

Abstract
Membrane type 1 (MT1)-matrix metalloproteinase (MT1-MMP) is a membrane-tethered MMP that has been shown to play a key role in promoting cancer cell invasion. MT1-MMP is highly expressed in bone metastasis of prostate cancer (PC) patients and promotes intraosseous tumor growth of PC cells in mice. The majority of metastatic prostate cancers harbor loss-of-function mutations or deletions of the tumor suppressor PTEN (phosphatase and tensin homologue deleted on chromosome ten). However, the role of PTEN inactivation in MT1-MMP expression in PC cells has not been examined. In this study, prostate epithelial cell lines derived from mice that are either heterozygous (PTEN+/-) or homozygous (PTEN-/-) for PTEN deletion or harboring a wild type PTEN (PTEN+/+) were used to investigate the expression of MT1-MMP. We found that biallelic loss of PTEN is associated with posttranslational regulation of MT1-MMP protein in mouse PC cells. PTEN-/- PC cells display higher levels of MT1-MMP at the cell surface when compared to PTEN+/+ and PTEN+/- cells and consequently exhibited enhanced migratory and collagen-invasive activities. MT1-MMP displayed by PTEN-/- cells is differentially O-glycosylated and exhibits a slow rate of turnover. MT1-MMP expression in PTEN-/- cells is under control of the PI3K/AKT signaling pathway, as determined using pharmacological inhibitors. Interestingly, rapamycin, an mTOR inhibitor, up-regulates MT1-MMP expression in PTEN+/+ cells via PI3K activity. Collectively, these data in a mouse prostate cell system uncover for the first time a novel and complex relationship between PTEN loss-mediated PI3K/AKT activation and posttranslational regulation of MT1-MMP, which may play a role in PC progression.
Keywords: matrix metalloproteinases, prostate cancer, PTEN, glycosylation, posttranslational modification
1. Introduction
The matrix metalloproteinase (MMP) family of zinc-dependent endopeptidases comprises secreted and membrane-anchored proteases that are major mediators of cellular proteolysis [1]. MMPs play key roles in normal and pathological conditions due to their ability to accomplish the proteolytic processing of multiple proteins thereby influencing cell behavior. The members of the membrane-anchored MMP subfamily, referred to as membrane-type MMPs (MT-MMPs), comprise type I transmembrane and glycosylphosphatidylinositol (GPI)-containing MMPs that by their specific structural organization and subcellular localization play a pivotal role in pericellular proteolysis [2]. Membrane-anchoring confers these MMPs with a unique set of regulatory mechanisms that serve to maintain a pool of active protease at the cell surface including internalization, autocatalytic processing and ectodomain shedding [3]. In addition, there is evidence that post-translational modifications such as O-glycosylation, homodimerization, phosphorylation, and/or palmytoilation can influence MT-MMP function by regulating intracellular trafficking, insertion into membrane microdomains, stability, endocytosis, substrate interaction, and/or inhibitor binding [3-11].
In cancer, members of the MT-MMP subfamily have been shown to promote tumor cell migration and invasion through extracellular matrices. In particular, the transmembrane MT-MMPs, MT1-(MMP14), MT2-(MMP15) and MT3-(MMP16) MMP, were described as a triad of MMPs that support the invasive behavior of cancer cells [12]. MT1-MMP has been shown to facilitate invasion of tumor cells through interstitial collagen by accomplishing proteolysis of collagen I [13]. Consistently, MT1-MMP expression promotes intraosseous growth of human prostate cancer (PC) cells and induces an osteolytic response in a mouse model of PC intraosseous growth [14]. Human PC cell lines expressing MT1-MMP exhibit enhanced migratory and invasive activity in vitro and increased tumorigenicity and metastatic potential in mice [15-18]. Furthermore, MT1-MMP was shown to induce an epithelial-to-mesenchymal transition phenotype when ectopically expressed in LNCaP cells [19]. These experimental results together with evidence showing expression of MT1-MMP in primary tumors [20-22] and in bone metastasis [14] of PC suggest a role for MT1-MMP in PC progression.
PTEN (phosphatase and tensin homologue deleted on chromosome ten) is a phosphatase that its main function is to dephosphorylate phosphatidylinositol 3,4,5-trisphosphate (PIP3), generated by phosphoinositide 3-kinase (PI3K), which results in the formation of PIP2. Dephosphorylation of PIP3 by PTEN negatively regulates the activation AKT/Pkb pathway whereas inactivation of PTEN leads to accumulation of PIP3, which promotes AKT phosphorylation. Active AKT modulates multiple downstream effectors leading to protein synthesis, cell proliferation, and cell survival. Consequently, deregulated activation of the PI3K/AKT pathway upon loss of PTEN function can contribute to cancer development and progression. Accumulating evidence indicate that loss of heterozygosity and somatic mutations of the tumor suppressor gene PTEN is one of the most frequent genetic alterations found in PC patients [23-25]. PTEN deletions are associated with 15-30% of localized cancers [26, 27] and with poor clinical outcome [28, 29]. The rate of PTEN mutations increases to more than 50% in metastatic PC [30]. Consistent with these findings, homozygous, conditional deletion of PTEN by Cre recombinanse under control of the probasin or prostate specific antigen (PSA) promoter in mouse prostate epithelium promotes PC development and invasion closely recapitulating the stages of human prostate cancer progression [31, 32]. Therefore, these mouse models have been useful to investigate some of the cellular and molecular mechanisms contributing to PC progression initiated by loss of PTEN function. With this in mind, we set to investigate the consequences of PTEN loss on expression of transmembrane MT-MMPs, with focus on MT1-MMP, in prostate tissues and prostate epithelial cell lines derived from wild type mice and mice with prostate-specific deletion (heterozygous or homozygous) of PTEN [33]. The data presented here show that loss of PTEN in mouse PC cells is associated with a differential profile of secreted and membrane-tethered MMPs, in particular gelatinases and MT-MMPs (MT1-, MT2-, and MT3-MMP). The data also show that activation of the PI3K/AKT pathway upon loss of PTEN in mouse prostate epithelial cells is associated with increased expression of MT1-MMP at the cell surface and with differential O-glycosylation and reduced turnover of the protease.
2. Materials and Methods
2.1. Reagents and antibodies
LY 294,002, PD 98,059, rapamycin, cycloheximide, mitomycin C, benzyl 2-acetamido-2-deoxy-α-D-galactopyranoside (BGN), and concanavalin A (ConA) were obtained from Sigma (St. Louis, Missouri). AKT inhibitor IV was obtained from Calbiochem (San Diego, CA). All other chemicals were of highest available grade and were purchased from standard sources. A mouse monoclonal antibody (mAb) against the catalytic domain of MT1-MMP (3G4), which cross reacts with mouse MT1-MMP, was a kind gift from Dr. Alex Strongin (Burnham Research Institute, La Jolla, CA). The same antibody, referred to as 3G4, was also purchased from Millipore (Billerica, MA, Cat.# MAB1767). Rabbit polyclonal antibody (pAb) against MT1-MMP cytosolic domain (Cat.# ab28209) was obtained from Abcam (Cambridge, MA). A pAb against MT3-MMP (pAb318) was raised against a synthetic peptide derived from the hinge region of MT3-MMP [34] and cross-reacts with both human and mouse MT3-MMP (unpublished data). Mouse IgG, TrueBlot™ ULTRA, was purchased from eBioscience (Cat.# 18-8817-38, San Diego, CA). The mAb against β-actin was obtained from Sigma (St. Louis, Missouri). Antibodies to pAkt T308 (Cat. # 9275S), pAkt S473 (Cat. # 9271S), total Akt (Cat.# 9272), phosphorylated p70 S6 kinase T389 (Cat. # 9205), and total p70 S6 kinase (Cat. # 9202) were all btained from Cell Signaling (Danvers, CA). Rat mAb against mouse transferrin receptor (Cat.# Sc-59112) and goat anti-Rat secondary antibody (Cat.# Sc-2006) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA).
2.2. Cell lines
Cells were isolated from mice harboring a prostate-specific deletion of floxed PTEN in prostate epithelium that was achieved by expressing Cre recombinase under the control of a probasin promoter (PB). Mouse genotypes are PtenloxP/lox;PPB-cre4–/– (wild-type), PtenloxP/+;PB-cre4T/–(heterozygous), and PtenloxP/lox;PPB-cre4T/-(homozygous) and referred to as PTEN+/+, PTEN+/-, and PTEN-/-, respectively, as described [33]. PTEN-/- and PTEN+/- mouse prostate epithelial cells were isolated from anterior prostates of 8-10 week-old corresponding mice [33] using the method previously described [35]. Ptenlox/lox cells were isolated from anterior prostates of 8 week-old mice, and are referred here to as PTEN+/+ cells. Cells were clonally selected using serial dilution method [36] and PTEN status was confirmed by genotyping and Western blotting. Three distinct clones were isolated for each cell type. Detailed characterization of these mouse cell lines is the focus of another manuscript (Chen Y-Q, et al, under preparation). The mouse cell lines were cultured in advanced Dulbecco's modified Eagle's medium (DMEM) (Invitrogen, Carlsbad, MA) supplemented with 5% fetal bovine serum (FBS) (Invitrogen), L-glutamine (Invitrogen), and penicillin/streptomycin (Invitrogen). HT1080 cells were obtained from the American Type Culture Collection (ATCC, Rockville, MD) and were cultured in DMEM supplemented with 10% FBS and antibiotics.
2.3. Semi-quantitative RT-PCR
Total RNA was extracted with RNeasy® mini kit (Qiagen, Valencia, CA), according to the manufacturer instructions. RT-PCR was performed with 2 μg of each total RNA sample, collected from cells growing in complete media, using SuperScript™ III reverse transcriptase (Invitrogen) to synthesize cDNA and subsequently Taq DNA polymerase (Invitrogen) was used to perform amplification of DNA. The sequences of the primers (IDT, Coralville, IA) for mouse PTEN, MMPs and TIMPs are provided in Supplemental Table 1. The housekeeping GAPDH mRNA was used as an internal control. The amplified fragments were resolved by 2% agarose gels and detected by ethidium bromide staining.
2.4. Real-time PCR
RNA was collected from cells growing in complete media. The sequence of the primers specific for mouse MT1-, MT2-, and MT3-MMP and GAPDH are described in Supplemental Table 1. Real-time PCR was performed on 50 ng of cDNA using the ABsolute Blue QPCR SYBR Green master mix kit according to the manufacturer's protocol (Thermo Scientific, Rockford, IL). PCR conditions included polymerase activation at 95 °C for 10 min followed by 40 cycles at 95 °C for 30 s, 55 °C for 30s, and 72 °C for 30 s. Each assay included a negative control (DEPC-treated water, Ambion), a no-template control. The threshold cycle (Ct) of each sample was recorded as a quantitative measure of the amount of PCR product in the sample using a Stratagene (La Jolla, CA) MX 3000P™ PCR machine. The MT1-, MT2-, and MT3-MMP signals were normalized against the relative quantity of human GAPDH and the relative amount of MT-MMPs' mRNA was expressed as the percentage of GAPDH. Data processing and statistical analyses were performed using Microsoft Excel (Microsoft Corp., Redmond, WA).
