

Abstract
The focus is to expand the original design of fast photochemical oxidation of proteins (FPOP) and introduce SO4−•, generated by 248 nm homolysis of low mM levels of persulfate, as a radical reactant in protein footprinting. FPOP is a chemical footprinting approach to footprinting proteins and protein complexes by “snapshot” reaction with free radicals. The radical used until now is the OH radical, and it provides a measure of residue-resolved solvent accessibility of the native protein. We show that FPOP can accommodate other reagents, increasing its versatility. The new persulfate FPOP system is a potent, non-specific, and tunable footprinting method; 3–5 times less persulfate is needed to give the same global levels of modification as seen with OH radicals. Although solvent-exposed His and Tyr residues are more reactive with SO4−• than with •OH, oxidation of apomyoglobin and calmodulin shows that •OH probes smaller accessible areas than SO4−•, with the possible exception of histidine. His64, an axial ligand in the heme-binding pocket of apomyoglobin, is substantially up-labeled by SO4−• relative to •OH. Nevertheless, the kinds of modification and residue selectivity for both reagent radicals are strikingly similar. Thus, the choice of these reagents relies on the physical properties, particularly the membrane permeability, of the radical precursors.
INTRODUCTION
Chemical footprinting1–3 of proteins has more sensitivity and speed than X-ray crystallography and NMR spectroscopy although it has lower resolution. It is enabled by modern mass spectrometry that allows effective analysis of the footprint products in presence differential experiment (e.g., in the presence and absence of ligand). The outcome brings insight on the binding site(s) and changes in protein conformation provided the labeling of the apo state, for example, is different than that of the holo state. The most general and informative chemical reactions currently used for protein footprinting are hydrogen/deuterium exchange (HDX)4 and hydroxyl radical oxidation.5 They are complementary because •OH samples predominantly side-chains6, whereas HDX reveals backbone amide hydrogen accessibility.7, 8
Hydroxyl radicals probe solvent accessibility because they have comparable size to solvent water molecules and high reactivity with a significant fraction of amino-acid side chains. An advantages of •OH footprinting is that the modifications are irreversible, enabling a “bottom-up” proteomics methodology of proteolysis and LC/MS/MS for their detection and quantification. Interest is increasing in other chemical probes, including the highly reactive methylene carbene9–11, that engender similarly stable modifications. Complementary structural information of protein complexes might be obtainable by taking advantage of the different chemical selectivity and reactivity of accessible protein residues.
We argued that FPOP occurs on the μsec timescale for •OH labeling reactions,12 affording near mM levels of •OH from the homolytic cleavage of H2O2 upon irradiation by a 17 ns flash of 248 nM KrF excimer laser. Constituent Gln acts as a radical scavenger, ensuring that the radical have disappeared by ~ 1 μsec. The strength of this footprinting is that the yield of protein modification by •OH on a profoundly short exposure is high. A μsec labeling timescale prevents sampling of protein conformation that are altered by the modification, as will be the case for timescales that are longer.13
Herein we describe a new footprinting agent for FPOP, an ionic sulfate radical anion, SO4−•. One reason for choosing SO4−• is its reduction potential is 2430 mV, which is higher than that of •OH (1900 mV),14 making it a potentially useful oxidant for proteins. For example, the formation of protein cross-links can be triggered by slow interaction with SO4−•15, 16, and protein oxidation using SO4−• can occur in the metal-catalyzed oxidation reactions.17 The reactant SO4−•, like •OH, should rapidly oxidize proteins, which is an advantage of FPOP using •OH. Reactions of SO4−• with amino acids and some dipeptides in aqueous solution are known albeit on a continuous timescale rather than pulsed as we demonstrate here. This reactivity of SO4−• is determined by its high electron affinity and its capability to oxidize aromatic and Met side chains to the corresponding radical cations, which subsequently undergo either fragmentation or hydration.18, 19 The sulfate radical anion can also oxidize carboxyl anions, including zwitterionic amino acids in aqueous solution, to acyloxyl radicals (RCO2•), which rapidly decarboxylate to give carbon-centered radicals.14, 20 Radicals formed by SO4−• hydrogen abstraction also occur for some amino acids such as Leu and Ser.20
The precursor for SO4−• is the stable and water-soluble Na2S2O8; S2O82− as an aqueous ion has a UV band maximum at 215 nm (we measured the molar absorptivity in PBS to be 24 M−1 cm−1 at 215 nm. The photolysis of S2O82− in water at 308 nM gives SO4−• radical anions with a quantum yield of 0.55.21
We will compare the amino-acid residue reactions of laser-initiated sulfate radical anions with those of hydroxyl radicals after tuning the oxidation levels so that they nearly match. Suitable proteins for the test are apomyoglobin (aMb), Ca2+-free calmodulin (CaM), and a mixture of peptides, bradykinin and angiontensin II. We sought (1) to identify the types of residue modifications and their frequency, (2) to ascertain whether SO4−• may sample more completely a subset of residues than does •OH, and (3) to determine whether persulfate FPOP exclusively modifies solvent accessible residues, and on a timescale fast enough to sample native conformation only.
More than any other method of oxidative protein footprinting, FPOP is highly tunable both in yield (by altering starting material levels of quencher and labeling precursor), timescale, and targeted chemistry. Besides hydroxyl radicals and sulfate radical anions, there are several potential peroxy-22 and diazirine23 species that when formed by UV photolysis, generate either other radicals or carbenes, which may be selected for their physical properties or residue specificity. These opportunities make clear another outcome of this study; that is, a method for validating methods using new reactive reagents for protein footprinting.
