

Abstract
Purpose
The present study investigates the efficacy of compartmental targeting in grafted tumours treated by meta-tetra (hydroxyphenyl) chlorin (mTHPC) mediated photodynamic therapy (PDT). The therapeutic efficacy was further related to a regional photoinduced distribution of apoptosis and mTHPC biodistribution profile.
Methods and Materials
Mice bearing EMT6 tumours were subjected to a single irradiation (10 J/cm2) of red laser light (652 nm) at different intervals after a single (0.3 mg/kg or 0.15 mg/kg) or double intravenous (2 × 0.15 mg/kg) injection(s) of mTHPC. Efficiency of the treatment was evaluated by monitoring tumour regrowth. mTHPC pharmacokinetics were assessed by HPLC analysis of excised organs. Regional distribution of apoptosis in tumour sections was investigated with a newly developed co-labelling immunohistochemistry technique.
Results
A fractionated double injection protocol of mTHPC with 24h and 3h drug-light interval (DLI) yielded 100% tumour cure with tumours presenting a massive apoptosis of neoplastic cells along with a distortion of vessels. The best efficiency for a single injection (0.3 mg/kg) was about 54% tumour cures and corresponded to a DLI of 3h. At this DLI tumours showed apoptosis of endothelial cells in residual vessels. Concentrations of mTHPC observed in plasma and tumour for the fractionated injection were not statistically different and were less than the total drug dose in each compartment.
Conclusions
The present work suggests that clinical PDT protocols with mTHPC could be greatly improved by fractionation of the drug administration. Time points should be chosen based on the intratumoral spatio-temporal drug distribution.
Keywords: Apoptosis, compartmental targeting, mTHPC, photodynamic therapy
INTRODUCTION
Photodynamic therapy (PDT) is a therapeutic strategy for the treatment of localized tumours that are accessible to light irradiation. It is based on the combined action of a photosensitizer (PS), light and molecular oxygen, which leads to the generation of toxic reactive oxygen species (ROS). The tumoricidal effect of PDT is triggered by a direct damage of malignant cells and/or indirect vascular damage accompanied by an inflammatory response1. mTHPC is a second generation photosensitizer, approved in the EU in 2001 for the palliative treatment of head and neck cancers. It is the most potent photosensitizer characterized by a low required light dose and reduced skin photosensitivity as compared to other PS2.
Compartmental targeting of the tumour microvasculature or parenchyma is closely related to the PS distribution, governed by the pharmacokinetic and cell/tissue binding properties of the PS. Photosensitizer distribution can be modulated by the drug-light interval (DLI) which is defined as the time that separates PS administration from light irradiation3. PDT with short DLIs mainly damages endothelial cells4-6 whereas longer DLI’s favour direct neoplastic cell photodamage3, 4, 7, 8.
Most pharmacokinetic studies are based on the assessment of drug content in various organs, including the tumour, but very little is known about drug transfer from the vessels to the tumour cells. Recently, Foster and co-workers first described the dynamics of intratumoral spatial distribution of mTHPC in mammary carcinoma EMT6 tumours with confocal fluorescence microscopy9. mTHPC was confined to the vicinity of the vasculature for the first 3-6h, followed by progressive migration to tissue at increasing distance from the vessels. At 24h post injection, mTHPC fluorescence could only be detected in tumour cells remote from the perfused vessels.
Some efforts have been undertaken to target both compartments (vessels and tumour cells) simultaneously. Fractionated photosensitizer and/or light administration has been studied with different photosensitizers with heterogeneous results4-6. The possible advantage of fractionation of light and/or drug lies in reduced administered doses since the moderate photooxidative insult has been shown to favour apoptotic cell death10.
Apoptosis is highly recommended in clinical situations with regard to the absence of a strong inflammatory reaction11,12. Photoinduced programmed cell death is characterized by signalling convergent to mitochondria, which induces a cascade of caspase activation. The final cleavage of vital substrates is realised by caspase effectors, particularly caspase-3. Evidence for the occurrence of apoptosis in PDT-exposed tumours is reviewed in Oleinick et al.,13 but the discrimination between apoptosis of endothelial and parenchyma cells has not been adequately addressed. At early time points post-Photofrin PDT, apoptosis occurred in tumour endothelial cells lining occluded blood vessels, whereas at longer time points, apoptosis became more widespread14.
In the present study, we attempted to evaluate the effect of compartmental targeting according to spatial intratumoral mTHPC distribution. The primary objective was to evaluate therapeutic efficacy of a dual PS injection, in order to target both compartments during a single illumination. The secondary objective was to establish the spatial distribution of PDT induced apoptosis as a function of illumination and drug administration regime. For this purpose we developed a new approach based on the co-labelling of apoptotic cells and vessels by cleaved caspase-3 and murine collagen IV antibodies, respectively.