2.5. Immunoblot analyses
Cultured cells were solubilized with ice-cold lysis buffer (25 mM Tris-HCl [pH 8.0], 100 mM NaCl, 1% IGEPAL CA-630, 5 mM EDTA) supplemented with protease inhibitor cocktail without EDTA (Complete, Mini, EDTA-free, Roche, Indianapolis, IN). For immunoblot analyses of phosphorylated proteins, the cells were lysed with ice-cold RIPA buffer (0.05 M Tris-HCl, pH 7.4, 0.15 M NaCl, 0.25% deoxycholic acid, 1% NP-40, 1 mM EDTA) supplemented with 1 mM phenylmethylsulfonyl fluoride (PMSF), 2 mM sodium metavanadate (NaVO3), 1 mM sodium floride (NaF), and protease inhibitor cocktail. The protein concentration in the lysates was determined by the BCA procedure (Thermo Scientific). An aliquot of each lysate was mixed with Laemmli 4× SDS-sample buffer with 1% β-mercaptoethanol, boiled (95 °C, 5 min), and resolved by 10% or 7.5% SDS-PAGE (depending on the experiment) followed by immunoblot analyses using various antibodies. Tissue lysates obtained from prostates isolated from wild type and PTEN knock out mice were provided by Dr. Yong Chen (Wake Forest University), and prepared as previously described [33]. A total of 35 μg of tumor lysates and 60 μg of normal lysates were used for detection of MT1-MMP by immunoblotting. Trial experiments showed that a higher amount of protein was required to clearly detect MT1-MMP in normal prostate lysates and therefore accurately determine its molecular mass relative to MT1-MMP expressed in prostate tumor lysates. Briefly, normal and tumor prostate lysates were incubated (4 °C, overnight) with 30 μl of Protein G agarose beads (Thermo Scientific) to remove endogenous mouse IgG. After a brief centrifugation (10,000 g, 3 min) at 4 °C, the supernatants were collected, mixed with Laemmli SDS-sample buffer, and resolved by 8% SDS-PAGE under reducing conditions. The protein was then subjected to immunoblot analyses using the mAb MAB1767 (3G4), which was used at 1:1000 dilution. The blots were then incubated (room temperature, 90 min) with a 1:2000 dilution of the TrueBlot™ ULTRA antibody against mouse IgG (eBioscience) followed by ECL detection of the antigen.
2.6. Cell surface biotinylation
Cells in 6-well plates were treated with the cell impermeable EZ-link-sulfo-NHS-biotin (Thermo Scientific), as described previously [37]. The cells were then lysed with 0.2 ml/well of cold lysis buffer on ice. After a brief centrifugation (15 min, 16,000 g), equal protein amounts of the supernatants were incubated with immobilized neutravidin protein beads (Thermo Scientific) at 4 °C, overnight. The beads were washed five times with harvest buffer (0.5% SDS, 60 mM Tris/HCl, pH 7.5, 2 mM EDTA) supplemented with 2.5% Triton X-100 (final concentration), and the biotinylated proteins were eluted with Laemmli 2× SDS-sample buffer, boiled, and resolved by reducing 10% SDS-PAGE followed by transfer to a nitrocellulose membrane. The proteins were detected with anti-MT1-MMP or anti-transferrin receptor antibodies.
2.7. Immunoprecipitation
Cells were solubilized with cold lysis buffer as described above. After determination of protein concentration, 0.5 mg of protein from each lysates were incubated (4 °C, overnight) with either 5 μg of pAb ab28209 against the cytosolic domain of MT1-MMP or non-immune rabbit IgG in lysis buffer. The mixtures were then incubated (4 °C, 4 h) with Protein A conjugated agarose beads (Thermo Scientific). The bound proteins were eluted with Laemmli SDS-sample buffer, boiled, and resolved by reducing 7.5% SDS-PAGE followed by immunoblot analysis using mAb 3G4 to MT1-MMP.
2.8. MT1-MMP deglycosylation and turnover
For assessment of MT1-MMP O-glycosylation, the cells were incubated (48 h, 37 °C) with 2 mM of benzyl 2-acetamido-2-deoxy-α-D-galactopyranoside (BGN) in serum-free media followed by lysis with ice-cold lysis buffer. The lysates were then subjected to immunoblot analyses as described above. For evaluation of MT1-MMP turnover, the cells were incubated with 50 μg/ml of cycloheximide (CHX) in complete media and at each designated time point (0-8 h) the cells were lysed with ice-cold lysis buffer. The lysates were then resolved by reducing 10% SDS-PAGE followed by immunoblot analyses. Densitometric analyses of the bands in the blots were performed with ImageJ Software (Version: 1.42q, by Wayne Rasband, NIH, Bethesda, MD), and represented in graphs as percentage of control: 100%, time zero.
2.9. Pro-MMP-2 activation
PTEN+/+, PTEN-/- and HT1080 cells were treated with 10 μg/ml of ConA overnight in serum-free media. The cells were then lyses with ice-cold lysis buffer without EDTA and the lysates were clarified by centrifugation and mixed with Laemmli sample buffer without reducing agents and heating. Pro-MMP-2 activation was monitored by gelatin zymography, as described previously [38].
2.10. Cell migration and invasion assays
Migration was assessed using a scratch wound assay. Briefly, confluent cultures of PTEN+/+ and PTEN-/- cells on 6-well plates were gently scratched with a sterile yellow pipette tip and incubated in complete media supplemented with 5 μg/ml mitomycin C to inhibit cell proliferation. After 8 h incubation at 37 °C, the cultures were photographed. Invasion assays using collagen type I were performed essentially as described [39]. Briefly, a solution of 2.7 mg/ml (final concentration) of purified rat type I collagen (BD Bioscience, San Jose, CA) in 0.02 N acetic acid was prepared as described by the manufacturer. The collagen solution (1 ml/well) was then poured into the inserts of Transwell®-6 well plate (Corning) and the plates were incubated (1h, 37 °C) to allow formation of a gel. Cells (1.5 × 105 cells/well) were seeded in complete medium on top of the collagen layer. The plates were placed in a tissue culture incubator at 37 °C and incubated for 14 days with media changes every other day. Some wells received 5 μM BB94 at the time of cell seeding, which was freshly added every other day. At the end of the incubation period, the media were removed and the gels were fixed with 4 % paraformaldehyde and freezed in Tissue-Tek® O.C.T™ Compound (Sakura Finetek USA Inc., Torrance, CA) at -80°C. Five μM-thick frozen sections were obtained from each gel and toluidine blue staining was performed for microscopic evaluation.
2.11. Treatment with pharmacological inhibitors
Cells at 80% confluence were treated with various doses of either LY 294,002, PD 98,059, Akt Inhibitor IV or rapamycin. Briefly, cells were plated in Advanced DMEM supplemented with 5% FBS in either 6 well plates or 60 mm dishes, and allowed to adhere for 18 h. The cells were then washed with warm PBS and then incubated in serum-free Advanced DMEM media supplemented with either LY 294,002 (0, 25, or 50 μM final concentration), PD 98,059 (0, 25, or 50 μM final concentration), Akt Inhibitor IV (0, 1, or 5 μM final concentration) for 18 h or rapamycin (0, 5, or 10 nM final concentration) for 48 h. In other experiment, serum starved PTEN-/- cells in 6-well plates were untreated or treated with either LY 294,002 (25 μM) or PD 98,059 (25 μM) for 18 h in serum free media. Rapamycin (10 nM) was then added to some wells containing cells that were treated or not with LY 294,002 or PD 98,059 for an additional 18 h incubation. At the end of the treatments, the cells were lysed for immunoblot analyses, as described above. Lysates were examined for MT1-MMP, pAkt (T308), total Akt, phospho p70 S6 kinase (T389), total p70 S6 kinase, and β-actin using specific antibodies. Cells in duplicate wells, for each treatment condition, were extracted for total RNA isolation, as described previously.
3. Results
3.1. PTEN loss in mouse prostate epithelial cells is associated with differential expression profile of MT-MMPs and enhanced gelatinase expression
To investigate the relationship between PTEN loss and MT-MMP expression in prostate cancer, we utilized immortalized prostate epithelial cells derived from PTEN+/+, PTEN+/-, and PTEN-/- mice bearing a prostate specific PTEN deletion in the epithelium. As shown in Supplemental Figure 1, semi-quantitative PCR analyses showed a differential profile of MT-MMPs in these cells. While the three cell lines express relatively similar levels of MT1-MMP, loss of PTEN was associated with downregulation of MT2-MMP and upregulation of MT3-MMP mRNA, under the experimental conditions. No expression of MT5-MMP or the GPI-anchored MT-MMPs (MT4- and MT6-MMP) was detected in either of the cell lines (data not shown). Biallelic loss of PTEN is also associated with upregulation of gelatinases, MMP-2 and MMP-9. mRNAs for the secreted mouse collagenases, MMP1a, MMP-1b, and MMP-13, were not detected in any of these cell lines, under these conditions (Supp. Fig.1). Analyses of TIMP mRNAs showed that both TIMP-1 and TIMP-3 were upregulated in PTEN+/- and PTEN-/- cells when compared to PTEN+/+ cells. In contrast, no significant changes were observed in TIMP-2 mRNA. TIMP-4 mRNA was not detected in any of the cell lines (Supp. Fig.1). Since MT1-, MT2-, and MT3-MMP have been shown to represent a triad of MMPs that drive the invasive phenotype of cancer cells [12], the levels of mRNA expression of these MT-MMPs in PTEN+/+ and PTEN-/- cells were further quantified by real-time PCR (Fig. 1A). These results were in general agreement with the semi-quantitative PCR (Supp. Fig.1) and showed that mRNA levels of MT1-MMP did not differ significantly in PTEN+/+ and PTEN-/- cells. In contrast, loss of PTEN correlated with enhanced expression of MT3-MMP mRNA and downregulation of MT2-MMP mRNA. Next, we examined the protein levels of MT1-MMP by immunoblotting in the three cell lines (Fig. 1B). Low levels of MT1-MMP were detected in both PTEN+/+ and PTEN+/- cells. In contrast, PTEN-/- cells exhibited significantly higher amounts of MT1-MMP protein. Levels of MT2-MMP protein could not be determined due to the unreliability of the commercially available MT2-MMP antibodies when using mouse cells. MT3-MMP protein was consistently undetectable in the PTEN-/- cells in spite of upregulation of mRNA and availability of antibodies readily recognizing recombinant mouse MT3-MMP (data not shown). It should be mentioned, that the lack of detection of MT3-MMP protein is not unique to the mouse PTEN cellular system as we have also found a similar lack of correlation between MT3-MMP mRNA and protein expression results in multiple human and mouse cell lines (manuscript in preparation). Taken together these data demonstrate that mouse prostate epithelial cells derived from PTEN null prostate tumors exhibit a differential profile of MMPs and TIMPs with MT1-MMP being the major MT-MMP expressed upon loss of PTEN. Furthermore, these data show that MT1-MMP enhanced expression in PTEN-/- cells is due to posttranscriptional regulation.