EXPERIMENTAL PROCEDURES
Reagents
Bovine β-Lactoglobulin A (BLG), 30% hydrogen peroxide, sodium persulfate, angiotensin II, bradykinin, L-glutamine, L-methionine, catalase, urea, ethylene glycol-bis(2-aminoethylether)-N,N,N',N'-tetraacetic acid (EGTA), acetonitrile, formic acid, proteomics grade trypsin, and phosphate buffered saline (PBS) were purchased from the Sigma Aldrich Chemical Company (St. Louis, MO). Bovine CaM was from Oceanbiologics (Corvallis, OR). The proteins were used without further purification. Purified water (18 MΩ) was obtained from a Milli-Q Synthesis system (Millipore, Billerica, MA).
Oxidative-modification labeling
Each 50 μL sample was prepared in PBS (10 mM phosphate buffer, 138 mM NaCl, 2.7 mM KCl, pH 7.4 at 25 °C); final protein concentration of 10 μM. The bradykinin and angiotensin II used for peptide mixtures were also prepared at these concentrations. Apo-CaM samples included 100 μM EGTA for chelation of adventitious calcium. Glutamine was added to a final concentration of 20 mM in normal FPOP samples. Hydrogen peroxide or Na2S2O8 was added to a final concentration of 15 or 3 mM, respectively, just before FPOP infusion except for BLG, where [Na2S2O8] was 5 mM. The apparatus and procedure for laser flash photolysis of samples were detailed previously.12 The laser power was adjusted to 44 mJ/pulse. The fraction of sample masked from irradiation was set to 20% by adjusting the infusion flow rate and laser-pulse frequency; this ensured that no protein was “double-shot”.12, 13
Samples from FPOP were collected in a microcentrifuge tube containing 20 μL of 70 mM Met, and immediately purified from excess H2O2 or Na2S2O8 by Millipore Ziptip (Billerica, MA) 0.6 μL bed solid-phase extraction. C4 tips were used for BLG, aMb, and CaM samples; C18 tips were for samples containing peptide mixtures. Each 10 μL Ziptip accommodates up to 45 μL in aspirate with careful pipette action. One slow aspiration/dispense cycle was sufficient to load the Ziptip with good recovery; this precaution minimized post-FPOP oxidation.
Except for BLG, four treatments per protein or peptide mixture were applied in triplicate. “Native” samples were solutions of analyte, buffer, and Gln, and were not laser irradiated. “Peroxide” sample solutions included H2O2 and were subjected to flash photolysis. “Persulfate” sample solutions included Na2S2O8 and were subjected to flash photolysis. “Persulfate control” sample solutions were prepared identically to “Persulfate” but were not laser irradiated; persulfate exposure was 5 min prior to desalting. BLG samples were processed similarly but at two persulfate levels and in duplicate.
Global mass spectrometry of FPOP-labeled β-lactoglobulin
For BLG samples, ZiptipC4 eluent was diluted 5-fold in 50% acetonitrile and immediately infused onto a Waters Ultima Global quadrupole time-of-flight (Milford, MA), operating in V mode at 12,000 FWHM resolving power at 838.8 m/z ((CF3COONa)6Na+ calibrant ion).
Proteolysis and LC/MS/MS of FPOP-labeled apomyoglobin, calmodulin, and peptide mixture
Apomyoglobin samples were trypsin-digested for 18 h at 37 ° C; CaM samples were trypsin-digested 6 h at 37 ° C; each was digested at a 1:10 trypsin: protein weight ratio. The peptides were desalted using ZiptipC18 (Millipore), and the 10 μL eluent was diluted 20-fold with purified water. A small sample (2 μL) was loaded by autosampler (Eksigent nanoLC, Dublin, CA) onto a capillary column having a laser-pulled tip (Sutter Instruments, Novato, CA), bomb-packed with C18 reverse phase material (Magic, 0.075mm × 200mm, 5μm, 300Å, Michrom, Auburn, Ca). The gradient was from 1% solvent B (97% acetonitrile, 3% water, 0.1% formic acid) to 60% solvent B over 60 min at an eluent flow of 260 nL/min. The LC was coupled to the nanospray source of an LTQ-Orbitrap mass spectrometer (Thermo Fisher, Waltham, MA), for aMb and peptide mixture samples, or an LTQ-FT mass spectrometer (Thermo Fisher), for CaM. Mass spectra of eluting peptides were obtained at high mass resolving power (100,000 for ions of m/z 400) with the FT mass spectrometer component, while MS/MS experiments on the six most abundant eluting ions per high resolution scan were performed in the LTQ at a normalized collision energy of 35% of maximum, using a 2 Da isolation width and wide-band activation. Ions submitted to MS/MS were placed in a dynamic exclusion list for 8 s. A blank run followed every sample acquisition.