MATERIALS AND METHODS
Chemicals and reagents preparation
Cell culture was performed in phenol red-free RPMI-1640 medium (Invitrogen, Cergy-Pontoise, France) supplemented with 9% foetal calf serum (PAN Biotech GmbH, Aidenbach, Germany), 1% 200mM glutamine and 1% penicillin (10.000 UI/mL)-streptomycin (10.000 μg/mL) (Invitrogen, Cergy-Pontoise, France). Meta-tetra-hydroxyphenyl-chlorin (mTHPC) was provided by Biolitec AG (Jena, Germany). Antibodies to mouse collagen IV (rabbit polyclonal) were purchased from Novotec (Varilhes, France) and goat antirabbit IgG coupled to Fluorescein Isothiocyanate (FITC) from Beckman Coulter (Roissy, France). Sheep anti-FITC antibodies coupled to an alkaline phosphatase were purchased from Roche (Meylan, France), and rabbit antibodies anti-cleaved caspase-3 from Cell Signaling (Danvers, MA). Biotinylated goat anti-rabbit IgG and streptavidin alkaline phosphatase were purchased from DakoCytomation (Trappes, France). All solutions were prepared according to standard procedures provided by Sigma-Aldrich (Saint Quentin Fallavier, France). AFA (Alcohol Formaline Acetic, Labonord, Templemars, France) fixing agent was composed of 75% (v/v) absolute ethanol, 20% (v/v) 30% formaline and 5% (v/v) 100% acetic acid. The 0.1 mg/mL collagenase (Eurobio, Les Ulis, France) solution was prepared in CaCl2 solution at 4 g/1000. Buffer A containing 0.1M PBS, 0.3% (w/v) nuclease-free BSA, 0.1% (w/v) sodium azide, 0.06% (w/v) n-ethyl-maleimide and 20% (v/v) of glycerol was used to dilute primary antibodies. Fastblue and Fasted were composed of 96% (v/v) 0.1M Tris (pH 8.2), 2% (v/v) Naphthol (Sigma-Aldrich, Saint Quentin Fallavier, France) ASMX, 2% (v/v) MRX50PA (Brij 35, Saponine, Tween 20, MgCl2 1M, Eau BM, Calbiochem, France), 0.05% (v/v) 1M Levamisole and respectively 0.3% (w/v) Fastblue or 0.1% (w/v) Fastred. Equilibration buffer (pH 9.4) consisted of 12% (v/v) 1M Tris HCl (pH 7.4), 2.4% (v/v) NaCl 5M and 6% (v/v) MgCl2 1M in sterile water. Polyvinyl alcohol (PVA) medium was prepared with 80% (v/v) 50mM Tris HCl buffer (pH 8.5), 20% (v/v) glycerol and 13% (w/v) PVA.
Animal model and tumour system
Female BALB/c mice (Harlan, Gannat, France) (18-20g, 8-10 weeks) were used in compliance with the French Animal Scientific Procedures Act (April 1988). Animals were kept under standard conditions and provided with food and water ad libitum. Animals were inoculated subcutaneously into the right flank with exponentially growing EMT6 cells (0.1 mL of 5 × 105 cells/mL in 0.9% NaCl). Experiments were initiated 10-12 days later when tumours reached 4-5 mm diameter.
Photosensitizer administration and photodynamic treatment
mTHPC was dissolved in ethanol/polyethylene glycol 400/water solution (2/3/5, v/v/v). The treatment was carried out under anaesthesia with intraperitoneal injection of a ketamine (Ketalar®, 50 mg/mL)/xylazine (Rompun® 2%) mixture (90/10 mg/kg b.w). Irradiation was performed at 652 nm by an argon-ion-laser-pumped dye laser (Spectra-Physics, Les Ulis, France) to a total light dose of 10 J/cm2 at a fluence rate of 30 mW/cm2. Irradiation was performed through the skin overlaying the tumour with a spot size of 1 cm. Different treatment regimes are schematized in Figure 1. Irradiation was performed at 3h or 24h after a single injection of 0.15 or 0.3 mg/kg. A 6h drug-light interval (DLI) was applied after a single injection of 0.3 mg/kg. A separate set of experiments consisted of an injection of 0.15 mg/kg, followed 21h later by a second injection of 0.15 mg/kg and a single irradiation 3h after the second injection. Mice received 100 μL mTHPC solution via the tail vein. Controls animals received an intravenous (I.V.) injection of 0.3 mg/kg b.w. mTHPC without irradiation (“no light” group).
Figure 1.
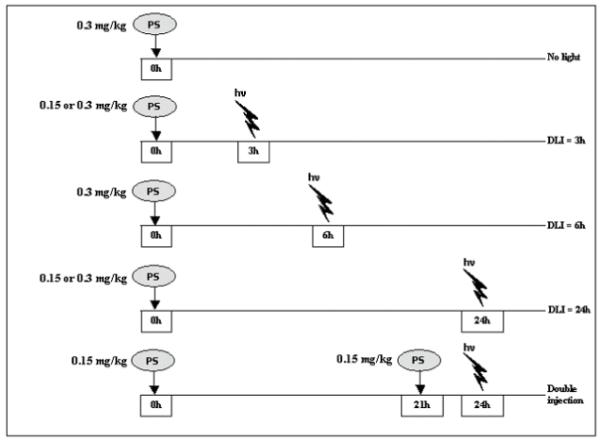
Time line sketch illustrating treatment protocols for mTHPC-PDT in control (drug only) and PDT-treated mice. In the control group, mice received an I.V. injection of mTHPC (0.3 mg/kg). In PDT-treated groups, mTHPC was administered 3, 6 and/or 24h before irradiation with different concentrations. «hv» - irradiation,«PS» - photosensitizer.