Fig 1.
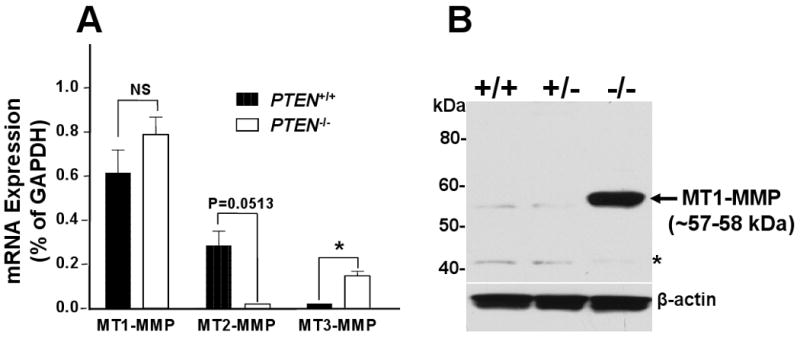
Differential expression of MT-MMP mRNA and MT1-MMP protein in mouse prostate epithelial cells with different PTEN status. (A) Total RNA from PTEN+/+ (black bar) and PTEN-/- (white bar) cells cultured in complete media were used for cDNA synthesis and subsequently real time PCR to quantify MT1-, MT2-, and MT3-MMP mRNA. Ct value of each gene was normalized by the GAPDH Ct value. Results shown are representative of two independent experiments. *, p <0.05; Student's t test. NS, not statistically significant. (B) Cell lysates from PTEN+/+, PTEN+/-, and PTEN-/- cells were resolved by reducing 10% SDS-PAGE followed by immunoblotting analysis using mAb 3G4 against the catalytic domain of MT1-MMP (Mr ∼57-59 kDa). β-actin was used as a loading control. Asterisk shows non-specific bands.
3.2. Enhanced MT1-MMP expression on the surface of PTEN-/- cells and in extracts of PTEN null prostate tumors
Active MT1-MMP is displayed at the cell surface where it accomplishes its proteolytic tasks. Therefore, we compared the levels of MT1-MMP in PTEN+/+, PTEN+/-, and PTEN-/- cells by surface biotinylation. As shown in Figure 2A, a major form of biotinylated MT1-MMP of ∼57-58 kDa was detected in all three cell lines. However, PTEN-/- PC cells displayed significantly higher levels of surface MT1-MMP (Fig. 2A, lane 5). No signal was observed in lysates of non-biotinylated cells (Fig. 2A, lanes 2, 4, and 6) demonstrating the specificity of the detection. We also noticed a slight difference in MT1-MMP electrophoretic migration between MT1-MMP forms expressed by these cell lines with MT1-MMP in PTEN-/- cells showing a slightly higher molecular weight. The mobility shift was clearly evident when lysates of PTEN+/+ and PTEN-/- cells (Fig. 2B, lane 1 and 2, respectively) were electrophoresed side by side in 7.5% SDS-polyacrylamide gels. The differences in MT1-MMP molecular weight were also detected after immunoprecipitation of lysates with a pAb to the cytosolic tail of MT1-MMP and detection with a mAb to the catalytic domain (Fig. 2C).
Fig. 2.
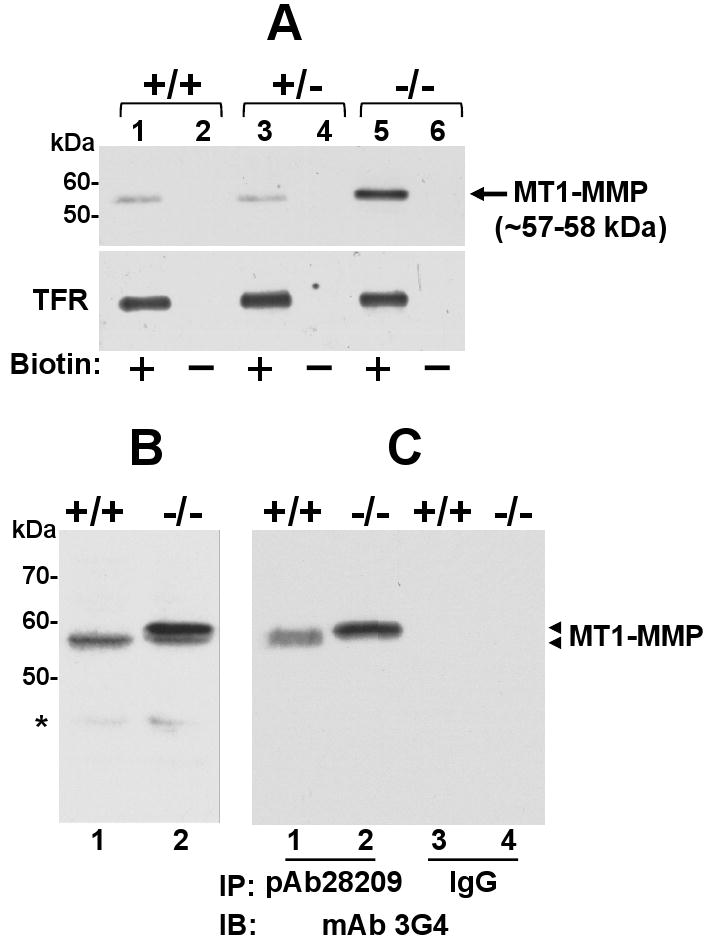
Elevated surface expression of MT1-MMP in PTEN-/- cells. (A) PTEN+/+ (lanes 1 and 2), PTEN+/- (lane 3 and 4), and PTEN-/- cells (lane 5 and 6) were seeded on 6-well plates and incubated with (+) or without (-) cell-impermeable biotin for 30 min on ice. Cell lysates from each cell were then incubated with avidin-conjugated beads and biotinylated proteins were resolved by reducing 10% SDS-PAGE followed by immunoblot analysis using mAb 3G4. The same blot was reprobed with an antibody against transferrin receptor (TFR), as a control. (B) Lysates from PTEN+/+ and PTEN-/- cells were resolved by reducing 7.5% SDS-PAGE followed immunoblotting analysis using mAb 3G4. (C) Lysates from PTEN+/+ and PTEN-/- cells were immunoprecipitated with pAb28209 to the cytosolic tail of MT1-MMP or non-immune rabbit IgG and protein-A conjugated agarose beads. The immunoprecipitates were resolved by reducing 7.5% SDS-PAGE followed immunoblotting analysis using mAb 3G4. Arrowheads on the right in panes B and C indicate the different species of MT1-MMP detected. Asterisk shows non-specific bands.
Next, we examined the levels of expression of MT1-MMP in normal prostates from wild type mice and prostate adenocarcinomas from PTEN null mice. Immunohistochemistry was not reliable due to the limitations of the anti-mouse MT1-MMP antibodies; therefore we performed immunoblot analyses of tissue extracts. This approach also allowed for assessing possible differences in the molecular mass of MT1-MMP in normal and cancerous mouse prostates. The results depicted in Figure 3 show that prostate tumors from PTEN null mice contain significantly higher levels of MT1-MMP than extracts from normal prostates. Furthermore, as observed with the isolated PTEN-/- cells, tumor-associated MT1-MMP exhibits a slight increase in molecular mass, which resembles that observed in MT1-MMP expressed by PTEN-/- PC cells when resolved under the same conditions (Fig. 3, lane 2). Therefore the tumor results validated the finding with the isolated prostate cells.
Fig. 3.
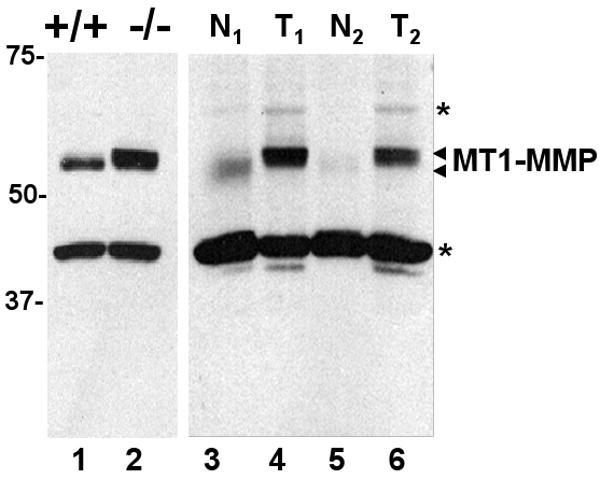
Enhanced expression of MT1-MMP in lysates of wild type and PTEN-/- mouse prostate tissues. Mouse PTEN-/- prostate tumor (T1 and T2, lanes 4 and 6) and normal (N1 and N2, lanes 3 and 5) prostate lysates from different mice and lysates from cultured PTEN+/+ (lane 1) and PTEN-/- (lane 2) cells were resolved by reducing 8% SDS-PAGE followed by immunoblot analyses using mAb 3G4 to the catalytic domain. The antigen was detected using the TrueBlot™ ULTRA antibody against mouse IgG followed by chemiluminescence. Asterisks show non-specific bands.