LC/MS/MS data analysis
The peak alignment algorithm of the Rosetta Elucidator data management system (Rosetta Biosoftware) was used to generate tables of extracted ion chromatogram features. Manually validated Mascot error-tolerant search results were paired to their tabulated mass spectral features by a custom Excel VBA program, which also augmented the search results with a 5 ppm threshold mass list of anticipated peroxide and persulfate FPOP products not found by Mascot. Augmented hits were included only if a product-ion spectrum verified the accurate mass match. A third modification discovery method utilized a correlation algorithm to compare unidentified product-ion spectra to exemplary CID fragment spectra of unmodified tryptic peptides of aMb, CaM, and of bradykinin and angiontensin II.24
RESULTS AND DISCUSSION
Optimal Sodium Persulfate FPOP Conditions
The mass spectrum bounding the 15th charge state of β-lactoglobulin (BLG) obtained when 15 mM Na2S2O8 was mixed with PBS-buffered 10 μM BLG and incubated 5 min at room temperature (Figure 1a) shows insignificant protein oxidation. The base peak corresponds to unmodified BLG; the very low levels of modified and/or ESI adducts of BLG are superimposable with the mass spectrum of a stock solution BLG (data not shown). Thus, short-time exposure to low levels of Na2S2O8 does not oxidize BLG. Apomyoglobin, CaM, bradykinin, and angiontensin II also do not show Na2S2O8 oxidation over short-time exposures, although ESI MS revealed that CaM in its starting state was already oxidized to show +16 and +32 Da adducts.
Figure 1.

The ESI-QTOF mass spectra of the 15th charge state of β-lactoglobulin subjected to different labeling conditions. Spectrum (a) is of the 15 mM Na2S2O8 control, absent only laser irradiation; (b) is of 15 mM Na2S2O8 FPOP; (c) is of 5 mM Na2S2O8 FPOP; (d) is of 15 mM H2O2 FPOP.
Irradiating the flowing solution containing the same level of Na2S2O8 with the KrF excimer laser beam, however, gives rise to a high yield of modified BLG (Figure 1b). We see that the dominant persulfate radical modification products correspond to successive +16 Da additions (or +1.07 m/z addition for the 15th charge state). These are the dominant products of •OH reactions in other protein footprinting chemistry5 and is what we observed with 15 mM H2O2 FPOP (Figure 1d). Further, FPOP of 15 mM Na2S2O8 gives a much higher yield of modified protein than the standard FPOP of 15 mM H2O2. Solely by reducing the concentration of Na2S2O8 to 5 mM, the modification distribution seen in Figure 1c is moderated to nearly the same global protein outcome as with standard H2O2 FPOP.
A more rigorous analysis of this product distribution, presented in the Supporting Information section, shows it is well modeled by a Poisson distribution of 0, +16, +32, … Da states, indicating the labeling has sampled a single (native) conformation. β-lactoglobulin is known to be conformationally sensitive to oxidation.25 It follows that 5 mM Na2S2O8 FPOP labeling, like 15 mM H2O2 FPOP, labels faster than most secondary and tertiary protein motions26, 27 and corroborates the theoretical prediction of labeling at the μs level.
Residue-Resolved Modification Measurement by LC/MS/MS
Owing to the stable covalent label imparted by •OH and the high percentage of solvent-accessible residues that may be labeled, we chose a “bottom-up” proteomics approach for identification and quantitation of products of footprinting, typically using trypsin proteolysis and online LC-MS/MS.5 We used this strategy because the global pattern of persulfate modification indicates a similar promiscuity in residue reaction as with •OH labeling.
Site-specific labeling was established by LC/MS/MS analysis. The aggregate of all modifications at each site was determined (Tables 1, Supporting Information Tables 1 and 2) as a “fraction-modified” metric, allowing comparisons of amino-acid types that are modified by persulfate and peroxide treatments (Figure 2). The fraction-modified metric is calculated, per residue, by summing, in the numerator, signals for all detected peptides having a modification at that residue and by summing, in the denominator, signals for all peptides, modified and unmodified, having the same 1° sequences as the numerator set of peptides. The fraction-modified metric is implicitly normalized across samples because all variations in post-FPOP sample handling, proteolysis, de-salting, on-line chromatography, and MS that affect a modified peptide’s signal will likely influence signals for its unmodified and modified “siblings” in the same proportion, with the exception of ionization efficiency.28
Table 1.
