

Abstract
Duchenne muscular dystrophy (DMD) is a devastating primary muscle disease with pathological changes in skeletal muscle that are ongoing at birth. Progressive deterioration in striated muscle function in affected individuals ultimately results in early death due to cardio-pulmonary failure. Since affected individuals can be identified prior to birth by prenatal genetic testing for DMD, gene replacement treatment could be started in utero. This approach offers the possibility of preventing pathological changes in muscle that begin early in life. To test in utero gene transfer utilizing the AAV8 vector in the mdx mouse model of DMD, a minidystrophin gene driven by the human cytomegalovirus promoter was delivered systemically by intraperitoneal injection to the fetus at embryonic day 16. Treated mdx mice studied at 9 weeks after birth demonstrated widespread expression of recombinant dystrophin in skeletal muscle, restoration of the dystrophin associated glycoprotein complex in dystrophin-expressing muscle fibers, improved muscle pathology, and functional benefit to the transduced diaphragm compared to untreated littermate controls. These results support the potential of the AAV8 vector to efficiently cross the blood vessel barrier to achieve systemic gene transfer to skeletal muscle in utero in a mouse model of muscular dystrophy, to significantly improve the dystrophic phenotype and to ameliorate the processes that lead to exhaustion of skeletal muscle regenerative capacity.
INTRODUCTION
Duchenne muscular dystrophy (DMD) is an X-linked recessive muscle disorder affecting 1 in 3500 live male births.1 The disease is characterized by progressive loss of muscle function leading to premature death, most commonly caused by cardiac and respiratory failure.2 Although improvements in medical management and treatment with glucocorticoids have significantly improved life expectancy and the quality of life of DMD patients, a specific treatment addressing the genetic defect causing DMD is not yet clinically available. A crucial challenge of DMD therapeutic research is to develop approaches with functional benefit to widespread muscle tissues. Preclinical gene replacement therapy has shown promising results in postnatal dystrophic mice3–6 and dogs7,8 although widespread gene delivery often requires large vector doses and issues of immunity induced by viral vectors remain.
Among gene delivery vectors, adeno-associated viral (AAV) vectors hold great promise for gene therapy because of their safety, low toxicity and stable transgene expression.9 In particular, AAV serotype 8 (AAV8) vectors demonstrate robust muscle transduction after systemic delivery to postnatal mice and hamsters.10,11 However, there are very few studies testing gene delivery and functional efficacy of therapeutic transgenes carried by AAV8 vectors in dystrophic animal models.12
In utero gene delivery for muscle gene replacement therapy offers several advantages. Compared to gene delivery later in life, gene transfer to the fetus provides an opportunity to target a higher percentage of cells when the tissue mass is small. In humans, myotube formation is observed between gestation weeks 7 and 14 and by week 20, muscle fibers are arranged in discrete bundles.13 In mice, secondary myotubes begin to form on the scaffold of the primary myotubes from embryonic day 14 (E14) to E17.14–19 Therefore, most murine in utero muscle gene delivery studies have been performed at E15 or E16 when secondary myotubes are forming.
For genetic diseases such as DMD where the clinical signs and symptoms are often not detected until affected children are 2–5 years old, fetal DNA diagnostics and gene transfer in utero provide a unique opportunity to treat at the earliest stage of the disease. The immaturity of the basal lamina may play an important role in widespread gene delivery to fetal muscle.20,21 Finally, the minimal transduction of liver by AAV8 vectors when delivered systemically in utero is a desirable outcome of muscle-targeted gene delivery and limits the possibility of liver toxicity from gene delivery.22
Since most primary muscle disorders affect multiple muscle groups including the diaphragm, an important challenge for muscle gene therapy is to achieve transgene expression in widespread muscle tissues. In our previous study of the biodistribution of an AAV8 vector carrying the lacZ gene after systemic administration in utero, we demonstrated successful widespread gene expression in various muscle tissues including high levels of expression in diaphragm and intercostal muscles.22 This biodistribution study suggested that AAV8 could be an ideal vector for the treatment of primary muscle disorders in utero. Therefore, we explored the therapeutic potential of an AAV8 vector carrying a minidystrophin gene when delivered systemically in utero to the dystrophic mdx mouse model of DMD.
MATERIALS AND METHODS
Production of AAV8 minidystrophin vector
The cloning and construction of the canine minidystrophin cDNA has been previously described.23 Briefly, the AAV8 vector carrying a canine minidystrophin expression cassette driven by the human cytomegalovirus (HCMV) promoter (AAV8 minidystrophin) was generated by the triple-plasmid transfection method using 3 plasmids comprising the AAV-CMV-minidystrophin vector plasmid, the mini-adeno helper plasmid, and the AAV8 packaging plasmid containing the AAV2 Rep gene and AAV8 Cap gene, as described previously.24,25 AAV8 minidystrophin viral particles were purified by double CsCl gradient centrifugation and dialyzed 3 times against PBS containing 5% sorbitol. The titer of vector genomes (vg) was determined by a standard DNA dot-blot assay.