3.3. Differential MT1-MMP O-glycosylation and turnover in PTEN+/+ and PTEN-/- cells
Several possibilities may explain small differences in mobility shift between MT1-MMP forms within the range of 57-60 kDa. These include latent (60 kDa) vs. active species (57 kDa), O-glycosylation and/or phosphorylation [3, 7-9, 40]. Because the available antibodies to mouse MT1-MMP are directed to the catalytic domain (mAb 3G4) or the cytosolic tail (pAb ab28209), immunoblot analyses were not suitable to discriminate between latent and active MT1-MMP forms. However, it is unlikely that the larger MT1-MMP form displayed by PTEN-/- cells represents the latent species (∼60 kDa) because trafficking to the cell surface has been shown to be coupled with activation of the pro-form in the Trans Golgi network [41]. Previous studies showed that MT1-MMP is O-glycosylated at four residues (Thr291, Thr299, Thr300, and Ser301, human notation) located within the hinge region [8], which are conserved in mouse MT1-MMP (data not shown). Therefore, we hypothesized that PTEN+/+, PTEN+/-, and PTEN-/- cells express different MT1-MMP glycoforms. To test this hypothesis, the cell lines were treated with benzyl 2-acetamido-2-deoxy-α-D-galactopyranoside (BGN). BGN is a competitive inhibitor of galactosyltransferases by preventing addition of galactose to N-acetyl-galactosamine moieties O-linked to serine and threonine residues, thus blocking elongation of O-glycosyl chains. After BGN treatment, the cell lysates were subjected to immunoblot analyses. As shown in Figure 4, MT1-MMP displayed by untreated PTEN-/- cells (Fig. 4, lane 3) exhibits a higher molecular weight when compared to PTEN+/+ and PTEN+/- cells (Fig. 4, lanes 1 and 2, respectively). BGN treatment resulted in a mobility shift in MT1-MMP in all the cell lines. However, the decrease in MT1-MMP molecular mass was more pronounced in BGN-treated PTEN-/- cells (Fig. 4, lane 3 vs. lane 6). These data indicate that the high molecular mass of MT1-MMP displayed by PTEN-/- cells is due to differential O-glycosylation.
Fig. 4.
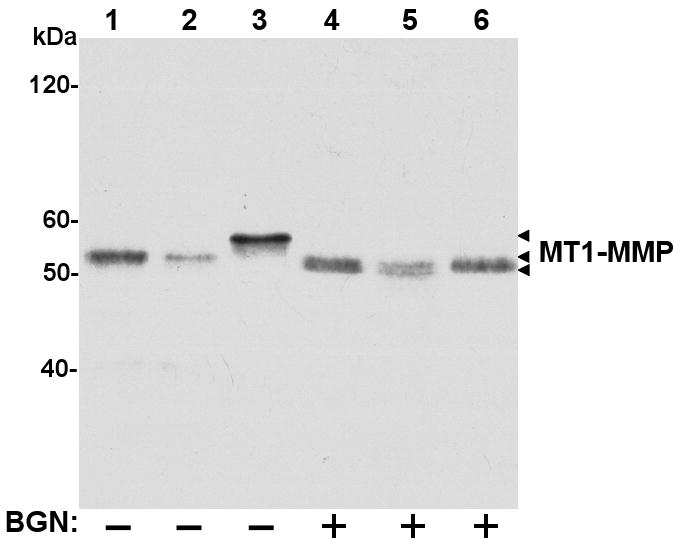
MT1-MMP is differentially O-glycosylated in PTEN+/+, PTEN+/- and PTEN-/- cells. PTEN+/+ (lanes 1 and 4), PTEN+/- (lanes 2 and 5), and PTEN-/- cells (lanes 3 and 6) were incubated with (+) or without (-) the O-glycosylation inhibitor BGN (2 mM) for 48 h. The cells were then lysed, and the lysates were resolved by reducing 7.5% SDS-PAGE followed immunoblot analysis using the mAb 3G4. Arrowheads on the right indicate the different species of MT1-MMP detected.
O-glycosylation of MT1-MMP's hinge region was postulated to protect the active protease from autocatalytic cleavage within the hinge region [7]. Consistently, reduced O-glycosylation was associated with enhanced degradation of the active MT1-MMP [7]. This observation suggested the possibility that the high levels of MT1-MMP detected in PTEN-/- cells were due in part to reduced autocatalytic processing of the active protease and consequently accumulation of enzyme at the cell surface. To test this hypothesis, we measured the turnover of MT1-MMP in PTEN+/+ and PTEN-/- cells. To this end, cells were treated with cycloheximide (CHX) to stop de novo protein synthesis. At each time point after initiation of CHX treatment, the cells were lysed, and the lysates were resolved by SDS-PAGE followed by immunoblot analyses (Fig. 5A and C). The intensity of the MT1-MMP band at each time was then quantified by densitometry (Fig. 5B and D). These studies showed that MT1-MMP protein levels decreased as a function of time in the presence of CHX in both cell lines. However, MT1-MMP expressed by PTEN-/- cells displayed a slower rate of turnover when compared to MT1-MMP produced by PTEN+/+ cells, under the same experimental conditions in two independent experiments. The rate of MT1-MMP turnover at the cell surface is dictated in part by a balance between autocatalytic processing and internalization [3]. However, the contribution of each of these processes in regulating the levels of MT1-MMP in PTEN-/- cells could not be determined. First, detection of the autocatalytic degradation fragment of MT1-MMP was not possible because the anti-catalytic domain antibodies recognizing mouse MT1-MMP do not identify the 44-kDa processed form, which lacks the catalytic domain, while the anti-cytosolic domain antibodies yielded non-specific bands overlapping the 44-kDa region and thus were unreliable. Second, endocytosis studies of MT1-MMP in PTEN+/+ and PTEN-/- cells using a reversible biotinylation assay were inconclusive due to the low levels of internalized protease in particular in PTEN+/+ cells, which display very low levels of surface MT1-MMP. Regardless, the data shown here suggest that the enhanced surface expression of MT1-MMP in mouse PTEN-/- prostate cancer cells is due in part to reduced protease turnover.
Fig. 5.
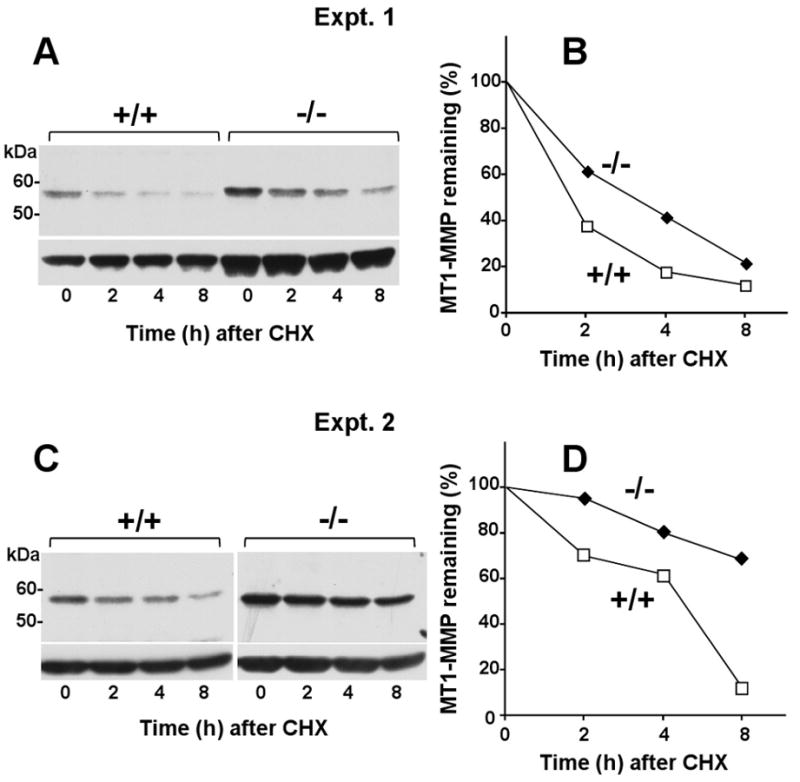
Reduced turnover of MT1-MMP in PTEN-/- cells. (A-D) PTEN+/+ and PTEN-/- cells were incubated with 50 μg/ml of cycloheximide (CHX) for 8 h. At each time point (0, 2, 4, and 8 h), the cells were lysed and the lysates were resolved by reducing 10% SDS-PAGE followed by immunoblotting analysis using mAb 3G4. A and B show blots derived from two independent experiments. C and D show the results of the densitometry analyses, as described in Materials and Methods. Values represent the relative amount of MT1-MMP at each time point relative to time zero (100%).
3.4. Functional analyses of MT1-MMP-dependent activity
MT1-MMP is a major activator of pro-MMP-2 (pro-gelatinase A) [42]. We therefore examined the expression and activation of pro-MMP-2 in PTEN+/+ and PTEN-/- cells by gelatin zymography. As shown in the zymogram of Figure 6A, PTEN+/+ cells display no detectable levels of gelatinases, in agreement with the PCR data. In contrast, PTEN-/- cells express high levels of pro-MMP-2 and lower levels of pro-MMP-9, under unstimulated conditions. As MT1-MMP-dependent pro-MMP-2 activation usually requires stimulation with the lectin ConA [43], the cells were treated with ConA and pro-MMP-2 activation was monitored by gelatin zymography of cell lysates. As a reference, we used fibrosarcoma HT1080 cells known to efficiently activate pro-MMP-2 in response to ConA [43]. These studies showed that ConA-treated PTEN-/- cells displayed a modest but readily detectable processing of pro-MMP-2 to its active form of ∼62 kDa in (Fig. 6A). ConA treatment also induced expression but not activation of pro-MMP-9 in PTEN-/- cells by an unknown mechanism. A limited activation of pro-MMP-2 in response to ConA was also reported in PC3 cells even after ectopic expression of recombinant MT1-MMP or pro-MMP-2 [18, 44] suggesting that levels of expression are not the only factor regulating MT1-MMP activity. It is also possible that MT1-MMP O-glycosylation may affect the recruitment of TIMP-2 to the cell surface and consequently compromise pro-MMP-2 activation [8].
Fig. 6.