Apomyoglobin Fraction Modified per Residuea
| residue | SASA (Å2)b | k•OH (M−1 sec−1)c | native | peroxide | persulfate | persulfate control |
|---|---|---|---|---|---|---|
| E6 | 56.1 | 2.3 ×108 | 0 | 0.13 +/− 0.04% | 0.069 +/− 0.007% | 0.0003 +/− 0.0002% |
| W7 | 15.3 | 1.3 ×1010 | 1.41 +/− 0.09% | 13.9 +/− 0.9% | 15.3 +/− 0.6% | 2.2 +/− 0.2% |
| W14 | 6.8 | 1.3 ×1010 | 0.84 +/− 0.06% | 12.5 +/− 0.6% | 4.6 +/− 0.5% | 1.57 +/− 0.07% |
| E18 | 65.5 | 2.3 ×108 | 0.000049 +/− 0.000009% | 2.07 +/− 0.06% | 2.4 +/− 0.2% | 0.004 +/− 0.002% |
| D20 | 65.8 | 7.5 ×107 | 0 | 0.20 +/− 0.01% | 0.247 +/− 0.009% | 0 |
| I21 | 62.1 | 1.8 ×109 | 0 | 0.45 +/− 0.03% | 0.05725 +/− 0.00009% | 0 |
| H24 | 7.7 | 4.8 ×109 | 0.0047 +/− 0.0003% | 0.70 +/− 0.05% | 0.32 +/− 0.02% | 0.0060 +/− 0.0004% |
| Q26 | 22.5 | 5.4 ×108 | 0 | 0.07 +/− 0.01% | 0.07 +/− 0.01% | 0.00010 +/− 0.00006% |
| E27 | 49.0 | 2.3 ×108 | 0 | 0.71 +/− 0.03% | 0.24 +/− 0.01% | 0.00010 +/− 0.00006% |
| I30 | 7.9 | 1.8 ×109 | 0 | 0.054 +/− 0.006% | 0.002 +/− 0.002% | 0 |
| T34 | 48.8 | 5.1 ×108 | 0.0042 +/− 0.0009% | 0.43 +/− 0.04% | 0.08 +/− 0.03% | 0.008 +/− 0.001% |
| H36 | 54.5 | 4.8 ×109 | 0.0023 +/− 0.0007% | 2.4 +/− 0.2% | 1.7 +/− 0.8% | 0.009 +/− 0.004% |
| P37 | 81.9 | 6.5 ×108 | 0 | 0.38 +/− 0.04% | 0.06 +/− 0.01% | 0.0002 +/− 0.0002% |
| E41 | 93.5 | 2.3 ×108 | 0 | 0.105 +/− 0.005% | 0.14 +/− 0.04% | 0 |
| H64 | 41.8 | 4.8 ×109 | 0.0022 +/− 0.0003% | 0.81 +/− 0.01% | 4.5 +/− 0.6% | 0.042 +/− 0.009% |
| V67 | 86.1 | 8.5 ×108 | 0.00034 +/− 0.00005% | 0.41 +/− 0.02% | 0.071 +/− 0.009% | 0.00031 +/− 0.00009% |
| T70 | 79.3 | 5.1 ×108 | 0.016 +/− 0.006% | 0.09 +/− 0.02% | 0.03 +/− 0.02% | 0.009 +/− 0.003% |
| H81 | 129.3 | 4.8 ×109 | 0.029 +/− 0.003% | 3.9 +/− 0.1% | 7 +/− 1% | 0.49 +/− 0.03% |
| H82 | 5.1 | 4.8 ×109 | 0.0052 +/− 0.0005% | 0.61 +/− 0.04% | 0.69 +/− 0.06% | 0.011 +/− 0.001% |
| E83 | 112.0 | 2.3 ×108 | 0.018 +/− 0.002% | 0.36 +/− 0.06% | 0.73 +/− 0.02% | 0.026 +/− 0.004% |
| L86 | 1.9 | 1.7 ×109 | 0 | 0.37 +/− 0.03% | 0.0004 +/− 0.0003% | 0 |
| L89 | 43.1 | 1.7 ×109 | 0.010 +/− 0.002% | 1.06 +/− 0.08% | 0.08 +/− 0.02% | 0.0077 +/− 0.0005% |
| Q91 | 72.9 | 5.4 ×108 | 0 | 0.29 +/− 0.02% | 0.14 +/− 0.02% | 0.04 +/− 0.03% |
| H93 | 52.0 | 4.8 ×109 | 0.0023 +/− 0.0004% | 1.9 +/− 0.1% | 1.63 +/− 0.07% | 0.004 +/− 0.002% |
| H113 | 80.6 | 4.8 ×109 | 0 | 1.9 +/− 0.1% | 2.0 +/− 0.4% | 0.009 +/− 0.004% |
| H116 | 63.9 | 4.8 ×109 | 0 | 0.2 +/− 0.1% | 1.7 +/− 0.5% | 0 |
| H119 | 30.9 | 4.8 ×109 | 0 | 0.95 +/− 0.07% | 0.55 +/− 0.06% | 0 |
| Q128 | 66.5 | 5.4 ×108 | 0 | 0.34 +/− 0.03% | 0.018 +/− 0.002% | 0 |
| M131 | 0.9 | 8.5 ×109 | 3.7 +/− 0.3% | 25 +/− 1% | 24 +/− 3% | 5.9 +/− 0.4% |
| K133 | 67.6 | 3.5 ×108 | 0 | 0.09 +/− 0.01% | 0.0001 +/− 0.0001% | 0 |
| E136 | 76.3 | 2.3 ×108 | 0 | 0.083 +/− 0.007% | 0.137 +/− 0.006% | 0 |
| L137 | 40.4 | 1.7 ×109 | 0 | 0.27 +/− 0.03% | 0.003 +/− 0.001% | 0 |
| F138 | 19.8 | 6.9 ×109 | 0 | 0.70 +/− 0.03% | 0.141 +/− 0.007% | 0.0009 +/− 0.0006% |
| F151 | 26.7 | 6.9 ×109 | 0 | 36 +/− 2% | 7.9 +/− 0.4% | 0 |
The numerator is the sum all signal of peptides modified at a residue. The denominator is the sum of all signal of peptides, modified and unmodified, having the same sequences as the numerator set of peptides.
Solvent accessible surface area (SASA) was calculated as the sum of non-hydrogen side chain and α-carbon areas using the online calculator at http://molbio.info.nih.gov/structbio/basic.html, using a 1.4 Å probe 29. The myoglobin crystal structure 1WLA.pdb was used with heme atoms omitted 30.
Figure 2.