In utero administration of AAV8 minidystrophin vector
AAV8 minidystrophin was administered intraperitoneally in utero into E16 pups of timed pregnant mdx female mice as described previously22 according to a protocol approved by the University of Pittsburgh Institutional Animal Care and Use Committee. The vector was injected at a dose of 6.4 × 1011 vg/pup. In order to identify the injected pup, a fluorescent marker, 2% orange fluorescent FluoSpheres (Invitrogen, Carlsbad, CA, USA), was injected into one of the limbs permitting identification of injected pups several days after birth by observation under a fluorescent microscope. The vector-treated mice were analyzed at 9 weeks of age in parallel with age-matched untreated mdx littermate and C57BL/10 controls.
Ex vivo functional analysis of diaphragm
Ex vivo functional analysis was performed on diaphragm 9 weeks after birth following in utero treatment with AAV8 minidystrophin vector. Diaphragm specific force (peak isometric tetanic force normalized for muscle cross-sectional area) and force generation during repetitive isovelocity lengthening activations were performed as previously described.26 Analysis of variance with Tukey’s post hoc test for multiple comparisons was used to identify statistical differences between groups with respect to specific force and residual force following lengthening activations (P<0.05).
Histology and Immunohistochemistry
Muscle samples were snap frozen and sectioned using a cryostat. To assess morphology, sections were stained with hematoxylin and eosin (H&E). For immunohistochemistry, the sections were blocked with 2% bovine serum albumin and washed with phosphate-buffered saline 3 times after each step. Immunostaining for α-sarcoglycan and β-dystroglycan was performed as described previously.27,28 For dystrophin staining, sections were incubated with a rabbit anti-dystrophin antibody (Invitrogen, Eugene, OR, USA) at a dilution of 1:800, followed by AffiniPure Donkey Anti-Rabbit IgG (Jackson ImmunoResearch Inc., West Grove, PA, USA) at a dilution of 1:1000. Cell nuclei were stained with Hoechst dye. Uninjected mdx littermate and C57BL/10 normal muscles were used as negative and positive controls, respectively, for immunostaining.
Quantification of fibers with centrally-placed nuclei
Muscle sections stained for dystrophin and Hoechst dye were counted to score the number of fibers with centrally-placed nuclei in dystrophin positive and dystrophin negative fibers in muscle sections from treated mice. A similar analysis of uninjected mdx littermates and C57BL/10 mice was performed for disease and normal controls, respectively. The percentage of fibers with centrally-placed nuclei was than calculated from analysis of approximately 300 muscle fibers in randomly selected fields. Analysis of variance with Tukey’s post hoc test for multiple comparisons was used to identify statistical differences between groups (P<0.05).
Analysis of vector genomes by real time PCR
Total DNA was isolated from the muscle tissues of treated and untreated mdx and C57BL/10 mice by ethanol precipitation (Wizard genomic DNA purification kit, Promega, Madison, WI, USA) and the vector genomes calculated as described previously.22 PCR primers for the minidystrophin gene had the following sequence: forward primer: CACCCATAAGGAAAGGCTTCTAGAA and reverse primer: GAGATCTTGCCATTGTTTCATCAG. The TaqMan probe sequence was: ATTCCAAGGGAGTAAGAGA. The results were presented as copies of vector particles per 1000 nuclei.
RESULTS
Expression of recombinant dystrophin and improvement in muscle morphology after intraperitoneal administration of AAV8 minidystrophin in utero
A dose of 6.4 × 1011 vg of an AAV8 vector carrying a canine mini-dystrophin cDNA driven by the HCMV promoter was injected intraperitoneally per fetus on E16. Pups were delivered naturally at full term. At 9 weeks of age, experimental mice were sacrificed and diaphragm, upper limb, and lower limb muscles were collected from the injected pups and the uninjected littermates. To assess the gene transfer efficiency, cryosections of these tissues were analyzed for expression of dystrophin (Figure 1). The treated diaphragm exhibited the highest level of transgene expression. Limb muscles also demonstrated widespread dystrophin expression, while in the control mdx muscles we observed only rare revertant dystrophin positive fibers.
Figure 1. Restoration of dystrophin and amelioration of dystrophic pathology in AAV8 minidystrophin treated mdx diaphragm, upper limb and lower limb muscles.

Tissues were collected at 9 weeks after birth following an intraperitoneal injection of AAV8 minidystrophin into E-16 pups of pregnant mdx mice and analyzed for dystrophin expression. Dystrophin immunohistochemistry (upper panels) and H & E staining (lower panels) were evaluated in (a) diaphragm, (b) upper limb, and (c) lower limb muscles. Uninjected mdx littermates and C57BL/10 tissues were used as negative and positive controls, respectively, for immunohistochemistry and histology. Dys, dystrophin; α-SG, α-sarcoglycan; β-DG, β-dystroglycan. H&E, hematoxylin and eosin. Bar = 100 μm.