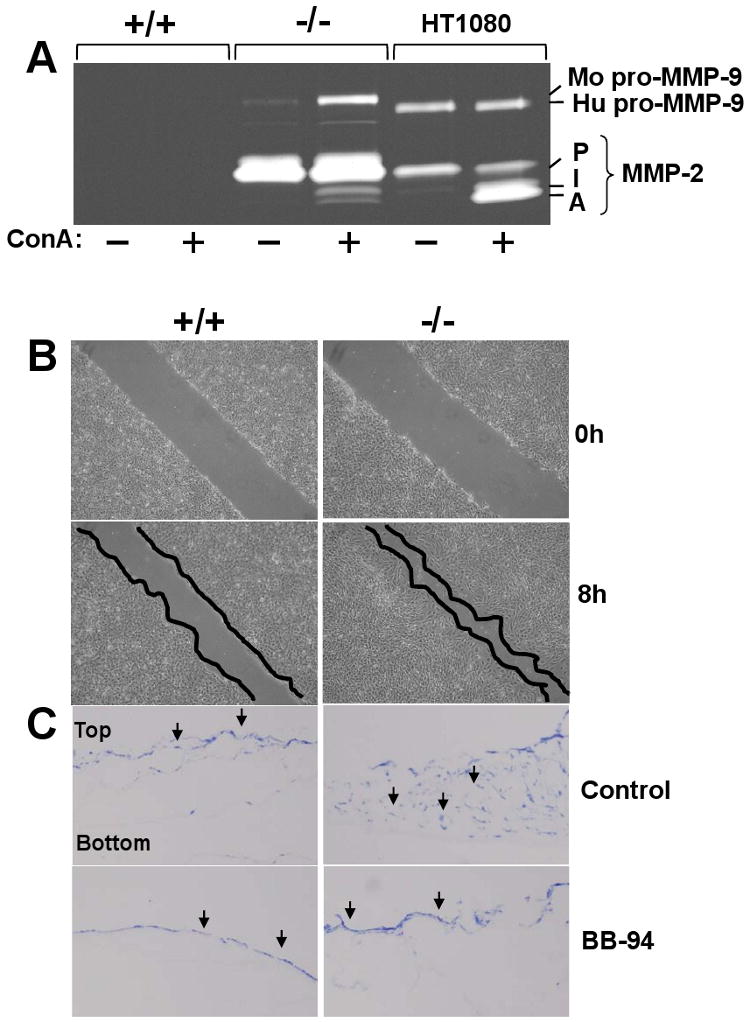
Pro-MMP-2 activation, migration and invasiveness of PTEN-/- cells. (A) Cultures of PTEN+/+, PTEN-/-, and HT1080 cells in 6-well plates were untreated (-) or treated (+) with ConA and the lysates were subjected to gelatin zymography. P, pro-MMP-2 (∼72 kDa), I, intermediate form of MMP-2 (∼66 kDa), A, active MMP-2 (∼62 kDa). Mo, mouse, Hu, human. (B) Confluent cultures of PTEN+/+ and PTEN-/- cells in 6-well plates were scratched, and incubated in complete media supplemented with 5 μg/ml mitomycin C for 8 h. The cell monolayers were then photographed. (C) PTEN+/+ and PTEN-/- cells were seeded on top of a type I collagen gel in the presence or absence of BB-94. After 14 days, 5 μm-thick frozen sections were stained with toluidine blue to identify the cells, indicated as black arrows. The experiments presented in panels A, B, and C were repeated at least three times with similar results.
MT1-MMP expression and activity has been linked to enhanced tumor cell migration and invasion [6, 13]. Therefore, we compared the migratory and invasive activity of PTEN+/+ and PTEN-/- cells using a scratch wound assay for cell migration and collagen I gels for cell invasion. Migration assays showed that PTEN-/- cells were able to close the gap in the scratched monolayer more efficiently than PTEN+/+ cells after 8 h (Fig. 6B). Invasive activity was tested by culturing PTEN+/+ and PTEN-/- cells atop gels of collagen I, as described [13]. As shown in Figure 6C, PTEN-/- cells readily invaded the collagen I gel when compared to PTEN+/+ cells, under the same conditions. This process was blocked by the broad-spectrum MMP inhibitor, BB-94 (Fig. 6C) indicating an MMP-dependent process. While BB94 inhibits all members of the MMP family, including MT1-MMP, we interpret the collagen-invasive of PTEN-/- cells most likely as a result of MT1-MMP activity based on previous observations indicating a dependence on MT1-MMP for invasion of collagen I [13, 45]. Moreover, these cells do not express soluble collagenases, MT3-MMP or MT2-MMP (Suppl. Fig. 1), the latter also implicated in collagen invasion (47).
3.5. The PI3K/AKT pathway regulates MT1-MMP expression in PTEN-/- cells
Loss of PTEN results in deregulation of the PI3K/AKT and MAPK pathways and their downstream effectors. To determine the role of these pathways in MT-MMP (MT1-, MT2-, and MT3-MMP) expression, PTEN+/+ and PTEN-/- cells were treated with established pharmacological inhibitors of PI3K, MAPK, AKT and mTOR. We found that neither LY 294,002, PD 98,059, nor rapamycin treatment had a significant effect on MT-MMP mRNA expression levels in both PTEN+/+ and PTEN-/- cells, as determined by semi-quantitative PCR (Suppl. Fig. 2). We then examined the effects of these inhibitors on MT1-MMP protein expression by immunoblot analyses (Fig. 7). These studies showed that treatment with the MAPK inhibitor PD98,059 had no significant effect on MT1-MMP protein levels in both PTEN+/+ and PTEN-/- cells (Fig. 7A). In contrast, inhibition of PI3K activity with LY294,002 caused a dose-dependent reduction of MT1-MMP protein level in PTEN-/- cells while in PTEN+/+ cells reduction of MT1-MMP was only evident at an inhibitor concentration of 50 μM (Fig. 7B). Treatment with AKT inhibitor IV, a cell-permeable benzimidazole compound that inhibits activation of all AKT isoforms, also reduced MT1-MMP protein levels in both cell lines at doses of 5 μM (Fig. 7C). As expected, this inhibitor caused a marked decrease in pAKT (Fig. 7C). One of the downstream targets of activated AKTs is mTOR, a phosphoinositide-3-kinase-related kinase that is a component of the mTOR complex 1 (mTORC1) and 2 (mTORC2) [46]. Once activated, mTORC1 promotes protein synthesis and consequently cell growth and proliferation via phosphorylation of the translational regulators 4E-BP and 40S ribosomal protein S6 kinases [47]. To examine the role of mTORC1 in regulation of MT1-MMP expression, PTEN+/+ and PTEN-/- cells were treated with rapamycin, a known pharmacological inhibitor of mTORC1. Immunoblot analyses showed that rapamycin treatment completely inhibited S6K phosphorylation in both cell lines at the doses used here (5 and 10 μM), consistent with mTOR inhibition (Fig. 7D). Surprisingly, rapamycin treatment significantly increased the levels of MT1-MMP protein in PTEN-/- cells while had minimal effect in PTEN+/+ cells (Fig. 7D). As shown in Supplemental Figure 2, rapamycin treatment had no significant effect on MT1-MMP mRNA levels in both cell lines when compared to untreated cells, suggesting a posttranslational effect of rapamycin on MT1-MMP regulation. Previous studies demonstrated that rapamycin treatment induced activation of survival pathways via PI3K activity in cancer cells by a feedback mechanism [48]. Therefore, we examined the effects of rapamycin on MT1-MMP protein levels in the absence or presence of LY 294,002 or PD 98,059 in PTEN-/- cells. These studies showed that administration of rapamycin in the presence of LY 294,002 (Fig. 7E, lane 5), but not PD 98,059 (Fig. 7E, lane 6), inhibited rapamycin-induced expression of MT1-MMP (Fig. 7E, lane 4). Thus, PI3K activity is required for the upregulation of MT1-MMP protein levels caused by rapamycin treatment in PTEN-/- cells. Taken together these results suggest a role for the PI3K/AKT pathway in posttranslational regulation of MT1-MMP in mouse prostate cancer PTEN+/+ cells
Fig. 7.
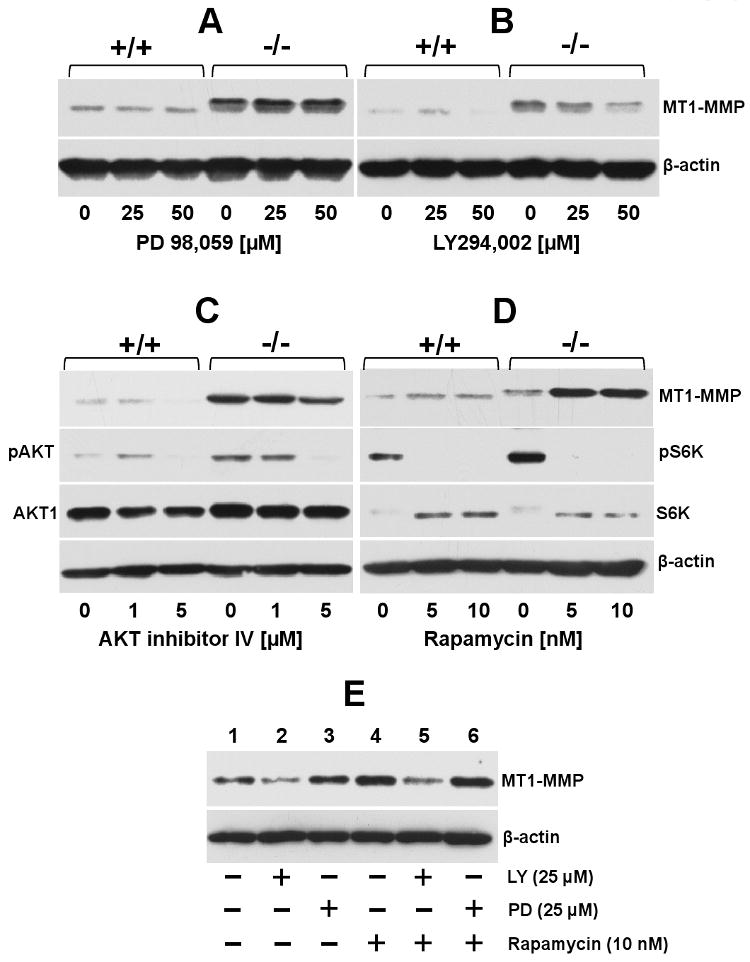
Effect of pharmacological inhibitors to PI3K, AKT, MAPK and mTOR on MT1-MMP expression. Serum starved PTEN+/+ and PTEN-/- cells were incubated (18 h) with (+) or without (-) the indicated doses of either PD 98,059 (A), LY 294,002 (B), Akt inhibitor IV (C) or rapamycin (D) in serum-free media. (E) Serum starved PTEN-/- cells were untreated (-) or treated (+) with either LY 294,002 or PD 98,059 for 18 h. Then, the cultures received rapamycin (+) or vehicle (-) and were incubated for 18 h incubation. At the end of the incubation period, the cells were lysed, and the lysates were resolved by reducing 10% SDS-PAGE followed by immunoblot analyses with the appropriate antibodies. The data presented in panels A-E were repeated at least three times with similar results.