Assessment of tumor response
Following treatment, orthogonal diameters of tumours were measured 3 times weekly. The tumour volume was calculated using V = Dd2/2 where D is the longest diameter and d is the diameter perpendicular to D. Tumours were monitored until they reached the ethically-mandated maximum volume of 1 cm3. Animals were considered to be cured when they remained tumour free more than 180 days after PDT. Each experimental group included between 5 (control and 0.15 mg/kg groups) and 11 animals.
Histological studies
Three to four animals were used for each treatment condition. Animals were sacrificed 24h after PDT by anaesthetic overdose and cervical dislocation. Tumors were harvested and placed for 24h in AFA fixing agent. Samples were routinely processed and embedded in paraffin. Necrosis was assessed by Hematoxylin-Eosin-Safran (HES) coloration. Apoptosis distribution was addressed by a co-labelling of vessels by collagen IV and apoptotic cells by cleaved caspase-3.
HES staining of necrosis
Three deparaffinized sections of 5 μm thickness obtained serially at 0.2 mm intervals were stained with HES for light microscopic examination and calculation of tumour size. Each section was imaged with the DMD 108 microscope (Leica Microsystems, Wetzlar GmbH, Germany) at magnifications of 40x to 100x. The ratio of necrosis area/tumour area was measured by tracing the demarcation of the tumour and the necrosis on the computer screen using ImageJ™ software and expressed as a percentage.
Co-labelling of blood vessels and apoptosis
Deparaffinized sections were washed and subjected to heat induced epitope retrieval by incubation in 10 mM EDTA (pH 8.0) for 10 min (120°C) followed by 2h cool-down. After washing, the samples were hydrated for 30 min in Tris Buffered Saline Tween (TBST, pH 7.6). Before staining, slides were treated at 37°C for 20 min with a 0.1 mg/mL collagenase solution. Primary antibodies were diluted in buffer A. Mouse collagen IV was detected with a polyclonal rabbit anti-mouse antibody at 4°C overnight followed by a goat anti-rabbit IgG coupled to FITC for 1h. Sheep anti-FITC antibody coupled to an alkaline phosphatase was then applied for 1h and revealed by Fastblue 40 min later. Staining was fixed for 5 min in an equilibration buffer (pH 9.4). After washing with osmosed water and TBST, sections were hydrolysed with 0.1N HCL for 20 min and incubated with rabbit anti-cleaved caspase 3 at 4°C overnight. Biotinylated goat anti-rabbit IgG was used for 1h followed by 1h incubation with streptavidin alkaline phosphatase and followed by Fastred for 40 min. Finally, slides were fixed with PVA. Negative controls were realized by using antibody solvent without primary antibody. After drying, slides were observed with Axioscop II microscope (Zeiss, Jena, Germany), and images were registered with Axiovision 4.4™ software. Apoptotic cells in each tumor were counted in ten consecutive fields away from the necrotic areas at 1000x magnification and were presented as the mean of the total calculated number of apoptotic cells.
Pharmacokinetic study
After anaesthesia, animals were subjected to a single or double injection of mTHPC with subsequent removal of blood samples by intracardiac puncture at different times after injection. Plasma was collected after centrifugation at 3000 rpm for 5 min at 4°C and kept at −80°C. For biodistribution studies, selected organs (skin, muscle, tumour) were removed, washed in 0.9% NaCl, and frozen at −80°C. mTHPC tissue concentrations were determined by high-performance liquid chromatography (HPLC) as previously described15. Briefly, all tissue samples were minced, weighed, and freeze dried. Ten to twenty milligrams of each powdered sample were diluted in 1.5 mL methanol/DMSO (3/5, v/v), mixed for 3 to 5 s at 451.58 g, and finally incubated (60 °C) under continuous shaking for 12h. Solutions were spun at 16.000 g in a centrifuge for 5 min, and 1mL of the supernatant was analyzed by HPLC. Each group consisted of at least 4 animals.
Statistics
Statistical analysis of tumour response between groups was carried out using Kaplan-Meier analysis and non-parametric Fischer’s test with StatView 5.0 system for Windows™ (SAS institute Inc., SAS Campus Drive, NC, USA). Non-parametric Mann Whitney’s U test was used for pharmacokinetic, apoptosis and necrosis measurements with a significance level of p < 0.05. Results are presented as mean ± SE.