With (a), the relative difference between persulfate and peroxide fraction modified, of CaM, aMb, bradykinin, and angiotensin II residues, are determined per residue; these values are averaged per amino-acid type. The error bars denote the average pairwise-comparison standard error. In (b) the maximum fraction modified among all same-amino acid residues is plotted. Black bars denote H2O2 FPOP labeling; diagonal-pattern bars denote Na2S2O8 FPOP labeling.
We observed no detectable modifications of proteins in their mass spectra following control experiments for which no laser irradiation was used, consistent with global observations (e.g., Figure 1a). Furthermore, we found that the fraction-modified levels of persulfate control residues are statistically identical to those of the “native” control (absent persulfate) fraction-modified levels and, with the exception of Met, are near the limit of detection. These results follow from examination of Tables 1 and Supporting Information Tables 1 and 2, and they show one general trend necessary for validating persulfate FPOP as a viable labeling strategy.
Selectivity of Na2S2O8 FPOP
From these tables and figure 2, it is clear that Na2S2O8 FPOP is a non-specific labeling method that samples many of the same residues with comparable reactivity as does H2O2 FPOP. Both methods reliably label over one half of the 20 common amino acids. Comparing reactivity on a residue-by-residue basis (Figure 2a) shows that peroxide FPOP more readily labels aliphatic residues as well as Phe, Thr, Gln, and Lys. If equivalent levels of labeling are expected for 5 mM persulfate vs. 15 mM hydrogen peroxide, as witnessed at the protein global level, it seems unlikely that the large-difference contributors underlying this trend (for example, aMb I21 in table 1) are explained solely by the use of 3 mM [Na2S2O8] instead of 15 mM. These variations also arise from different inherent reactivities of •OH and SO4−•, and different molecular sizes. In particular, Trp is exceptionally sensitive to both labeling methods; aMb W7 and W14 are equivalently labeled by H2O2 FPOP, butW7 is 3-fold more oxidized than W14 with Na2S2O8 FPOP. The solvent-accessible surface area values are small but non-zero, 15 and 7 Å2, respectively, for W7 and W14, so that the difference between labeling methods for this residue type may be a function exclusively of size differences in the reactant probes.
The amino acid labeling levels are listed from left-to-right in both graphs of Figure 2 in order of decreasing observable peroxide reactivity per amino-acid group, as established by the maximum observed fraction modified per residue of each group (Figure 2b). These maximum fraction values should pertain to the most fully solvent-exposed side-chain residues of each amino acid group, permitting a comparison of the inherent reactivity of SO4−• vs. •OH. The order of SO4−• reactivity is slightly different: M > Y = W > F = E = H > S > P > D = T > K = Q > L = V = I. In either case, the most reactive residues are consistent with amino acid rate data and with the •OH labeling products from water radiolysis31, with the exception of Glu. Persulfate FPOP is more reactive with His, and Tyr and equally reactive to Met, Trp, Glu, and Ser. Despite the negative charge of SO4−• and its precursor, we observed no increased affinity for basic residues other than His, possibly owing to attenuation of electrostatic interaction by the phosphate-buffered saline solution. Trypsin digestion may also bias against detecting modified Lys and Arg.
Chemistry of Na2S2O8 FPOP
With the exception of the +34 Da His modification (Supporting Information) many modifications discernable for peroxide FPOP were also found among persulfate FPOP replicates (Table 2). Furthermore, these modifications comprise the “usual suspects” in •OH labeling by peroxide-initiated and water-radiolysis experiments.5 The commonality of the sets of modifications produced by each FPOP method suggests that the dominant chemical pathways of SO4−• labeling are analogous to the better understood •OH mechanisms.32 A likely explanation for these similar outcomes is that both processes begin with an initial hydrogen abstraction reaction at aliphatic sites. Additionally, transfer of radical to water (equation 1) may compete, giving rise to the common •OH footprinting products.
Table 2.
Observed Residue Modifications of Persulfate and Peroxide FPOP
| Amino Acid | Net Modifications (Da) | ||||||
|---|---|---|---|---|---|---|---|
| Asp | −44 | ||||||
| Gln | +16 | ||||||
| Glu | −44 | −30 | −28 | −18 | |||
| Glya | −44 | ||||||
| His | −23 | −22 | −10 | +5 | +16 | +32 | +34b |
| Ile | +14 | +16 | |||||
| Leu | −2 | +16 | |||||
| Lys | +14 | +16 | |||||
| Met | +16 | ||||||
| Phe | +16 | +30 | +46 | ||||
| Pro | +14 | +16 | |||||
| Ser | −2 | +16 | |||||
| Thr | −2 | ||||||
| Trp | −14 | +4 | +16 | +32 | +48 | ||
| Tyr | +16 | +32 | +34b | ||||
| Val | +14 | +16 | |||||
Only C-terminus G153 of myoglobin exhibited carbon dioxide loss.
Only observed among persulfate FPOP replicates.
| (1) |
We can’t rule out other novel products of SO4−• not detected in these experiments; that 85% of all +2 and higher charge state species of significant abundance were identified as aMb modified and unmodified peptides indicates that any such products are of low-yield.
