stowe RP, Pierson DL. Incidence of Epstein-Barr virus in astronaut saliva during spaceflight. Aviation, Space & Environmental Medicine. 1999;70:1211. [PubMed] [Google Scholar]
- Pierson DL, Stowe RP, Phillips TM, Lugg DJ, Mehta SK. Epstein-Barr virus shedding by astronauts during space flight. Brain, Behavior, and Immunity. 2005;19:235–242. doi: 10.1016/j.bbi.2004.08.001. [DOI] [PubMed] [Google Scholar]
- Prachova K, Roubal J. Effects of hydrocortisone on the synthesis of Epstein-Barr virus antigens in P3HR-1 cells. Acta Virologica. 1981;25:163–166. [PubMed] [Google Scholar]
- Rabin BS. Stress, immune function, and health: the connection. Wiley-Liss; New York: 1999. [Google Scholar]
- Sarid O, Anson O, Yaari A, Margalith M. Epstein-Barr virus specific salivary antibodies as related to stress caused by examinations. Journal of Medical Virology. 2001;64:149–156. doi: 10.1002/jmv.1030. [DOI] [PubMed] [Google Scholar]
- Schuster C, Chasserot-Golaz S, Urier G, Beck G, Sergeant A. Evidence for a functional glucocorticoid responsive element in the Epstein-Barr virus genome. Molecular Endocrinology (Baltimore Md) 1991;5:267–272. doi: 10.1210/mend-5-2-267. [DOI] [PubMed] [Google Scholar]
- Spanjaard RA, Chin WW. Reconstitution of ligand-mediated glucocorticoid receptor activity by trans-acting functional domains. Molecular Endocrinology. 1993;7:12–16. doi: 10.1210/mend.7.1.8446102. [DOI] [PubMed] [Google Scholar]
- Speck SH, Chatila T, Flemington E. Reactivation of Epstein-Barr virus: regulation and function of the BZLF1 gene. Trends In Microbiology. 1997;5:399–405. doi: 10.1016/S0966-842X(97)01129-3. [DOI] [PubMed] [Google Scholar]
- Stowe RP, Pierson DL, Feeback DL, Barrett AD. Stress-induced reactivation of Epstein-Barr virus in astronauts. Neuroimmunomodulation. 2000;8:51–58. doi: 10.1159/000026453. [DOI] [PubMed] [Google Scholar]
- Ye J, Gradoville L, Daigle D, Miller G. De novo protein synthesis is required for lytic cycle reactivation of Epstein-Barr virus, but not Kaposi’s sarcoma-associated herpesvirus, in response to histone deacetylase inhibitors and protein kinase C agonists. Journal of Virology. 2007;81:9279–9291. doi: 10.1128/JVI.00982-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang HY, Qu G, Deng ZW, Yao TH, Glaser R. Epstein-Barr virus DNA in nasopharyngeal biopsies. Virus Research. 1989;12:53–59. doi: 10.1016/0168-1702(89)90053-1. [DOI] [PubMed] [Google Scholar]