RESULTS
Tumour response to mTHPC-PDT treatment as a function of DLIs and mTHPC dose
The Kaplan-Meier plot shown in Figure 2 demonstrates that 100% of tumour-bearing mice were cured with two administrations of 0.15 mg/kg b.w. of mTHPC at 24h and 3h before a single irradiation. This regime appeared to be the most effective as compared to a single injection, irrespective of the administered dose or DLI. Indeed, with a single injection of 0.15 mg/kg b.w. at 3h or 24h before illumination, only 20% of mice were cured. A better efficacy was observed with a single injection of mTHPC at 0.3 mg/kg b.w. for DLI 3h and 6h with 54% and 40% cure rates, respectively. A DLI of 24h induced a cure rate of less than 20%, irrespective of the drug dose. There were no significant differences (p > 0.05) in growth delays between the different DLIs for all single injection treatments.
Figure 2.
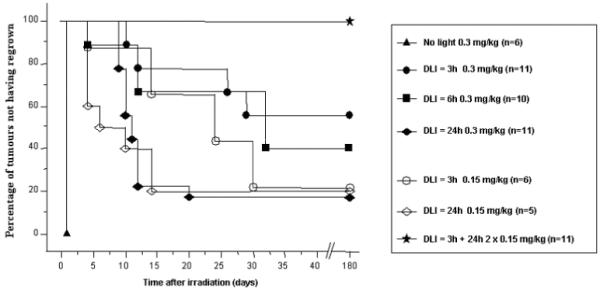
Regrowth of EMT6 tumors after mTHPC-based PDT.
Results are expressed as Kaplan-Meier plots, where the percentage of tumors not exhibiting regrowth is plotted as a function of time after PDT.
We noted that PDT damage to normal skin after a single (0.3 mg/kg, DLI 24h) or a double injection of 0.15 mg/kg mTHPC consisted of a minimal redness during 48h without subsequent scar formation (Figure 3). This deeply contrasted to the injection of 0.3 mg/kg and a 3h DLI, where irradiated skin demonstrated pronounced scab formation (Figure 3).
Figure 3.
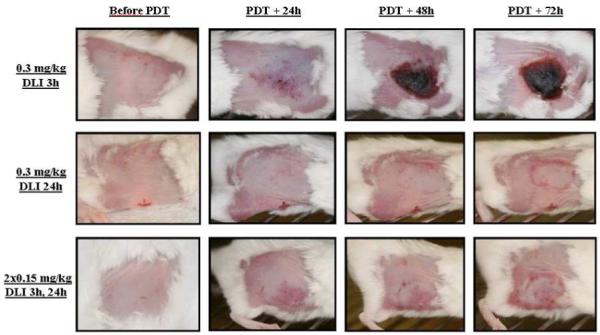
Representative photographs of skin damage in mice induced by photodynamic treatment with a single (0.3 mg/kg) or fractionated I.V. injections (0.15 mg/kg) of mTHPC.
Apoptosis and necrosis with compartmental targeting
Control tumours stained by HES (Figure 4A) presented a central necrotic area (30.8 ± 2.6 %) (Figure 5) surrounded by proliferating neoplastic cells with numerous mitotic figures. Consequently, for further observations, only non-necrotic areas were assessed. Anti-collagen IV staining (blue) of control tumours (Figure 4B) revealed the presence of viable vessels in the tissue without apoptotic cell staining (anti cleaved caspase-3 staining, red). HES staining of PDT treated tumours demonstrated in all experimental conditions a significant increase in central necrotic volume (Figure 5), 1.5 to 2.2 times larger than controls. Necrosis was maximal for a DLI of 6 h (66.5 ± 8.7 %), without however being significant (Figure 5). Colocalization of collagen IV (blue) and cleaved caspase 3 revealed 2 different profiles at a 3h DLI. Either important apoptosis in the vessels (82 ± 8.7) but not in the parenchyma (11±1.0) (Figure 6B) or a complete absence of vessels as well as apoptotic cells (Figure 6D) was observed. At a 6h DLI, vessel staining was absent, and there was significantly (p < 0.05) less parenchymal apoptosis (31 ± 6) (Figure 6F). For a DLI of 24h, apoptosis was significantly increased in the parenchyma (63 ± 3.2). Fractionated administration of mTHPC induced massive tumour destruction (Figure 7C) with exhaustive parenchymal apoptosis (583 ± 43) and distorted vessel structures (Figure 7D).
Figure 4.
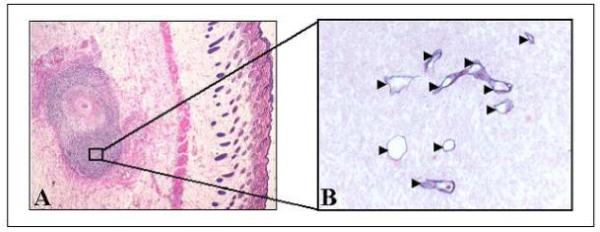
Immunohistochemical analysis of excised EMT6 controls 24h after an I.V. injection of mTHPC (0.3 mg/kg) in tumour-bearing mice. A, HES coloration (x40). B, immunohistochemical assessment of apoptosis with anti-cleaved caspase-3 (red) and microvasculature with anti-murine collagen IV (blue) antibodies (x400). Arrow heads indicate blood vessels.
Figure 5.