Solvent Accessibility
In comparing equilibrium states, as is typical for footprinting experiments5, changes in the fraction modified at a residue between states should solely reflect a change in solvent accessibility—all other biases are inherently normalized. The sensitivity of persulfate FPOP labeling to solvent accessibility can be assayed by comparing the solvent accessible surface area (SASA) calculated from X-ray and NMR structures to the per-residue fraction modified for any same-amino-acid set of residues of a protein. The restriction to same residue types stems from the inherent •OH (and presumably with SO4−•) reactivity difference between free amino acids.5 An analysis of the aMb His modification levels is illustrative of the promise of this approach (Figure 3). We find that its inherent reactivity is not too low to limit detection of the modifications and that a large range of SASA values are spanned by the 11 myoglobin His residues. The reactivity of His in Na2S2O8 FPOP correlates reasonably well with the calculated SASA (R2 = 0.83, Figure 3b). Although the correlation is not high, most of the uncertainty may be in the calculated SASA. When H64, an outlier, is excluded, a better correlation is obtained. The rationale for omitting this residue (Figure 4) is that it is an axial ligand of the heme iron. The correlation of aMb His labeling and SASA when using H2O2 FPOP is better still, even with H64 inclusion (plot not shown). We propose that S2O82− or SO4−• has an affinity for the heme binding pocket that H2O2 or •OH does not share; thus, the local concentration of labeling agent is higher than bulk. That H64 and H93, the second heme-ligating residue, are not equally labeled suggests a preferred orientation (e.g., a chelate bridge) and/or affinity for persulfate.
Figure 3.

The modification yields of aMb His residues are plotted against their calculated solvent accessible surface area (SASA), with least-squares straight-line fits shown. In plot (b) the myoglobin His64 was omitted; this improved the R2 fit from 0.63 in (a) to 0.83. The SASA values were calculated from the 1WLA.pdb crystal structure of myoglobin with its heme ignored by the calculator at http://molbio.info.nih.gov/structbio/basic.html, using a 1.4 Å probe.
Figure 4.
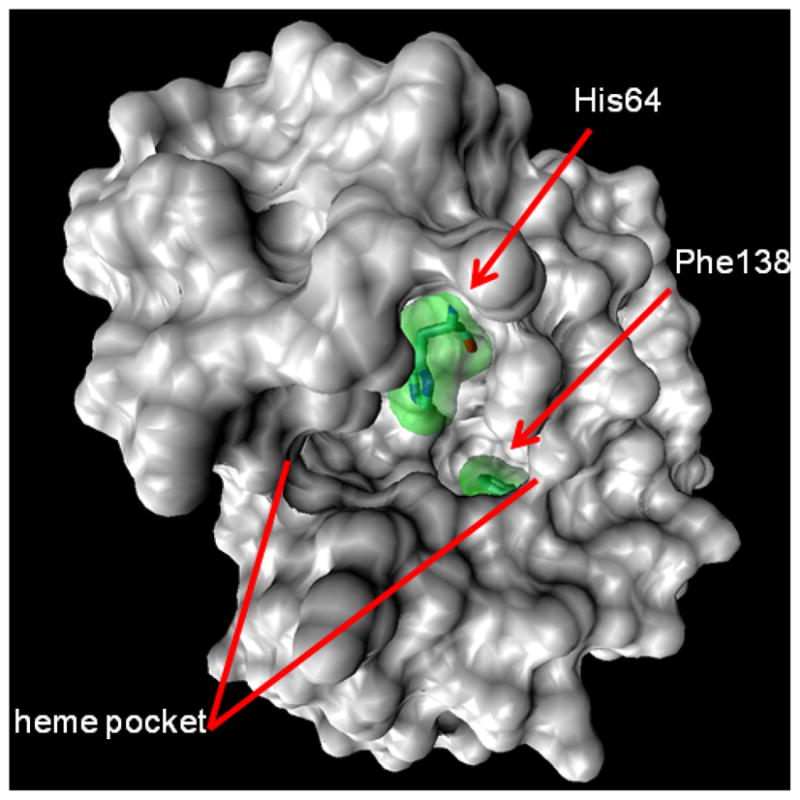
The 1.4 Å probe surface rendering of the myoglobin 1WLA.pdb crystal structure, showing the heme binding pocket, His64, and Phe138.
With the exception of Glu, all amino-acid residues of the same type show increasing yield with increasing SASA, although the data sets for Trp, Phe, Leu, Ile, and Thr are too small for proper correlation analysis. Their sampling may be improved with a different proteolysis methodology, to give shorter or longer peptides in regions of too few or too many trypsin cleavage sites, thus enabling informative production (MS/MS) spectra in these regions. Nevertheless the most compelling correlation is binary: never is a non-Met residue labeled that shouldn’t be. Although Met is extremely sensitive to oxidative labeling, it does not serve as the ideal residue for probing changes in solvent accessibility without careful controls5 because its inherent reactivity is high even with mild oxidizing agents33 and because it may serve as an oxidation sink for persistent protein hydroperoxides that can arise as intermediates in the formation of common oxidative-modification products initiated by fast •OR chemistry.34 Furthermore, many proteins are isolated with endogenous Met oxidation; we found, for example, that CaM samples exhibited significant Met oxidation “out of the box” (Supporting Information Table 1, “native” column).
The labeling of apomyoglobin F138 is 50-fold lower than that of F151, although the calculated SASA is only 25% less. This calculation is based on the myoglobin crystal structure 1WLA30 with the heme removed. This result quantitatively reproduces our previous finding12 that the H-helix moves to close the pocket in the apo state, burying F138. It also underscores the potential biases of using static solid-state structures and the merits of FPOP to determine SASAa of proteins in dilute solution. Finally, the myoglobin E18 is dominated by significant CO2 loss (−44 Da) signal in both peroxide and persulfate FPOP. Other acidic residues also exhibit this pathway, but their yields are significantly lower. This may be a consequence of local S2O82− and H2O2 affinity but probably not a consequence of error in SASA estimation as the magnitude of yield change is so large.