The dystrophic change of untreated mdx muscle tissue at 9 weeks of age is characterized by necrotic and regenerating muscle fibers, a mononuclear cell infiltrate and increased fibrous connective tissue. To evaluate the histology of muscle tissues, cryosections of diaphragm, upper limb and lower limb muscles collected from the injected pups at 9 weeks of age were stained with H&E. The treated muscles showed decreased fiber size variability, less mononuclear cellular infiltration, and reduced fibrosis compared to untreated mdx controls (Figure 1). Together, the morphological findings suggest skeletal muscle benefit from expression of recombinant dystrophin delivered by an AAV8 vector in utero to mdx mice.
Restoration of dystrophin-associated glycoprotein (DAG) complex in muscle after intraperitoneal administration of AAV8 minidystrophin in utero
The absence of dystrophin expression in mdx muscle disrupts the structural link between cytoplasmic actin filaments and the sarcolemmal DAG complex leading to the dysfunction and/or loss of the DAG complex from the sarcolemma. Dystrophin protein functions in conjunction with the DAG complex.29,30 Hence, we studied whether in utero gene transfer of an AAV8 vector carrying minidystrophin can restore the DAG complex. At 9 weeks of age the diaphragm, the upper limb, and the lower limb muscles from the injected pups, and the uninjected littermates were analyzed for α-sarcoglycan and β-dystroglycan expression. Expression of α-sarcoglycan and β-dystroglycan was restored in fibers expressing recombinant dystrophin (Figure 2) suggesting functional benefit of AAV8 vector-mediated minidystrophin gene delivery in utero to mdx mice.
Figure 2. Restoration of dystrophin associated glycoprotein (DAG) complex proteins in lower limb muscle sections after in utero gene transfer of AAV8 minidystrophin.

Gastrocnemius muscle from mice treated with AAV8 minidystrophin at E16 in utero at a dose of 6.4 × 1011 vector genomes per fetus was cryosectioned. Staining of serial sections with hematoxylin and eosin (H&E) and immunohistochemistry for dystrophin, α-sarcoglycan (α-SG) and β-dystroglycan (β-DG) are shown. Bar = 100 μm.
Decreased percentage of fibers harboring centrally-placed nuclei in muscle after intraperitoneal administration of AAV8 minidystrophin in utero
Cycles of degeneration and regeneration of muscle fibers result in the accumulation of muscle fibers harboring centrally-placed nuclei.31,32 To characterize the degree of this pathological process we calculated the percentage of muscle fibers with centrally-placed nuclei in mdx untreated fibers, AAV8 minidystrophin treated, dystrophin-positive fibers, AAV8 minidystrophin-treated, dystrophin-negative fibers, and C57BL/10 fibers in diaphragm and hindlimb muscles of 9-week-old mice (Figure 3). We observed a high percentage of fibers with centrally-placed nuclei in untreated mdx diaphragm (62.6% ± 4.7) and hindlimb muscle (71.8% ± 4.6), while C57BL/10 muscle showed very few fibers with centrally-placed nuclei in the diaphragm and hindlimb (1.1% ± 0.7 and 1.3% ± 0.5, respectively). The percentage of fibers with centrally placed nuclei in dystrophin-expressing fibers of AAV8 minidystrophin treated mdx mice was significantly less (P < 0.05) in diaphragm and hindlimb (21.7% ± 2.4 and 26.7% ± 2.7, rrespectively) compared to untreated control mdx mice. The improvements in mdx muscle pathology observed after AAV8 minidystrophin treatment in utero are indications of its potential benefit in a muscular dystrophy animal model.
Figure 3. Decreased percentage of fibers with centrally placed nuclei associated with recombinant minidystrophin expression in muscle tissues after intraperitoneal administration of AAV8 minidystrophin vector in utero.
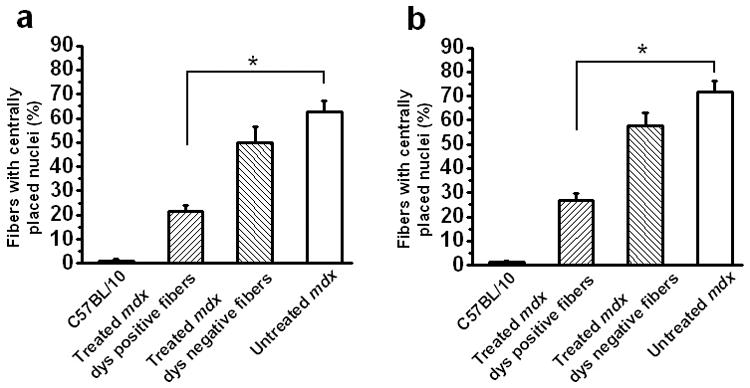
The percentage of fibers with centrally-placed nuclei was calculated from cross-sections of (a) diaphragm and (b) lower limb muscles treated by systemic in utero delivery of an AAV8 minidystrophin vector at E16 at a dose of 6.4 × 1011 vector genomes per fetus. Fibers from the C57BL/10 normal mice, the untreated mdx, and the treated dystrophin positive and dystrophin negative mdx, were analyzed. The data is shown as mean ± SE. Significant differences from untreated mdx mice are shown (*P<0.05). dys, dystrophin.