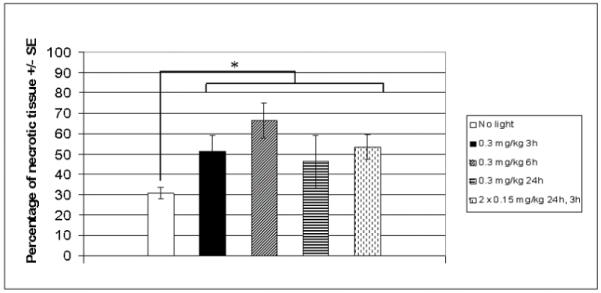
Necrosis observed in EMT6 tumours excised 24h after PDT with a single (0.3 mg/kg) or fractionated (2 × 0.15 mg/kg) mTHPC injections.
Tumours were processed by HES staining. The ratio of necrosis area/tumour area was measured by tracing the demarcation of the tumour and the necrosis regions using ImageJ™ software. Mean percentage of necrosis from 3 mice for each group ± SE is shown. Control mice received the solvent only. *p < 0.05 for all PDT treated groups versus control group.
Figure 6.
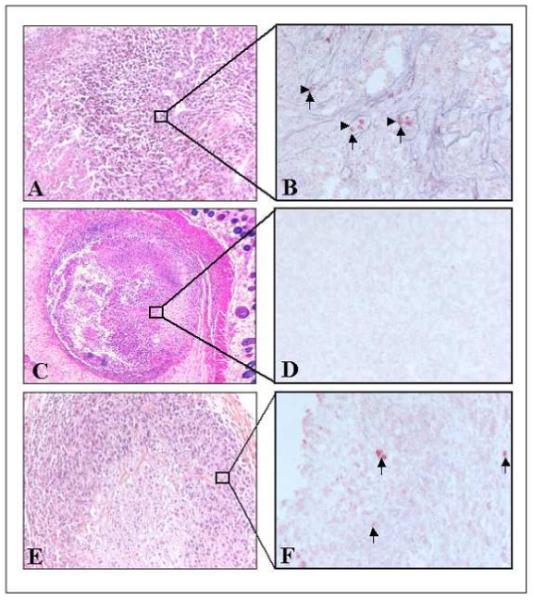
Immunohistochemical analysis of EMT6 tumours excised 24h after PDT with a single mTHPC injection (0.3 mg/kg) at DLIs of 3h (A-D) or 6h (E-F).
A-C-E, HES staining (x200 for A, E and x40 for C).
B-D-F, immunohistochemical assessment of apoptosis with anti-cleaved caspase-3 (red) and microvasculature with anti-murine collagen IV (blue) antibodies (x400). Arrow heads indicate blood vessels, and arrows indicate apoptotic cells.
Figure 7.
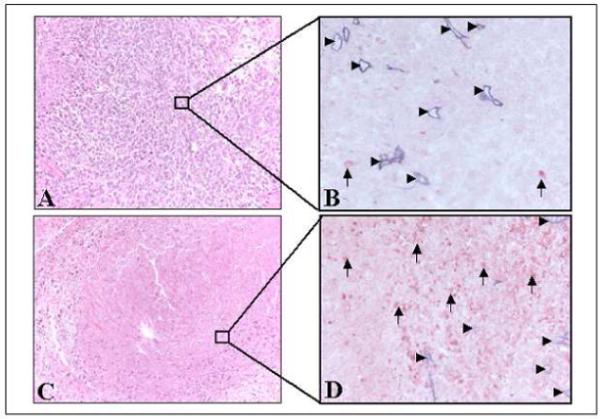
Immunohistochemical analysis of EMT6 tumours excised 24h after PDT with a single injection of mTHPC (A-B, 0.3 mg/kg) and a DLI of 24h or fractionated mTHPC injections (C-D, 2 × 0.15 mg/kg).
A-C, HES staining (x200). B-D, immunohistochemical assessment of apoptosis with anti-cleaved caspase-3 (red) and microvasculature with anti-murine collagen IV (blue) antibodies (x400). Arrow heads indicate blood vessels, and arrows indicate apoptotic cells.
mTHPC bio-distribution assessed by HPLC
HPLC analysis of mTHPC in plasma, skin, tumour, and muscle is presented in Figure 8. The highest plasma concentration is found 3h following 0.3 mg/kg mTHPC (0.13 ± 0.007 ng/mg tissue), whereas the highest tumour content is observed 24h post 0.3 mg/kg mTHPC administration (0.077 ± 0.014 ng/mg tissue, p < 0.05). Fractionated mTHPC injection with a combination of 24h and 3h DLIs before HPLC analysis demonstrated comparable concentrations in all compartments (respectively 0.059 ± 0.003 for plasma, 0.045 ± 0.013 for skin and 0.056 ± 0.01 ng/mg for tumour tissue, p > 0.05). The highest tumour-to-muscle ratio of 4 was observed 24h after 0.3 mg/kg mTHPC.
Figure 8.