4. Discussion
Conditional biallelic deletion of PTEN in mouse prostate epithelium leads to the development of PC in a process that resembles the spectrum of pathological stages observed in human PC, including development of invasive and metastatic adenocarcinoma. Inactivation of PTEN in these particular mouse models can therefore initiate a cascade of genetic and epigenetic events that results in the expression of a malignant phenotype that ultimately leads to tumor cell dissemination. It should be noted however that distant metastasis in these models is rather limited and the tumors fail to develop skeletal metastasis [49, 50], a characteristic of advanced PC in human patients. Nonetheless, in spite of their inherent limitations, PTEN mouse models of PC provide an opportunity to examine the relationship between PTEN loss and genes known to be associated with tumor cell invasion. In this study, prostate tumors and cell lines derived from mice bearing a prostate-specific deletion of PTEN [33] and from wild type mice were used to investigate the expression of MMPs associated with cancer progression including gelatinases, MT1-MMP and its closely homologues MT2- and MT3-MMP. In particular, the MT-MMPs confer migratory and invasive activity to cancer cells [12, 39], and therefore may play a role in the invasive phenotype of PTEN null prostate tumors. Our data show that isolated PTEN null PC cells exhibit a profile of MMP expression that is consistent with the malignant phenotype of the prostate tumors, namely upregulation of MT1-MMP, MT3-MMP, and gelatinases. In addition, loss of PTEN was associated with enhanced protein expression of MT1-MMP in vivo and in vitro. We also noticed that the molecular mass of MT1-MMP displayed by PTEN null tumors and isolated cells was slightly but consistently higher than that found in wild type prostate tissues and their isolated epithelial cells. Studies with an O-glycosylation inhibitor indicated that the differences in molecular mass were consistent with differential O-glycosylation of MT1-MMP between wild type and PTEN null cells. Although not directly proven here, differential O-glycosylation is also likely to explain the mass differences in MT1-MMP observed in vivo. Nonetheless, to our knowledge, this is the first study to show a differential O-glycosylation of MT1-MMP that is associated with a loss of PTEN function in mouse prostate epithelial cells.
Posttranslational modification of MT1-MMP by O-linked glycosylation occurs at residues present within the hinge region [8], a stretch of 32 residues that link the active site with the hemopexin-like domain. Previous studies have shown that the hinge of MT1-MMP is highly susceptible to proteolysis, a process that has been postulated to be due to its amino acid composition and structural features [51]. Consistently, O-glycosylation of the hinge has been shown to regulate MT1-MMP stability, possibly by restricting its susceptibility to proteolytic attack [8]. Here we found that the differential O-glycosylation of MT1-MMP observed in PTEN-/- and PTEN+/+ cells is associated with a slower turnover of active protease in the cancer cells, consistent with a role for O-linked carbohydrates in controlling MT1-MMP stability, possibly by decreasing the rate of enzyme autolysis [7]. This observation suggests that O-glycosylation contributes to the high levels of MT1-MMP found at the surface of PTEN-/- cells. In turn, higher levels of surface MT1-MMP results in pro-MMP-2 activation and enhanced migratory and invasive activity, three cellular activities known to required MT1-MMP activity [4, 13, 37]. Collectively, these data uncover a novel relationship between the loss of a powerful tumor suppressor gene, PTEN, and a key post-translational modification, O-glycosylation, of a cancer-relevant protease, MT1-MMP, which can partly explain the association between PTEN loss and cancer invasion and metastasis. It is also tempting to speculate that the findings with MT1-MMP may reflect a general alteration in protein glycosylation as a consequence of PTEN inactivation. If so, it will be interesting to investigate whether PTEN inactivation in cancer cells, directly or indirectly, regulates the expression and function of glycosyltransferases, and consequently alter the profile of the cellular glycome [52]. Such potential alterations in glycosylation may have a profound impact in tumor cell behavior by regulating the function of cancer relevant proteins.
It is well established that loss of PTEN results in activation of the PI3K/AKT pathway and downstream effectors leading to cancer cell survival and expression of the malignant phenotype. Here we found that pharmacological inhibition of PI3K activity and Akt activation but not inhibition of MAPK decreased the level of MT1-MMP protein in mouse PTEN-/- prostate cancer cells. These results are consistent with previous studies that suggested a role for PI3K activity in regulation of MT1-MMP expression in both normal and cancer cells including prostate cancer cells [53-58]. Interestingly, mice lacking AKT1 exhibit dwarfism and alterations in skeletal development that resemble the bone phenotype of MT1-MMP null mice [59]. Moreover, delayed secondary ossification in AKT1 null mice has been ascribed to reduced expression of active MT1-MMP [60]. Taken together, these observations place the PI3K/AKT pathway as a master regulator of MT1-MMP expression during bone development. Given the role of AKT in regulation of MT1-MMP expression in cancer cells, AKT activation in PC patients bearing PTEN mutations may contribute to the development of skeletal metastasis through an MT1-MMP-dependent process [14]. Our data suggests that PTEN loss in mouse prostate epithelium is associated with upregulation of MT1-MMP protein expression and surface localization via activation of the PI3K/AKT pathway. It is noteworthy that while MT1-MMP protein levels were sensitive to inhibition of PI3K and AKT, no evident changes were observed in the molecular mass of MT1-MMP after inhibitor treatment of PTEN-/- cells. This finding suggests that alterations in the glycosylation machinery upon loss of PTEN in the mouse system are the result of secondary genetic modifications that cannot be overridden by direct inhibition of PI3K and AKT activity.
The PI3K/AKT pathway indirectly and directly activates mTORC1, which in turn positively regulates the translational machinery and thus promotes protein synthesis [61-63]. This suggests that mTORC1 activation, as a consequence of PTEN loss, may be behind the enhanced levels of MT1-MMP protein displayed by PTEN-/- cells. In this regard, it was interesting to find that rapamycin, a specific mTOR inhibitor, increased rather than decreased MT1-MMP protein in PTEN-/- cells. However, rapamycin had no apparent effect on the molecular mass of MT1-MMP. Unanticipated effects of rapamycin-mediated inhibition of mTOR in cancer cells have been reported [46]. For instance, rapamycin induces activation of multiple survival pathways including the PI3K/AKT pathway in various human cancer cell lines by a feedback activation mechanism [48, 64-68]. In addition, rapamycin was shown to induce the expression of Her2 and insulin growth factor receptor 1 in gastrointestinal cells [69]. In mouse PTEN-/- PC cells, the rebound effect of rapamycin treatment may lead to the activation of alternate pathways that augments MT1-MMP protein [70]. Our data show that PI3K but not MAPK inhibition blocks the rapamycin-induced expression of MT1-MMP in PTEN-/- cells, consistent with the reported effects of rapamycin on the PIK3/AKT pathway [70]. At present, the specific effectors and/or mechanisms mediating the upregulation of MT1-MMP protein levels in rapamycin-treated PTEN-/- cells remain to be elucidated and are under investigation. Although cancers are genetically and molecularly heterogeneous and rapamycin effects are context dependent, our data nonetheless unveils a potential side effect from mTOR inhibition in the context of PTEN inactivation that deserves further exploration, as upregulation of MT1-MMP expression may be counterproductive. Given the critical role that MT1-MMP plays in tumor invasion and the therapeutic potential of targeting mTOR, more studies are warranted to determine the effects of rapamycin or other mTOR inhibitors on the regulation of this key protease in several cancers. The results of these studies may have an impact in the development of therapies targeting complementary pathways including mTOR for more effective clinical responses [71]. In summary, the present studies demonstrate a differential regulation of MMP expression profile in cell lines established from prostates of wild type and PTEN null mice. Specifically, PTEN loss in this mouse model is associated with posttranslational upregulation and differential O-glycosylation of MT1-MMP. This finding unveils a novel effect of PTEN deregulation on the function of a tumor-associated protease that may partly explain the impact of PTEN loss in cancer progression.
Supplementary Material
Abbreviations
- PTEN
phosphatase and tensin homologue deleted on chromosome ten
- MMP
matrix metalloproteinase
- pAb
polyclonal antibody
- mAb
monoclonal antibody
- BGN
benzyl 2-acetamido-2-deoxy-α-D-galactopyranoside
- CHX
cycloheximide
- ConA
concanavalin A
Footnotes
This work was supported by NIH/NCI grant CA61986 to RF, and P01CA106742, R01CA107668 and AICR 07B087 grants to YQC.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Seaho Kim, Department of Pathology, School of Medicine and Karmanos Cancer Institute, Wayne State University, Detroit, MI 48201.
Wei Huang, Department of Pathology, School of Medicine and Karmanos Cancer Institute, Wayne State University, Detroit, MI 48201.
Emilio P. Mottillo, Department of Pathology, School of Medicine and Karmanos Cancer Institute, Wayne State University, Detroit, MI 48201
Anjum Sohail, Department of Pathology, School of Medicine and Karmanos Cancer Institute, Wayne State University, Detroit, MI 48201.
Yoon-Ah Ham, Department of Pathology, School of Medicine and Karmanos Cancer Institute, Wayne State University, Detroit, MI 48201.
M. Katie Conley-LaComb, Department of Pathology, School of Medicine and Karmanos Cancer Institute, Wayne State University, Detroit, MI 48201.
Chong Jai Kim, Department of Pathology, School of Medicine and Karmanos Cancer Institute, Wayne State University, Detroit, MI 48201.
Guri Tzivion, Department of Pathology, School of Medicine and Karmanos Cancer Institute, Wayne State University, Detroit, MI 48201.
Hyeong-Reh Choi Kim, Department of Pathology, School of Medicine and Karmanos Cancer Institute, Wayne State University, Detroit, MI 48201.
Shihua Wang, Department of Cancer Biology, Wake Forest University School of Medicine, Winston-Salem, NC 27157.
Yong Q. Chen, Department of Cancer Biology, Wake Forest University School of Medicine, Winston-Salem, NC 27157
Rafael Fridman, Department of Cancer Biology, Wake Forest University School of Medicine, Winston-Salem, NC 27157.