Improvement of ex vivo functional force generation properties of costal diaphragm after intraperitoneal administration of AAV8 minidystrophin in utero
To test whether widespread expression of minidystrophin in costal diaphragm could provide functional improvement, the force properties of treated mdx diaphragm, untreated mdx diaphragm and C57BL/10 control diaphragm were tested and compared. Nine weeks after birth, costal diaphragm was collected for ex vivo force measurements. Diaphragm specific force (peak isometric tetanic force normalized for muscle cross-sectional area) and force generation during repetitive isovelocity lengthening activations were performed as previously described.26 AAV8 minidystrophin-treated diaphragm exhibited a statistically significant (P < 0.05) 32.6% increase in specific force compared to paired littermate untreated mdx diaphragm [mdx untreated (n=11 mice): 14.7 ± 3.0 N/cm2; mdx treated (n=6 mice): 19.5 ± 1.5 N/cm2; C57BL/10 control (n=5 mice) 21.1 ± 2.7] (Figure 4a). Specific force of AAV8 minidystrophin-treated diaphragm approximated (statistically insignificant difference) that seen in age-matched C57BL/10 wild-type controls. After determining peak tetanic force the diaphragm was subjected to repetitive lengthening activations, a paradigm of mechanical stress.33 Residual diaphragm force following 10 repetitive lengthening activations, expressed as a percent of initial, was significantly greater in AAV8 minidystrophin-treated diaphragm than untreated littermate paired mdx diaphragm [mdx untreated (n=11 mice): 84.5 ± 5; mdx treated (n=6 mice): 99 ± 7.6; C57BL10 control (n=5 mice): 101 ± 7.1] (Figure 4b). These data demonstrate that AAV8 minidystrophin delivered systemically in utero provides significant functional improvements in the dystrophic mdx diaphragm as demonstrated by specific force generation and the ability to withstand eccentric muscle contraction.
Figure 4. Improvement in force generation properties in diaphragm after intraperitoneal administration of AAV8 minidystrophin vector in utero.
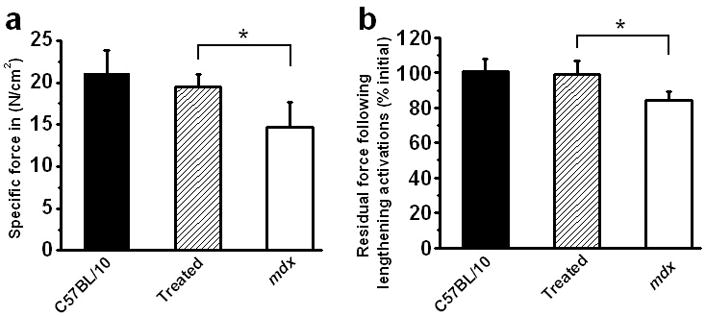
The diaphragm muscles were collected at 9 weeks of age from mice treated in utero and were analyzed ex vivo. Diaphragms of control C57BL/10 (n=5 mice), AAV8 minidystrophin vector-treated mdx (n=6 mice) and untreated mdx (n=11 mice) mice were analyzed for (a) specific force (N/cm2) and (b) residual force following 10 repetitive lengthening activations divided by initial force and expressed as a percentage. The data is shown as mean ± SE. Significant differences from untreated mdx mice are shown (*P<0.05).
Quantification of AAV vector genomes in muscle tissues after intraperitoneal administration of AAV8 minidystrophin in utero
To determine viral vector gene transfer efficiency and compare with the previous marker gene study in normal mice, we quantified the number of viral particles in individual tissues by real time PCR. The highest levels of viral vector particles were observed in diaphragm. Diaphragm had 227.77 ± 85.07 viral particles per 1000 nuclei respectively. Upper and lower limbs had 38.70 ± 22.59 and 33.90 ± 13.82 viral particles per 1000 nuclei respectively. These findings are similar to and confirm our previous study in normal mice in whom a similar dose of AAV8 carrying a lacZ gene was delivered by the same route in utero.22 The results thus demonstrate the efficacy of AAV8 minidystrophin to successfully deliver the therapeutic gene into dystrophic muscle tissues that was comparable to gene delivery to normal muscle tissue.
DISCUSSION
This study demonstrates the potential of fetal gene therapy to correct a genetic defect that is lethal in humans, in a preclinical model. Other disorders where preclinical fetal gene transfer studies have shown partial correction of a genetic defect include mucopolysaccharidosis type VII,34,35 Crigler-Najjar Syndrome Type 1,36α-thalassemia,37 hemophilia B,38 cystic fibrosis,39 and Pompe disease.40 In this study, the improvements in muscle pathology, which are also reflected in functional benefit in the diaphragm, suggest that dystrophin gene transfer in utero for the treatment of dystrophin deficiency has the potential to preserve muscle regenerative capacity by gene replacement at this very early stage.