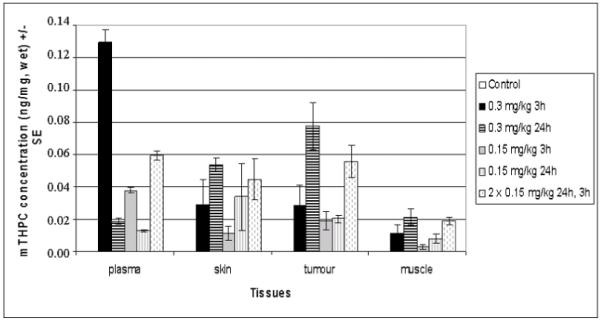
mTHPC content in different tissues of EMT6 tumour-bearing mice.
Mice were subjected to single (0.15 or 0.3 mg/kg) or fractionated (2 × 0.15 mg/kg) mTHPC injections. Three or 24h later plasma, skin, tumour, and muscle were excised and processed for HPLC analysis. Control mice received solvent only. Date are presented as the mean concentration of mTHPC (ng/mg, wet) ± SE based on 3 mice for control group and 5 mice for other groups.
DISCUSSION
Several teams have applied the concept of compartmental targeting to enhance photodynamic efficacy16-21. Zhou et al.22 noticed the importance of the choice of DLI to obtain a good treatment efficacy together with a maximal protection of normal tissues. Many subsequent investigations commented on the impact of DLI on the photodynamic effect, with significant improvement of efficacy with shorter time intervals, corresponding to high plasma levels of the drug23-25. Veenhuizen et al. investigated the impact of a double injection of 0.3 mg/kg mTHPC at 1-3 and 48h before illumination6. Although mTHPC levels in the tumour were the highest at the longer DLIs, response was better at shorter time intervals and high plasma levels. The combination of 2 injections with the aim to obtain high drug levels in both tumour and vascular compartments did not significantly improve results. Identical observations were made by the same group when both injections were separated by a time interval of 72h, despite the fact that the total drug dose was doubled4. Using a so-called vascular photosensitizer, Dolmans et al. demonstrated that a double injection of this drug at 15 minutes and 4h before illumination was significantly better than the single drug dose at any of those time points5. They attributed this positive effect to the fact that fluorescence studies indicated a more homogeneous staining of both endothelial and perivascular structures following a double injection.
In a previous study, we used high resolution confocal fluorescence imaging to simultaneously map microscopic intratumoral mTHPC localization with respect to perfused vasculature as a function of the time after injection9. A progressive gradient of PS fluorescence was observed from the lumen, to endothelial cells, parenchyma adjacent to the vessels, and finally tumour cells remote from the vascular structures. Three hours post injection, maximal mTHPC fluorescence was detected in the periluminal structures (within 15 μm). After 24h, fluorescence was about three times higher 140 μm from the vessel, corresponding to parenchyma localisation. We therefore chose those two time points to evaluate the impact of fractionated mTHPC delivery on PDT-induced regional distribution of apoptosis and overall tumour regrowth.
With regard to the tumour regrowth curves (Figure 2), it appears that irrespective of the single drug dose used, longer time intervals (24h) are less effective, producing < 20% tumour cure. These results are in agreement with previous mTHPC studies4-6. A 100% cure rate was observed when two separate injections of 0.15 mg/kg were administered 3 and 24h before illumination (Figure 2). A positive impact of drug fractionation has never been mentioned previously for mTHPC-PDT. This should probably be attributed to the time points that were chosen in the earlier studies, 48 and 72h4, 6. Indeed from our intratumor drug distribution studies, it appears that at those longer time points, maximal mTHPC fluorescence within the tumour is extremely remote from the vessels, thus indicating drug accumulation in regions that are potentially hypoxic and result in reduced PDT efficacy9.
Irrespective of DLI, necrosis strongly contributes to cell death as observed through HES staining (Figure 5). This result is anticipated considering that mTHPC is a strong mediator of photoinduced necrosis26. Regional distribution of apoptotic features induced by PDT has been poorly documented. An immunohistochemistry study by Engbrecht et al. demonstrated that Photofrin® mediated PDT induced apoptosis firstly in the endothelial cells bordering occluded blood vessels and became more widespread at later time points14. This observation relied on a double fluorescent staining of endothelial cells (anti-PECAM: Platelet Endothelial Cell Adhesion Molecule) and apoptosis via the TUNEL technique (terminal deoxynucleotidyl transferase-mediated dUTP nick end labelling). A more recent study of Henderson et al10 investigated tumour grafts treated by 2-[1-hexyloxyethyl]-2-devinyl pyropheophorbide-a (HPPH)-PDT with high and low fluences and fluence rates (48 and 128 J/cm2, 14 and 112 mW/cm2). They demonstrated that extent and regional distribution of tumour and vascular damage, particularly apoptosis, correlated with fluence and fluence rate used during treatment.
To enable a new approach for simultaneous co-labelling of vessels and apoptosis in the same section, cleaved caspase-3 is targeted for apoptosis and murine collagen IV is used to mark basal membranes of vessels. The advantage of this new method is that, on the same sample, apoptosis and vascular structures can be observed along with classical morphopathological features. When the PDT effect is extremely poor (24h), vessels are intact and only parenchyma cells show apoptotic features (Figure 7 A, B). Intermediate cure rates obtained with DLIs of 3 and 6h mostly indicate vascular destruction, since collagen IV staining is absent along with a very limited staining of cleaved caspase-3 (Figure 6 C, D, E, F). A fractionated injection results in vascular effects with a poor collagen IV staining, together with massive apoptosis within the bulk tumour (Figure 7 C, D).