References
- 1.Massova I, Kotra LP, Fridman R, Mobashery S. Matrix metalloproteinases: structures, evolution, and diversification. FASEB J. 1998;12:1075–1095. [PubMed] [Google Scholar]
- 2.Zucker S, Pei D, Cao J, Lopez-Otin C. Membrane type-matrix metalloproteinases (MT-MMP) Curr Top Dev Biol. 2003;54:1–74. doi: 10.1016/s0070-2153(03)54004-2. [DOI] [PubMed] [Google Scholar]
- 3.Osenkowski P, Toth M, Fridman R. Processing, shedding, and endocytosis of membrane type 1-matrix metalloproteinase (MT1-MMP) J Cell Physiol. 2004;200:2–10. doi: 10.1002/jcp.20064. [DOI] [PubMed] [Google Scholar]
- 4.Itoh Y. MT1-MMP: a key regulator of cell migration in tissue. IUBMB Life. 2006;58:589–596. doi: 10.1080/15216540600962818. [DOI] [PubMed] [Google Scholar]
- 5.Itoh Y, Ito N, Nagase H, Evans RD, Bird SA, Seiki M. Cell surface collagenolysis requires homodimerization of the membrane-bound collagenase MT1-MMP. Mol Biol Cell. 2006;17:5390–5399. doi: 10.1091/mbc.E06-08-0740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Itoh Y, Seiki M. MT1-MMP: a potent modifier of pericellular microenvironment. J Cell Physiol. 2006;206:1–8. doi: 10.1002/jcp.20431. [DOI] [PubMed] [Google Scholar]
- 7.Remacle AG, Chekanov AV, Golubkov VS, Savinov AY, Rozanov DV, Strongin AY. O-glycosylation regulates autolysis of cellular membrane type-1 matrix metalloproteinase (MT1-MMP) J Biol Chem. 2006;281:16897–16905. doi: 10.1074/jbc.M600295200. [DOI] [PubMed] [Google Scholar]
- 8.Wu YI, Munshi HG, Sen R, Snipas SJ, Salvesen GS, Fridman R, Stack MS. Glycosylation broadens the substrate profile of membrane type 1 matrix metalloproteinase. J Biol Chem. 2004;279:8278–8289. doi: 10.1074/jbc.M311870200. [DOI] [PubMed] [Google Scholar]
- 9.Nyalendo C, Michaud M, Beaulieu E, Roghi C, Murphy G, Gingras D, Beliveau R. Src-dependent phosphorylation of membrane type I matrix metalloproteinase on cytoplasmic tyrosine 573: role in endothelial and tumor cell migration. J Biol Chem. 2007;282:15690–15699. doi: 10.1074/jbc.M608045200. [DOI] [PubMed] [Google Scholar]
- 10.Anilkumar N, Uekita T, Couchman JR, Nagase H, Seiki M, Itoh Y. Palmitoylation at Cys574 is essential for MT1-MMP to promote cell migration. FASEB J. 2005;19:1326–1328. doi: 10.1096/fj.04-3651fje. [DOI] [PubMed] [Google Scholar]
- 11.Zhao H, Sohail A, Sun Q, Shi Q, Kim S, Mobashery S, Fridman R. Identification and role of the homodimerization interface of the glycosylphosphatidylinositol-anchored membrane type 6 matrix metalloproteinase (MMP25) J Biol Chem. 2008;283:35023–35032. doi: 10.1074/jbc.M806553200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hotary K, Li XY, Allen E, Stevens SL, Weiss SJ. A cancer cell metalloprotease triad regulates the basement membrane transmigration program. Genes Dev. 2006;20:2673–2686. doi: 10.1101/gad.1451806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hotary KB, Allen ED, Brooks PC, Datta NS, Long MW, Weiss SJ. Membrane type I matrix metalloproteinase usurps tumor growth control imposed by the three-dimensional extracellular matrix. Cell. 2003;114:33–45. doi: 10.1016/s0092-8674(03)00513-0. [DOI] [PubMed] [Google Scholar]
- 14.Bonfil RD, Dong Z, Trindade Filho JC, Sabbota A, Osenkowski P, Nabha S, Yamamoto H, Chinni SR, Zhao H, Mobashery S, Vessella RL, Fridman R, Cher ML. Prostate cancer-associated membrane type 1-matrix metalloproteinase: a pivotal role in bone response and intraosseous tumor growth. Am J Pathol. 2007;170:2100–2111. doi: 10.2353/ajpath.2007.060720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jennbacken K, Gustavsson H, Welen K, Vallbo C, Damber JE. Prostate cancer progression into androgen independency is associated with alterations in cell adhesion and invasivity. Prostate. 2006;66:1631–1640. doi: 10.1002/pros.20469. [DOI] [PubMed] [Google Scholar]
- 16.Udayakumar TS, Chen ML, Bair EL, Von Bredow DC, Cress AE, Nagle RB, Bowden GT. Membrane type-1-matrix metalloproteinase expressed by prostate carcinoma cells cleaves human laminin-5 beta3 chain and induces cell migration. Cancer Res. 2003;63:2292–2299. [PubMed] [Google Scholar]
- 17.Cao J, Chiarelli C, Kozarekar P, Adler HL. Membrane type 1-matrix metalloproteinase promotes human prostate cancer invasion and metastasis. Thromb Haemost. 2005;93:770–778. doi: 10.1160/TH04-08-0555. [DOI] [PubMed] [Google Scholar]
- 18.Wang X, Wilson MJ, Slaton JW, Sinha AA, Ewing SL, Pei D. Increased aggressiveness of human prostate PC-3 tumor cells expressing cell surface localized membrane type-1 matrix metalloproteinase (MT1-MMP) J Androl. 2009;30:259–274. doi: 10.2164/jandrol.108.006494. [DOI] [PubMed] [Google Scholar]
- 19.Cao J, Chiarelli C, Richman O, Zarrabi K, Kozarekar P, Zucker S. Membrane type 1 matrix metalloproteinase induces epithelial-to-mesenchymal transition in prostate cancer. J Biol Chem. 2008;283:6232–6240. doi: 10.1074/jbc.M705759200. [DOI] [PubMed] [Google Scholar]
- 20.Cardillo MR, Di Silverio F, Gentile V. Quantitative immunohistochemical and in situ hybridization analysis of metalloproteinases in prostate cancer. Anticancer Res. 2006;26:973–982. [PubMed] [Google Scholar]
- 21.Trudel D, Fradet Y, Meyer F, Harel F, Tetu B. Membrane-type-1 matrix metalloproteinase, matrix metalloproteinase 2, and tissue inhibitor of matrix proteinase 2 in prostate cancer: identification of patients with poor prognosis by immunohistochemistry. Hum Pathol. 2008;39:731–739. doi: 10.1016/j.humpath.2007.09.021. [DOI] [PubMed] [Google Scholar]
- 22.Upadhyay J, Shekarriz B, Nemeth JA, Dong Z, Cummings GD, Fridman R, Sakr W, Grignon DJ, Cher ML. Membrane type 1-matrix metalloproteinase (MT1-MMP) and MMP-2 immunolocalization in human prostate: change in cellular localization associated with high-grade prostatic intraepithelial neoplasia. Clin Cancer Res. 1999;5:4105–4110. [PubMed] [Google Scholar]
- 23.Dahia PL. PTEN, a unique tumor suppressor gene. Endocr Relat Cancer. 2000;7:115–129. doi: 10.1677/erc.0.0070115. [DOI] [PubMed] [Google Scholar]
- 24.Suzuki H, Freije D, Nusskern DR, Okami K, Cairns P, Sidransky D, Isaacs WB, Bova GS. Interfocal heterogeneity of PTEN/MMAC1 gene alterations in multiple metastatic prostate cancer tissues. Cancer Res. 1998;58:204–209. [PubMed] [Google Scholar]
- 25.Wang SI, Parsons R, Ittmann M. Homozygous deletion of the PTEN tumor suppressor gene in a subset of prostate adenocarcinomas. Clin Cancer Res. 1998;4:811–815. [PubMed] [Google Scholar]
- 26.McCall P, Witton CJ, Grimsley S, Nielsen KV, Edwards J. Is PTEN loss associated with clinical outcome measures in human prostate cancer? Br J Cancer. 2008;99:1296–1301. doi: 10.1038/sj.bjc.6604680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li J, Yen C, Liaw D, Podsypanina K, Bose S, Wang SI, Puc J, Miliaresis C, Rodgers L, McCombie R, Bigner SH, Giovanella BC, Ittmann M, Tycko B, Hibshoosh H, Wigler MH, Parsons R. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science. 1997;275:1943–1947. doi: 10.1126/science.275.5308.1943. [DOI] [PubMed] [Google Scholar]
- 28.Yoshimoto M, Cunha IW, Coudry RA, Fonseca FP, Torres CH, Soares FA, Squire JA. FISH analysis of 107 prostate cancers shows that PTEN genomic deletion is associated with poor clinical outcome. Br J Cancer. 2007;97:678–685. doi: 10.1038/sj.bjc.6603924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McMenamin ME, Soung P, Perera S, Kaplan I, Loda M, Sellers WR. Loss of PTEN expression in paraffin-embedded primary prostate cancer correlates with high Gleason score and advanced stage. Cancer Res. 1999;59:4291–4296. [PubMed] [Google Scholar]
- 30.Han B, Mehra R, Lonigro RJ, Wang L, Suleman K, Menon A, Palanisamy N, Tomlins SA, Chinnaiyan AM, Shah RB. Fluorescence in situ hybridization study shows association of PTEN deletion with ERG rearrangement during prostate cancer progression. Mod Pathol. 2009;22:1083–1093. doi: 10.1038/modpathol.2009.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Trotman LC, Niki M, Dotan ZA, Koutcher JA, Di Cristofano A, Xiao A, Khoo AS, Roy-Burman P, Greenberg NM, Van Dyke T, Cordon-Cardo C, Pandolfi PP. Pten dose dictates cancer progression in the prostate. PLoS Biol. 2003;1:E59. doi: 10.1371/journal.pbio.0000059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang S, Gao J, Lei Q, Rozengurt N, Pritchard C, Jiao J, Thomas GV, Li G, Roy-Burman P, Nelson PS, Liu X, Wu H. Prostate-specific deletion of the murine Pten tumor suppressor gene leads to metastatic prostate cancer. Cancer Cell. 2003;4:209–221. doi: 10.1016/s1535-6108(03)00215-0. [DOI] [PubMed] [Google Scholar]
- 33.Berquin IM, Min Y, Wu R, Wu J, Perry D, Cline JM, Thomas MJ, Thornburg T, Kulik G, Smith A, Edwards IJ, D'Agostino R, Zhang H, Wu H, Kang JX, Chen YQ. Modulation of prostate cancer genetic risk by omega-3 and omega-6 fatty acids. J Clin Invest. 2007;117:1866–1875. doi: 10.1172/JCI31494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhao H, Bernardo MM, Osenkowski P, Sohail A, Pei D, Nagase H, Kashiwagi M, Soloway PD, DeClerck YA, Fridman R. Differential inhibition of membrane type 3 (MT3)-matrix metalloproteinase (MMP) and MT1-MMP by tissue inhibitor of metalloproteinase (TIMP)-2 and TIMP-3 rgulates pro-MMP-2 activation. J Biol Chem. 2004;279:8592–8601. doi: 10.1074/jbc.M308708200. [DOI] [PubMed] [Google Scholar]
- 35.Barclay WW, Cramer SD. Culture of mouse prostatic epithelial cells from genetically engineered mice. Prostate. 2005;63:291–298. doi: 10.1002/pros.20193. [DOI] [PubMed] [Google Scholar]