Persulfate vs. peroxide FPOP: physical considerations and future prospects
Although protein footprinting at physiologic levels of analyte, ionic activity, and pH are possible with FPOP, the requirement of peroxy starting material begs the definition of “physiologic”. It is advantageous that persulfate FPOP requires 3–5 fold less starting material than peroxide FPOP to deliver the same levels of modification (Figure 1). On the other hand, excess Na2S2O8 must be removed non-enzymatically after labeling, and this may introduce another source of error. One potential application of FPOP is the study of membrane proteins in micelles, liposomes, lipoprotein particles, and synthetic protein-wrapped phospholipid bilayer discs.35 The lipid bilayer permeability of S2O82− is in all likelihood dramatically smaller than that of H2O2, commonly known to be membrane-permeable 36. Marla and coworkers 37 showed that ONOO− readily permeates large unilamellar vesicles composed of L-α-phosphatidylcholine, stearyl amine, and cholesterol, but that SO42− is membrane-impermeable. They argued the high peroxynitrate permeability may be due to its relatively high basicity (pKa = 6.8 38, 39), whereas sulfate is the conjugate base of a strong acid. Persulfate retains its anionic character in PBS-buffered solution and, thus, should be membrane-impermeable. By using H2O2 and Na2S2O82− in tandem FPOP experiments, a footprint may be acquired of extracellular-accessible and cytoplasmic-accessible residues in membrane particles imbedded with analyte protein. Finally, the thermal stability of S2O82− relative to H2O2 is advantageous for any temperature-varied FPOP experiment up to 50° C 40, 41.
CONCLUSIONS
The results demonstrate the viability of persulfate FPOP protein footprinting. The ideal footprinting experiment is of state comparisons whereby one seeks differences in labeling yield at identical sites for two treatments (e.g., apo vs. holo, native vs. unfolded) to reflect residue protection (or exposure with signal increase) owing to direct inter- or intramolecular interaction or allosteric change. If a detailed footprinting picture is required in which the “fraction modified” ascribes a solvent accessibility value, a thorough search and replicate quantitation for all FPOP modifications in addition to calibration with proteins of known structure will be required. The kinds of modifications with persulfate and peroxide, with one exception, are identical, and the promiscuity of SO4−• is similar and tunable like •OH. The choice for utilizing persulfate FPOP for non-residue-specific stable modification footprinting is best made considering its physical, rather than chemical properties. Most importantly, this study demonstrates the versatility of the FPOP method. That persulfate FPOP works serves as an invitation to try other UV-sensitive precursor molecules, such as L-photoleucine23, or to use the sulfate radical anion to produce other radicals and radical anions (e.g., CO3−• from HCO3− or •NO2 from NO2−). These approaches may give controllable reactive species capable of fast labeling, not only sampling native conformations is a non-specific or targeted way but also expanding the scope of FPOP.
Supplementary Material
Acknowledgments
The research was supported by the National Centers for Research Resources of the NIH, Grant No. 2P41RR000954. HC is very grateful to the generous support given by Prof. Graham Cooks and financial aid from Ohio University.
Footnotes
The research was supported by the National Centers for Research Resources of the NIH, Grant No. 2P41RR000954.
Contributor Information
Brian C. Gau, Washington University in St. Louis, St. Louis, Missouri 63130
Hao Chen, Ohio University, Athens, OH 45701.
Yun Zhang, Ohio University, Athens, OH 45701.
Michael L. Gross, Email: mgross@wustl.edu, Washington University in St. Louis, St. Louis, Missouri 63130
References
- 1.Hambly DM, Gross ML. In: The Encyclopedia of Mass Spectrometry: Ionization Methods. Gross ML, Caprioli RM, editors. Vol. 6 2006. [Google Scholar]
- 2.Guan JQ, Chance MR. Trends Biochem Sci. 2005;10:583–592. doi: 10.1016/j.tibs.2005.08.007. [DOI] [PubMed] [Google Scholar]
- 3.Galas DJ, Schmitz A. Nucl Acids Res. 1978;5:3157–3170. doi: 10.1093/nar/5.9.3157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen S, Engen JR. Cur Anal Chem. 2009;5:205–212. [Google Scholar]
- 5.Xu G, Chance MR. Chem Rev. 2007;107:3514–3543. doi: 10.1021/cr0682047. [DOI] [PubMed] [Google Scholar]