Historically, PDT protocols were established on the assumption that illumination should be performed at the highest tumour-to-normal drug ratios in order to protect healthy surrounding tissue and achieve a good tumouricidal effect27. These ratios are only observed at long DLIs. Recent preclinical work however suggests that better cure rates could be obtained at shorter time intervals, corresponding to high plasma levels of the drug8. From our work, it appears that the highest efficacy can be obtained when combining both propositions, aiming at the simultaneous destruction of tumour vasculature and neoplastic cells. The time point chosen for drug administration and illumination appears to be critical, since our study is the first to observe a benefit from mTHPC fractionation. It may be hypothesized that this is a consequence of the double injection resulting in a more homogeneous distribution of the drug in vascular structures as well as in tumour cells close to and more distant from vessels. These time points should not be determined solely on the basis of the plasma and bulk tumour drug levels obtained by extraction but rather by the temporal-spatial distribution of the photosensitizer within the tumour.
Although the excellent PDT efficacy obtained from dose fractionation at 24h and 3h may be unique to mTHPC due to its tight cell/tissue binding properties, the tumour response results clearly demonstrate the importance of DLI informed by detailed drug distribution studies like those performed for mTHPC9, 28.
Acknowledgements
The authors thank Dr Marie-Ange D’Hallewin for the critical reading of the manuscript and helpful remarks. The research was supported by the Alexis Vautrin Cancer Center, French Ligue Nationale contre le Cancer and by US NIH grant CA68409 (to S.M. and T.H.F.). This work was presented at the 13th Congress of ESP (Wroclaw, Poland, September 5-10, 2009).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
CONFLICTS OF INTEREST NOTIFICATION
The authors have no conflict of interest to declare.
REFERENCES
- 1.Dougherty TJ, Gomer CJ, Henderson BW, et al. Photodynamic therapy. J Natl Cancer Inst. 1998;90:889–905. doi: 10.1093/jnci/90.12.889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Triesscheijn M, Baas P, Schellens JH, et al. Photodynamic therapy in oncology. Oncologist. 2006;11:1034–1044. doi: 10.1634/theoncologist.11-9-1034. [DOI] [PubMed] [Google Scholar]
- 3.Chen B, Pogue BW, Hoopes PJ, et al. Vascular and cellular targeting for photodynamic therapy. Crit Rev Eukaryot Gene Expr. 2006;16:279–305. doi: 10.1615/critreveukargeneexpr.v16.i4.10. [DOI] [PubMed] [Google Scholar]
- 4.Cramers P, Ruevekamp M, Oppelaar H, et al. Foscan uptake and tissue distribution in relation to photodynamic efficacy. Br J Cancer. 2003;88:283–290. doi: 10.1038/sj.bjc.6600682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dolmans DE, Kadambi A, Hill JS, et al. Targeting tumor vasculature and cancer cells in orthotopic breast tumor by fractionated photosensitizer dosing photodynamic therapy. Cancer Res. 2002;62:4289–4294. [PubMed] [Google Scholar]
- 6.Veenhuizen R, Oppelaar H, Ruevekamp M, et al. Does tumour uptake of Foscan determine PDT efficacy? Int J Cancer. 1997;73:236–239. doi: 10.1002/(sici)1097-0215(19971009)73:2<236::aid-ijc13>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- 7.Jones HJ, Vernon DI, Brown SB. Photodynamic therapy effect of m-THPC (Foscan) in vivo: correlation with pharmacokinetics. Br J Cancer. 2003;89:398–404. doi: 10.1038/sj.bjc.6601101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Triesscheijn M, Ruevekamp M, Aalders M, et al. Outcome of mTHPC mediated photodynamic therapy is primarily determined by the vascular response. Photochem Photobiol. 2005;81:1161–1167. doi: 10.1562/2005-04-04-RA-474. [DOI] [PubMed] [Google Scholar]
- 9.Mitra S, Maugain E, Bolotine L, et al. Temporally and spatially heterogeneous distribution of mTHPC in a murine tumor observed by two-color confocal fluorescence imaging and spectroscopy in a whole-mount model. Photochem Photobiol. 2005;81:1123–1130. doi: 10.1562/2005-03-24-RA-471. [DOI] [PubMed] [Google Scholar]
- 10.Henderson BW, Gollnick SO, Snyder JW, et al. Choice of oxygen-conserving treatment regimen determines the inflammatory response and outcome of photodynamic therapy of tumors. Cancer Res. 2004;64:2120–2126. doi: 10.1158/0008-5472.can-03-3513. [DOI] [PubMed] [Google Scholar]