- 36.Barclay WW, Axanova LS, Chen W, Romero L, Maund SL, Soker S, Lees CJ, Cramer SD. Characterization of adult prostatic progenitor/stem cells exhibiting self-renewal and multilineage differentiation. Stem Cells. 2008;26:600–610. doi: 10.1634/stemcells.2007-0309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cho JA, Osenkowski P, Zhao H, Kim S, Toth M, Cole K, Aboukameel A, Saliganan A, Schuger L, Bonfil RD, Fridman R. The inactive 44-kDa processed form of membrane type 1 matrix metalloproteinase (MT1-MMP) enhances proteolytic activity via regulation of endocytosis of active MT1-MMP. J Biol Chem. 2008;283:17391–17405. doi: 10.1074/jbc.M708943200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Toth M, Gervasi DC, Fridman R. Phorbol ester-induced cell surface association of matrix metalloproteinase-9 in human MCF10A breast epithelial cells. Cancer Res. 1997;57:3159–3167. [PubMed] [Google Scholar]
- 39.Hotary K, Allen E, Punturieri A, Yana I, Weiss SJ. Regulation of cell invasion and morphogenesis in a three-dimensional type I collagen matrix by membrane-type matrix metalloproteinases 1, 2, and 3. J Cell Biol. 2000;149:1309–1323. doi: 10.1083/jcb.149.6.1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ludwig T, Theissen SM, Morton MJ, Caplan MJ. The cytoplasmic tail dileucine motif LL572 determines the glycosylation pattern of membrane-type 1 matrix metalloproteinase. J Biol Chem. 2008;283:35410–35418. doi: 10.1074/jbc.M801816200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wu YI, Munshi HG, Snipas SJ, Salvesen GS, Fridman R, Stack MS. Activation-coupled membrane-type 1 matrix metalloproteinase membrane trafficking. Biochem J. 2007;407:171–177. doi: 10.1042/BJ20070552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hernandez-Barrantes S, Bernardo M, Toth M, Fridman R. Regulation of membrane type-matrix metalloproteinases. Semin Cancer Biol. 2002;12:131–138. doi: 10.1006/scbi.2001.0421. [DOI] [PubMed] [Google Scholar]
- 43.Gervasi DC, Raz A, Dehem M, Yang M, Kurkinen M, Fridman R. Carbohydrate-mediated regulation of matrix metalloproteinase-2 activation in normal human fibroblasts and fibrosarcoma cells. Biochem Biophys Res Commun. 1996;228:530–538. doi: 10.1006/bbrc.1996.1694. [DOI] [PubMed] [Google Scholar]
- 44.Wilson MJ, Jiang A, Wiehr C, Wang X, Sinha AA, Pei D. Limited processing of pro-matrix metalloprotease-2 (gelatinase A) overexpressed by transfection in PC-3 human prostate tumor cells: association with restricted cell surface localization of membrane-type matrix metalloproteinase-1. J Androl. 2004;25:274–285. doi: 10.1002/j.1939-4640.2004.tb02788.x. [DOI] [PubMed] [Google Scholar]
- 45.Sabeh F, Li XY, Saunders TL, Rowe RG, Weiss SJ. Secreted versus membrane-anchored collagenases: relative roles in fibroblast-dependent collagenolysis and invasion. J Biol Chem. 2009;284:23001–23011. doi: 10.1074/jbc.M109.002808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chiang GG, Abraham RT. Targeting the mTOR signaling network in cancer. Trends Mol Med. 2007;13:433–442. doi: 10.1016/j.molmed.2007.08.001. [DOI] [PubMed] [Google Scholar]
- 47.Holz MK, Ballif BA, Gygi SP, Blenis J. mTOR and S6K1 mediate assembly of the translation preinitiation complex through dynamic protein interchange and ordered phosphorylation events. Cell. 2005;123:569–580. doi: 10.1016/j.cell.2005.10.024. [DOI] [PubMed] [Google Scholar]
- 48.Sun SY, Rosenberg LM, Wang X, Zhou Z, Yue P, Fu H, Khuri FR. Activation of Akt and eIF4E survival pathways by rapamycin-mediated mammalian target of rapamycin inhibition. Cancer Res. 2005;65:7052–7058. doi: 10.1158/0008-5472.CAN-05-0917. [DOI] [PubMed] [Google Scholar]
- 49.Ahmad I, Sansom OJ, Leung HY. Advances in mouse models of prostate cancer. Expert Rev Mol Med. 2008;10:e16. doi: 10.1017/S1462399408000689. [DOI] [PubMed] [Google Scholar]
- 50.Ratnacaram CK, Teletin M, Jiang M, Meng X, Chambon P, Metzger D. Temporally controlled ablation of PTEN in adult mouse prostate epithelium generates a model of invasive prostatic adenocarcinoma. Proc Natl Acad Sci U S A. 2008;105:2521–2526. doi: 10.1073/pnas.0712021105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Osenkowski P, Meroueh SO, Pavel D, Mobashery S, Fridman R. Mutational and structural analyses of the hinge region of membrane type 1-matrix metalloproteinase and enzyme processing. J Biol Chem. 2005;280:26160–26168. doi: 10.1074/jbc.M414379200. [DOI] [PubMed] [Google Scholar]
- 52.Ohtsubo K, Marth JD. Glycosylation in cellular mechanisms of health and disease. Cell. 2006;126:855–867. doi: 10.1016/j.cell.2006.08.019. [DOI] [PubMed] [Google Scholar]
- 53.Hess AR, Seftor EA, Seftor RE, Hendrix MJ. Phosphoinositide 3-kinase regulates membrane Type 1-matrix metalloproteinase (MMP) and MMP-2 activity during melanoma cell vasculogenic mimicry. Cancer Res. 2003;63:4757–4762. [PubMed] [Google Scholar]
- 54.Ispanovic E, Haas TL. JNK and PI3K differentially regulate MMP-2 and MT1-MMP mRNA and protein in response to actin cytoskeleton reorganization in endothelial cells. Am J Physiol Cell Physiol. 2006;291:C579–588. doi: 10.1152/ajpcell.00300.2005. [DOI] [PubMed] [Google Scholar]
- 55.Zahradka P, Harding G, Litchie B, Thomas S, Werner JP, Wilson DP, Yurkova N. Activation of MMP-2 in Response to Vascular Injury is Mediated by Phosphatidylinositol 3-Kinase-dependent Expression of MT-1 MMP (MMP-14) Am J Physiol Heart Circ Physiol. 2004 doi: 10.1152/ajpheart.00230.2004. [DOI] [PubMed] [Google Scholar]
- 56.Sroka IC, Nagle RB, Bowden GT. Membrane-type 1 matrix metalloproteinase is regulated by sp1 through the differential activation of AKT, JNK, and ERK pathways in human prostate tumor cells. Neoplasia. 2007;9:406–417. doi: 10.1593/neo.07193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhang D, Brodt P. Type 1 insulin-like growth factor regulates MT1-MMP synthesis and tumor invasion via PI 3-kinase/Akt signaling. Oncogene. 2003;22:974–982. doi: 10.1038/sj.onc.1206197. [DOI] [PubMed] [Google Scholar]
- 58.Suzuki A, Lu J, Kusakai G, Kishimoto A, Ogura T, Esumi H. ARK5 is a tumor invasion-associated factor downstream of Akt signaling. Mol Cell Biol. 2004;24:3526–3535. doi: 10.1128/MCB.24.8.3526-3535.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chen WS, Xu PZ, Gottlob K, Chen ML, Sokol K, Shiyanova T, Roninson I, Weng W, Suzuki R, Tobe K, Kadowaki T, Hay N. Growth retardation and increased apoptosis in mice with homozygous disruption of the Akt1 gene. Genes Dev. 2001;15:2203–2208. doi: 10.1101/gad.913901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ulici V, Hoenselaar KD, Agoston H, McErlain DD, Umoh J, Chakrabarti S, Holdsworth DW, Beier F. The role of Akt1 in terminal stages of endochondral bone formation: angiogenesis and ossification. Bone. 2009;45:1133–1145. doi: 10.1016/j.bone.2009.08.003. [DOI] [PubMed] [Google Scholar]
- 61.Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307:1098–1101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- 62.Laplante M, Sabatini DM. mTOR signaling at a glance. J Cell Sci. 2009;122:3589–3594. doi: 10.1242/jcs.051011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ma XM, Blenis J. Molecular mechanisms of mTOR-mediated translational control. Nat Rev Mol Cell Biol. 2009;10:307–318. doi: 10.1038/nrm2672. [DOI] [PubMed] [Google Scholar]
- 64.O'Reilly KE, Rojo F, She QB, Solit D, Mills GB, Smith D, Lane H, Hofmann F, Hicklin DJ, Ludwig DL, Baselga J, Rosen N. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res. 2006;66:1500–1508. doi: 10.1158/0008-5472.CAN-05-2925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wan X, Harkavy B, Shen N, Grohar P, Helman LJ. Rapamycin induces feedback activation of Akt signaling through an IGF-1R-dependent mechanism. Oncogene. 2007;26:1932–1940. doi: 10.1038/sj.onc.1209990. [DOI] [PubMed] [Google Scholar]
- 66.Wang X, Yue P, Kim YA, Fu H, Khuri FR, Sun SY. Enhancing mammalian target of rapamycin (mTOR)-targeted cancer therapy by preventing mTOR/raptor inhibition-initiated, mTOR/rictor-independent Akt activation. Cancer Res. 2008;68:7409–7418. doi: 10.1158/0008-5472.CAN-08-1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Carracedo A, Ma L, Teruya-Feldstein J, Rojo F, Salmena L, Alimonti A, Egia A, Sasaki AT, Thomas G, Kozma SC, Papa A, Nardella C, Cantley LC, Baselga J, Pandolfi PP. Inhibition of mTORC1 leads to MAPK pathway activation through a PI3K-dependent feedback loop in human cancer. J Clin Invest. 2008;118:3065–3074. doi: 10.1172/JCI34739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Shi Y, Yan H, Frost P, Gera J, Lichtenstein A. Mammalian target of rapamycin inhibitors activate the AKT kinase in multiple myeloma cells by up-regulating the insulin-like growth factor receptor/insulin receptor substrate-1/phosphatidylinositol 3-kinase cascade. Mol Cancer Ther. 2005;4:1533–1540. doi: 10.1158/1535-7163.MCT-05-0068. [DOI] [PubMed] [Google Scholar]
- 69.Lang SA, Hackl C, Moser C, Fichtner-Feigl S, Koehl GE, Schlitt HJ, Geissler EK, Stoeltzing O. Implication of RICTOR in the mTOR inhibitor-mediated induction of insulin-like growth factor-I receptor (IGF-IR) and human epidermal growth factor receptor-2 (Her2) expression in gastrointestinal cancer cells. Biochim Biophys Acta. doi: 10.1016/j.bbamcr.2010.01.009. [DOI] [PubMed] [Google Scholar]
- 70.Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129:1261–1274. doi: 10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Grant S. Cotargeting survival signaling pathways in cancer. J Clin Invest. 2008;118:3003–3006. doi: 10.1172/JCI36898. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.