This study demonstrates the ability of AAV8 to systemically transduce widespread muscles, including the diaphragm and therapeutic benefit in a DMD model in utero. Although we also achieved in utero dystrophin gene delivery in mdx mice with an adenoviral vector,21 the AAV8 vector provides markedly more evidence of morphological and functional benefit. One limitation of the AAV8 vector for the treatment of DMD in utero, however, is a low level of cardiac transduction.22 Another serotype of AAV, AAV9, offers higher levels of cardiac transduction when delivered postnatal,10 but has not been tested for in utero gene delivery.
Most previous muscle gene transfer studies with AAV8 have been performed in postnatal animals and showed that the AAV8 vector efficiently transduces muscle tissues of neonatal and adult animals. In a study of multiple AAV vector serotypes, Wang et al. showed that AAV8 was systemically delivered efficiently to muscle in neonatal and adult mice.11 Similarly, Inagaki et al. showed robust transduction of muscle tissue following systemic delivery of various doses of AAV8 vector in adult mice.10 Zincarelli et al. observed that tail vein injection of an AAV8-luciferase vector into 6–8 week old mice not only showed persistent transgene expression for at least 100 days, but also demonstrated uniform expression of luciferase in hindlimb, abdominal and thoracic regions.41
The biodistribution of AAV8 when delivered in utero was shown in our previous study of AAV8 gene delivery in utero. Intraperitoneal delivery of an AAV8 vector carrying a lacZ gene to fetal mice in utero resulted in widespread postnatal gene expression in multiple muscle tissues, including diaphragm, intercostal muscles, forelimb and hindlimb muscles with the highest expression seen in the diaphragm and intercostal muscles.22
Encouraged by these results with gene delivery of a marker gene in utero using an AAV8 vector, we performed the present in utero gene transfer study in a muscular dystrophy model with a therapeutic transgene and demonstrated for the first time that an AAV8 vector carrying a minidystrophin gene injected systemically in utero could restore muscle structure and function. The AAV8 vector carrying a mindystrophin gene was injected intraperitoneally into E16 mdx pups and muscle was analyzed 9 weeks after birth. We observed efficient transduction and restoration of dystrophin in diaphragm, forelimb and hindlimb muscles. In addition, immunostaining of the transduced muscles demonstrated restoration of the DAG complex, evidenced by expression of α-sarcoglycan and β-dystroglycan at the sarcolemma of those fibers expressing recombinant dystrophin.
Mdx muscle tissues undergo degeneration evidenced pathologically by necrosis and regeneration. The degree of regeneration is proportional to the percentage of fibers harboring centrally-placed nuclei. In this study we observed that the dystrophin-expressing fibers in treated muscle had significantly fewer centrally-placed nuclei compared to muscle fibers of untreated mdx mice. This finding suggested that recombinant dystrophin provided by systemic gene delivery in utero partially protected transduced muscle fibers from cycles of degeneration and regeneration. Furthermore, in treated mdx muscle, even among the fibers where recombinant dystrophin was not detected, there was a non-significant decrease in the number of fibers with centrally-placed nuclei compared to muscle of untreated mdx mice. It is possible that despite absence of immunohistochemical detection of dystrophin expressed by these fibers, dystrophin expression may have been present at a level below the detection threshold and may have provided functional benefit. Alternatively, there may be a ‘bystander’ benefit to non-transduced fibers or non-transduced regions of fibers from being in a treated muscle in close proximity to fibers and fiber segments that express recombinant dystrophin. Therefore, a significant potential benefit of in utero muscle gene transfer for DMD may be to reduce the degree of exhaustion of the satellite cell pool by achieving gene transfer early.
To date, AAV8 has successfully provided therapeutic benefit to skeletal muscle in two disease models in prior postnatal studies. Ziegler et al. showed that systemic administration of an AAV8 vector carrying the human acid α-glucosidase (GAA) gene to 2 month old presymptomatic GAA-deficient mice that model Pompe disease resulted in nearly complete correction of the lysosomal storage of glycogen in all the affected muscles.42 Another study also demonstrated correction of GAA deficiency in muscle tissue of immunodeficient GSD-II mice by treatment with an AAV8 vector.43 In another muscle disease model, systemic delivery of an AAV8 vector carrying a myostatin inhibitor in adult mdx mice enhanced muscle growth and also ameliorated the dystrophic phenotype.44
Only one previous study reported on AAV gene transfer in utero for treatment of a muscle disease. In a mouse model of Pompe disease, an AAV2 vector carrying the α-glucosidase gene was delivered intraperitoneally in utero and resulted in improvement in diaphragmatic in vitro isometric force-frequency studies 6 months after birth40 However, AAV8 has not been previously reported in therapeutic gene delivery studies in utero in a preclinical model of a muscle disease.