- 11.LaMuraglia GM, Schiereck J, Heckenkamp J, et al. Photodynamic therapy induces apoptosis in intimal hyperplastic arteries. Am J Pathol. 2000;157:867–875. doi: 10.1016/S0002-9440(10)64600-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wyllie AH. Apoptosis: an overview. Br Med Bull. 1997;53:451–465. doi: 10.1093/oxfordjournals.bmb.a011623. [DOI] [PubMed] [Google Scholar]
- 13.Oleinick NL, Morris RL, Belichenko I. The role of apoptosis in response to photodynamic therapy: what, where, why, and how. Photochem Photobiol Sci. 2002;1:1–21. doi: 10.1039/b108586g. [DOI] [PubMed] [Google Scholar]
- 14.Engbrecht BW, Menon C, Kachur AV, et al. Photofrin-mediated photodynamic therapy induces vascular occlusion and apoptosis in a human sarcoma xenograft model. Cancer Res. 1999;59:4334–4342. [PubMed] [Google Scholar]
- 15.Lassalle HP, Dumas D, Grafe S, et al. Correlation between in vivo pharmacokinetics, intratumoral distribution and photodynamic efficiency of liposomal mTHPC. J Control Release. 2009;134:118–124. doi: 10.1016/j.jconrel.2008.11.016. [DOI] [PubMed] [Google Scholar]
- 16.Aalders MC, Triesscheijn M, Ruevekamp M, et al. Doppler optical coherence tomography to monitor the effect of photodynamic therapy on tissue morphology and perfusion. J Biomed Opt. 2006;11:044011. doi: 10.1117/1.2337302. [DOI] [PubMed] [Google Scholar]
- 17.Chen B, Pogue BW, Goodwin IA, et al. Blood flow dynamics after photodynamic therapy with verteporfin in the RIF-1 tumor. Radiat Res. 2003;160:452–459. doi: 10.1667/RR3059. [DOI] [PubMed] [Google Scholar]
- 18.Chen B, Roskams T, de Witte PA. Antivascular tumor eradication by hypericin-mediated photodynamic therapy. Photochem Photobiol. 2002;76:509–513. doi: 10.1562/0031-8655(2002)076<0509:atebhm>2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 19.Karmakova T, Feofanov A, Pankratov A, et al. Tissue distribution and in vivo photosensitizing activity of 13,15-[N-(3-hydroxypropyl)]cycloimide chlorin p6 and 13,15-(N-methoxy)cycloimide chlorin p6 methyl ester. J Photochem Photobiol B. 2006;82:28–36. doi: 10.1016/j.jphotobiol.2005.08.006. [DOI] [PubMed] [Google Scholar]
- 20.Li LB, Luo RC. Effect of drug-light interval on the mode of action of Photofrin photodynamic therapy in a mouse tumor model. Lasers Med Sci. 2009;24:597–603. doi: 10.1007/s10103-008-0620-9. [DOI] [PubMed] [Google Scholar]
- 21.Olivo M, Chin W. Perylenequinones in photodynamic therapy: cellular versus vascular response. J Environ Pathol Toxicol Oncol. 2006;25:223–237. doi: 10.1615/jenvironpatholtoxicoloncol.v25.i1-2.140. [DOI] [PubMed] [Google Scholar]
- 22.Zhou CN, Yang WZ, Ding ZX, et al. The biological effects of photodynamic therapy on normal skin in mice--I. A light microscopic study. Adv Exp Med Biol. 1985;193:105–109. doi: 10.1007/978-1-4613-2165-1_12. [DOI] [PubMed] [Google Scholar]
- 23.Ferrario A, Kessel D, Gomer CJ. Metabolic properties and photosensitizing responsiveness of mono-L-aspartyl chlorin e6 in a mouse tumor model. Cancer Res. 1992;52:2890–2893. [PubMed] [Google Scholar]
- 24.Gomer CJ, Ferrario A. Tissue distribution and photosensitizing properties of mono-L-aspartyl chlorin e6 in a mouse tumor model. Cancer Res. 1990;50:3985–3990. [PubMed] [Google Scholar]
- 25.Harada M, Woodhams J, MacRobert AJ, et al. The vascular response to photodynamic therapy with ATX-S10Na(II) in the normal rat colon. J Photochem Photobiol B. 2005;79:223–230. doi: 10.1016/j.jphotobiol.2004.08.011. [DOI] [PubMed] [Google Scholar]
- 26.Marchal S, Fadloun A, Maugain E, et al. Necrotic and apoptotic features of cell death in response to Foscan photosensitization of HT29 monolayer and multicell spheroids. Biochem Pharmacol. 2005;69:1167–1176. doi: 10.1016/j.bcp.2005.01.021. [DOI] [PubMed] [Google Scholar]
- 27.Pervaiz S, Olivo M. Art and science of photodynamic therapy. Clin Exp Pharmacol Physiol. 2006;33:551–556. doi: 10.1111/j.1440-1681.2006.04406.x. [DOI] [PubMed] [Google Scholar]
- 28.Ball DJ, Vernon DI, Brown SB. The high photoactivity of m-THPC in photodynamic therapy. Unusually strong retention of m-THPC by RIF-1 cells in culture. Photochem Photobiol. 1999;69:360–363. doi: 10.1562/0031-8655(1999)069<0360:thpoti>2.3.co;2. [DOI] [PubMed] [Google Scholar]