- 6.Maleknia SD, Brenowitz M, Chance MR. Anal Chem. 1999;71:3965–3973. doi: 10.1021/ac990500e. [DOI] [PubMed] [Google Scholar]
- 7.Zhongqi Z, David LS. Protein Sci. 1993;2:522–531. [Google Scholar]
- 8.Katta V, Chait BT. J Am Chem Soc. 1993;115:6317–6321. [Google Scholar]
- 9.Gabriela EG, Ana C, Patricio OC, Fernando AG, José MD. Protein Sci. 2006;15:744–752. [Google Scholar]
- 10.Patricio OC, Daniela BU, José MD. Protein Sci. 2002;11:1353–1366. [Google Scholar]
- 11.Frederic MR, Raphael L, Richard W, Darshan P, Gerard O. Protein Sci. 2000;9:2506–2517. [Google Scholar]
- 12.Hambly DM, Gross ML. J Am Soc Mass Spectrom. 2005;16:2057–2063. doi: 10.1016/j.jasms.2005.09.008. [DOI] [PubMed] [Google Scholar]
- 13.Gau BC, Sharp JS, Rempel DL, Gross ML. Anal Chem. 2009;81:6563–6571. doi: 10.1021/ac901054w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Davies MJ, Dean RT. Radical-Mediated Protein Oxidation: From Chemistry to Medicine. Oxford University Press; 1997. [Google Scholar]
- 15.Fancy DA, Kodadek T. Proc Natl Acad Sci USA. 1999;96:6020–6024. doi: 10.1073/pnas.96.11.6020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rayshell M, Ross J, Werbin H. Carcinogenesis. 1983;4:501–507. doi: 10.1093/carcin/4.5.501. [DOI] [PubMed] [Google Scholar]
- 17.Bridgewater JD, Lim J, Vachet RW. Anal Chem. 2006;78:2432–2438. doi: 10.1021/ac051983r. [DOI] [PubMed] [Google Scholar]
- 18.Davies MJ, Gilbert BC, Norman ROC. J Chem Soc, Perkin Trans. 1984;2:503–509. [Google Scholar]
- 19.Davies MJ, Gilbert BC, McCleland CW, Thomas CB, Young J. J Chem Soc, Chem Commun. 1984:966–967. [Google Scholar]
- 20.Rustgi SN, Riesz P. Int J Radiat Biol. 1978;34:301–316. doi: 10.1080/09553007814550941. [DOI] [PubMed] [Google Scholar]
- 21.Ivanov KL, Glebov EM, Plyusnin VF, Ivanov YV, Grivin VP, Bazhin NM. J Photochem Photobiol, A. 2000;133:99–104. [Google Scholar]
- 22.Chatgilialoglu C, Lunazzi L, Macciantelli D, Placucci G. J Am Chem Soc. 1984;106:5252–5256. [Google Scholar]
- 23.Jumper CC, Schriemer D. 58th ASMS Conference on Mass Spectrometry and Allied Topics; Salt Lake City, UT. 2010. [Google Scholar]
- 24.Vidavsky I, Rempel DL, Gross ML. Proceedings of the 54th ASMS Conference on Mass Spectrometry and Allied Topics; Seattle, WA. 2006. [Google Scholar]
- 25.Venkatesh S, Tomer KB, Sharp JS. Rapid Commun Mass Spectrom. 2007;21:3927–3936. doi: 10.1002/rcm.3291. [DOI] [PubMed] [Google Scholar]
- 26.Chung HS, Ganim Z, Jones KC, Tokmakoff A. Proc Natl Acad Sci USA. 2007;104:14237–14242. doi: 10.1073/pnas.0700959104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chung HS, Khalil M, Smith AW, Ganim Z, Tokmakoff A. Proc Natl Acad Sci USA. 2005;102:612–617. doi: 10.1073/pnas.0408646102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fenn JB. J Am Soc Mass Spectrom. 1993;4:524–535. doi: 10.1016/1044-0305(93)85014-O. [DOI] [PubMed] [Google Scholar]
- 29.Gerstein M. Acta Crystallographica Section A. 1992;48:271–276. [Google Scholar]
- 30.Maurus R, Overall CM, Bogumil R, Luo Y, Mauk AG, Smith M, Brayer GD. Biochimica et Biophysica Acta (BBA) - Protein Structure and Molecular Enzymology. 1997;1341:1–13. doi: 10.1016/s0167-4838(97)00064-2. [DOI] [PubMed] [Google Scholar]
- 31.Xu G, Chance MR. Anal Chem. 2005;77:4549–4555. doi: 10.1021/ac050299+. [DOI] [PubMed] [Google Scholar]
- 32.Garrison WM. Chem Rev. 1987;87:381–398. [Google Scholar]
- 33.Shechter Y, Burstein Y, Patchornik A. Biochemistry. 1975;14:4497–4503. doi: 10.1021/bi00691a025. [DOI] [PubMed] [Google Scholar]
- 34.Saladino J, Liu M, Live D, Sharp JS. J Am Soc Mass Spectrom. 2009;20:1123–1126. doi: 10.1016/j.jasms.2009.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nath A, Atkins WM, Sligar SG. Biochemistry. 2007;46:2059–2069. doi: 10.1021/bi602371n. [DOI] [PubMed] [Google Scholar]
- 36.Rozga M, Bal W. Chemical Research in Toxicology. 2009 doi: 10.1021/tx900358j. [DOI] [PubMed] [Google Scholar]
- 37.Marla SS, Lee J, Groves JT. Proc Natl Acad Sci USA. 1997;94:14243–14248. doi: 10.1073/pnas.94.26.14243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pryor WA, Jin X, Squadrito GL. Proc Natl Acad Sci USA. 1994;91:11173–11177. doi: 10.1073/pnas.91.23.11173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Radi R, Beckman JS, Bush KM, Freeman BA. J Biol Chem. 1991;266:4244–4250. [PubMed] [Google Scholar]
- 40.Santos AM, Ph V, Graillat C, Guyot A, Guillot J. J Polymer Sci Polymer Chem. 1996;34:1271–1281. [Google Scholar]
- 41.Rice FO, Reiff OM. J Phys Chem. 1927;31:1352–1356. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.