In postnatal gene delivery studies, restoration of dystrophin expression has been correlated with muscle functional benefit after AAV8 gene delivery. In a study involving AAV8 carrying microdystrophin delivered systemically into the femoral artery in 3 – 4 week old mdx mice and non human primates, significant improvement in tetanic force and protection against eccentric contraction in the EDL muscle was shown.12 Since dystrophic patients usually die due to respiratory and cardiac failure,2 restoring dystrophin expression to the diaphragm, which plays a critical role in respiration and survival, will be highly important to DMD patients. In the study reported here, the diaphragm was collected 9 weeks following birth for ex vivo force measurements. AAV8 minidystrophin treated diaphragm exhibited a 32% improvement in specific force compared to diaphragms from untreated mdx littermates. In normal muscle, dystrophin provides resistance against contraction induced injury. We observed that the residual force generated following 10 repetitive lengthening activations was significantly improved by AAV8 minidystrophin gene delivery in utero compared to untreated mdx diaphragm.
In summary, systemic delivery of minidystrophin with an AAV8 vector in utero provides efficient transduction of diaphragm and limb muscles of the mdx mouse when studied after birth. The functional benefit demonstrated in transduced diaphragm muscle encourages further studies to test the persistence of vector expression and in utero gene delivery in large animal models of DMD.
Acknowledgments
This work was supported by Grant R01 AR050565 (PRC) and R01 AR45967 (XX) from the NIH. The authors take full responsibility for the contents of this paper, which do not represent the views of the Department of Veterans Affairs or the United States Government.
Footnotes
Work was done in Pittsburgh, PA, USA.
References
- 1.Emery AE. Population frequencies of inherited neuromuscular diseases--a world survey. Neuromuscul Disord. 1991;1:19–29. doi: 10.1016/0960-8966(91)90039-u. [DOI] [PubMed] [Google Scholar]
- 2.Emery AEH, Muntoni F. Duchenne Muscular Dystrophy. Oxford Univ. Press; Oxford, New York: 2003. [Google Scholar]
- 3.Gregorevic P, Allen JM, Minami E, Blankinship MJ, Haraguchi M, Meuse L, et al. rAAV6-microdystrophin preserves muscle function and extends lifespan in severely dystrophic mice. Nat Med. 2006;12:787–789. doi: 10.1038/nm1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gregorevic P, Blankinship MJ, Allen JM, Crawford RW, Meuse L, Miller DG, et al. Systemic delivery of genes to striated muscles using adeno-associated viral vectors. Nat Med. 2004;10:828–834. doi: 10.1038/nm1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu M, Yue Y, Harper SQ, Grange RW, Chamberlain JS, Duan D. Adeno-associated virus-mediated microdystrophin expression protects young mdx muscle from contraction-induced injury. Mol Ther. 2005;11:245–256. doi: 10.1016/j.ymthe.2004.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yoshimura M, Sakamoto M, Ikemoto M, Mochizuki Y, Yuasa K, Miyagoe-Suzuki Y, et al. AAV vector-mediated microdystrophin expression in a relatively small percentage of mdx myofibers improved the mdx phenotype. Mol Ther. 2004;10:821–828. doi: 10.1016/j.ymthe.2004.07.025. [DOI] [PubMed] [Google Scholar]
- 7.McClorey G, Moulton HM, Iversen PL, Fletcher S, Wilton SD. Antisense oligonucleotide-induced exon skipping restores dystrophin expression in vitro in a canine model of DMD. Gene Ther. 2006;13:1373–1381. doi: 10.1038/sj.gt.3302800. [DOI] [PubMed] [Google Scholar]
- 8.Cerletti M, Negri T, Cozzi F, Colpo R, Andreetta F, Croci D, et al. Dystrophic phenotype of canine X-linked muscular dystrophy is mitigated by adenovirus-mediated utrophin gene transfer. Gene Ther. 2003;10:750–757. doi: 10.1038/sj.gt.3301941. [DOI] [PubMed] [Google Scholar]
- 9.Daya S, Berns KI. Gene therapy using adeno-associated virus vectors. Clin Microbiol Rev. 2008;21:583–593. doi: 10.1128/CMR.00008-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Inagaki K, Fuess S, Storm TA, Gibson GA, McTiernan CF, Kay MA, et al. Robust systemic transduction with AAV9 vectors in mice: efficient global cardiac gene transfer superior to that of AAV8. Mol Ther. 2006;14:45–53. doi: 10.1016/j.ymthe.2006.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang Z, Zhu T, Qiao C, Zhou L, Wang B, Zhang J, et al. Adeno-associated virus serotype 8 efficiently delivers genes to muscle and heart. Nat Biotechnol. 2005;23:321–328. doi: 10.1038/nbt1073. [DOI] [PubMed] [Google Scholar]
- 12.Rodino-Klapac LR, Janssen PM, Montgomery CL, Coley BD, Chicoine LG, Clark KR, et al. A translational approach for limb vascular delivery of the micro-dystrophin gene without high volume or high pressure for treatment of Duchenne muscular dystrophy. J Transl Med. 2007;5:45. doi: 10.1186/1479-5876-5-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Emerson CP, Hauschka SD. Embryonic Origins of Skeletal Muscles. In: Engel AG, Franzini-Armstrong C, editors. Myology. McGraw-Hill; NewYork, USA: 2004. pp. 3–44. [Google Scholar]
- 14.Biressi S, Molinaro M, Cossu G. Cellular heterogeneity during vertebrate skeletal muscle development. Dev Biol. 2007;308:281–293. doi: 10.1016/j.ydbio.2007.06.006. [DOI] [PubMed] [Google Scholar]
- 15.Buckingham M, Bajard L, Chang T, Daubas P, Hadchouel J, Meilhac S, et al. The formation of skeletal muscle: from somite to limb. J Anat. 2003;202:59–68. doi: 10.1046/j.1469-7580.2003.00139.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Duxson MJ, Usson Y. Cellular insertion of primary and secondary myotubes in embryonic rat muscles. Development. 1989;107:243–251. doi: 10.1242/dev.107.2.243. [DOI] [PubMed] [Google Scholar]
- 17.Ontell M, Bourke D, Hughes D. Cytoarchitecture of the fetal murine soleus muscle. Am J Anat. 1988;181:267–278. doi: 10.1002/aja.1001810305. [DOI] [PubMed] [Google Scholar]
- 18.Ontell M, Kozeka K. Organogenesis of the mouse extensor digitorum logus muscle: a quantitative study. Am J Anat. 1984;171:149–161. doi: 10.1002/aja.1001710203. [DOI] [PubMed] [Google Scholar]
- 19.Ontell M, Kozeka K. The organogenesis of murine striated muscle: a cytoarchitectural study. Am J Anat. 1984;171:133–148. doi: 10.1002/aja.1001710202. [DOI] [PubMed] [Google Scholar]
- 20.Huard J, Feero WG, Watkins SC, Hoffman EP, Rosenblatt DJ, Glorioso JC. The basal lamina is a physical barrier to herpes simplex virus-mediated gene delivery to mature muscle fibers. J Virol. 1996;70:8117–8123. doi: 10.1128/jvi.70.11.8117-8123.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Reay DP, Bilbao R, Koppanati BM, Cai L, O’Day TL, Jiang Z, et al. Full-length dystrophin gene transfer to the mdx mouse in utero. Gene Ther. 2008;15:531–536. doi: 10.1038/gt.2008.8. [DOI] [PubMed] [Google Scholar]
- 22.Koppanati BM, Li J, Xiao X, Clemens PR. Systemic delivery of AAV8 in utero results in gene expression in diaphragm and limb muscle: treatment implications for muscle disorders. Gene Ther. 2009 doi: 10.1038/gt.2009.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang B, Li J, Qiao C, Chen C, Hu P, Zhu X, et al. A canine minidystrophin is functional and therapeutic in mdx mice. Gene Ther. 2008;15:1099–1106. doi: 10.1038/gt.2008.70. [DOI] [PubMed] [Google Scholar]
- 24.Xiao X, Li J, Samulski RJ. Production of high-titer recombinant adeno-associated virus vectors in the absence of helper adenovirus. J Virol. 1998;72:2224–2232. doi: 10.1128/jvi.72.3.2224-2232.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li J, Samulski RJ, Xiao X. Role for highly regulated rep gene expression in adeno-associated virus vector production. J Virol. 1997;71:5236–5243. doi: 10.1128/jvi.71.7.5236-5243.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Watchko JF, Johnson BD, Gosselin LE, Prakash YS, Sieck GC. Age-related differences in diaphragm muscle injury after lengthening activations. J Appl Physiol. 1994;77:2125–2133. doi: 10.1152/jappl.1994.77.5.2125. [DOI] [PubMed] [Google Scholar]
- 27.Bilbao R, Reay DP, Wu E, Zheng H, Biermann V, Kochanek S, et al. Comparison of high-capacity and first-generation adenoviral vector gene delivery to murine muscle in utero. Gene Ther. 2005;12:39–47. doi: 10.1038/sj.gt.3302392. [DOI] [PubMed] [Google Scholar]
- 28.Lu QL, Partridge TA. A new blocking method for application of murine monoclonal antibody to mouse tissue sections. J Histochem Cytochem. 1998;46:977–984. doi: 10.1177/002215549804600813. [DOI] [PubMed] [Google Scholar]
- 29.Blake DJ, Weir A, Newey SE, Davies KE. Function and genetics of dystrophin and dystrophin-related proteins in muscle. Physiol Rev. 2002;82:291–329. doi: 10.1152/physrev.00028.2001. [DOI] [PubMed] [Google Scholar]
- 30.Rando TA. The dystrophin-glycoprotein complex, cellular signaling, and the regulation of cell survival in the muscular dystrophies. Muscle Nerve. 2001;24:1575–1594. doi: 10.1002/mus.1192. [DOI] [PubMed] [Google Scholar]
- 31.Torres LF, Duchen LW. The mutant mdx: inherited myopathy in the mouse. Morphological studies of nerves, muscles and end-plates. Brain. 1987;110 (Pt 2):269–299. doi: 10.1093/brain/110.2.269. [DOI] [PubMed] [Google Scholar]
- 32.Bockhold KJ, Rosenblatt JD, Partridge TA. Aging normal and dystrophic mouse muscle: analysis of myogenicity in cultures of living single fibers. Muscle Nerve. 1998;21:173–183. doi: 10.1002/(sici)1097-4598(199802)21:2<173::aid-mus4>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 33.Watchko JF, O’Day TL, Hoffman EP. Functional characteristics of dystrophic skeletal muscle: insights from animal models. J Appl Physiol. 2002;93:407–417. doi: 10.1152/japplphysiol.01242.2001. [DOI] [PubMed] [Google Scholar]
- 34.Karolewski BA, Wolfe JH. Genetic correction of the fetal brain increases the lifespan of mice with the severe multisystemic disease mucopolysaccharidosis type VII. Mol Ther. 2006;14:14–24. doi: 10.1016/j.ymthe.2006.02.012. [DOI] [PubMed] [Google Scholar]
- 35.Shen JS, Meng XL, Maeda H, Ohashi T, Eto Y. Widespread gene transduction to the central nervous system by adenovirus in utero: implication for prenatal gene therapy to brain involvement of lysosomal storage disease. J Gene Med. 2004;6:1206–1215. doi: 10.1002/jgm.630. [DOI] [PubMed] [Google Scholar]
- 36.Seppen J, van der Rijt R, Looije N, van Til NP, Lamers WH, Oude Elferink RP. Long-term correction of bilirubin UDPglucuronyltransferase deficiency in rats by in utero lentiviral gene transfer. Mol Ther. 2003;8:593–599. doi: 10.1016/s1525-0016(03)00234-x. [DOI] [PubMed] [Google Scholar]
- 37.Han XD, Lin C, Chang J, Sadelain M, Kan YW. Fetal gene therapy of alpha-thalassemia in a mouse model. Proc Natl Acad Sci U S A. 2007;104:9007–9011. doi: 10.1073/pnas.0702457104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Waddington SN, Nivsarkar MS, Mistry AR, Buckley SM, Kemball-Cook G, Mosley KL, et al. Permanent phenotypic correction of hemophilia B in immunocompetent mice by prenatal gene therapy. Blood. 2004;104:2714–2721. doi: 10.1182/blood-2004-02-0627. [DOI] [PubMed] [Google Scholar]
- 39.Larson JE, Morrow SL, Happel L, Sharp JF, Cohen JC. Reversal of cystic fibrosis phenotype in mice by gene therapy in utero. Lancet. 1997;349:619–620. doi: 10.1016/S0140-6736(05)61567-X. [DOI] [PubMed] [Google Scholar]
- 40.Rucker M, Fraites TJ, Jr, Porvasnik SL, Lewis MA, Zolotukhin I, Cloutier DA, et al. Rescue of enzyme deficiency in embryonic diaphragm in a mouse model of metabolic myopathy: Pompe disease. Development. 2004;131:3007–3019. doi: 10.1242/dev.01169. [DOI] [PubMed] [Google Scholar]
- 41.Zincarelli C, Soltys S, Rengo G, Rabinowitz JE. Analysis of AAV serotypes 1–9 mediated gene expression and tropism in mice after systemic injection. Mol Ther. 2008;16:1073–1080. doi: 10.1038/mt.2008.76. [DOI] [PubMed] [Google Scholar]
- 42.Ziegler RJ, Bercury SD, Fidler J, Zhao MA, Foley J, Taksir TV, et al. Ability of adeno-associated virus serotype 8-mediated hepatic expression of acid alpha-glucosidase to correct the biochemical and motor function deficits of presymptomatic and symptomatic Pompe mice. Hum Gene Ther. 2008;19:609–621. doi: 10.1089/hum.2008.010. [DOI] [PubMed] [Google Scholar]
- 43.Sun B, Zhang H, Franco LM, Young SP, Schneider A, Bird A, et al. Efficacy of an adeno-associated virus 8-pseudotyped vector in glycogen storage disease type II. Mol Ther. 2005;11:57–65. doi: 10.1016/j.ymthe.2004.10.004. [DOI] [PubMed] [Google Scholar]
- 44.Qiao C, Li J, Jiang J, Zhu X, Wang B, Li J, et al. Myostatin propeptide gene delivery by adeno-associated virus serotype 8 vectors enhances muscle growth and ameliorates dystrophic phenotypes in mdx mice. Hum Gene Ther. 2008;19:241–254. doi: 10.1089/hum.2007.159. [DOI] [PubMed] [Google Scholar]