

Abstract
Type 2 diabetes mellitus (T2DM) is a major risk factor for cardiovascular disease (CVD) morbidity and mortality worldwide. Renin-angiotensin system (RAS) blockers have been indispensable in diminishing the macrovascular and microvascular complications of diabetes. In addition, cumulative evidence from retrospective studies pointed toward a beneficial effect of RAS agents in preventing the development and progression of T2DM. This disease-modifying potential of RAS blockers has been substantiated by recent prospective trials. Contemporary concepts regarding the natural history of T2DM and the pathophysiologic processes involved have increased our understanding of the mechanisms underlying the therapeutic potential of these agents in diabetes management. In addition to their established roles in the primary prevention of CVD in patients with diabetes, RAS blockers might be considered a suitable therapeutic choice for preventing the development of frank diabetes in high-risk patients.
Keywords: Type 2 diabetes mellitus, Diabetes natural history, Angiotensin-converting enzyme inhibitor, Angiotensin receptor blocker, Renin-angiotensin system, Impaired fasting glucose, Impaired glucose tolerance, Insulin resistance
Introduction
Diabetes mellitus afflicts an estimated 6.6% of the global adult population, or approximately 285 million individuals. This is projected to increase by more than 50% to a 7.8% worldwide prevalence in 20 years. Unless this trend is curbed, the diabetes pandemic will remain an enormous public health and economic burden. As a major risk factor for cardiovascular disease (CVD) and as a coronary artery disease (CAD)–equivalent, diabetes is associated with a multitude of macrovascular and microvascular complications. It is a leading cause of premature death, and approximately 6.8% of adult deaths worldwide are attributed to diabetes [1].
Renin-angiotensin system (RAS) agents, which include angiotensin-converting enzyme inhibitors (ACEIs) and angiotensin receptor blockers (ARBs), are mainstay therapies for hypertension. They are also known to prevent the macrovascular and microvascular complications in both hypertensive and normotensive individuals with diabetes. Recently, a growing body of evidence has emerged that suggests that RAS agents may have a direct role in preventing the development of abnormal glucose homeostasis and in directly altering the natural history of type 2 diabetes mellitus (T2DM). The promise of this therapeutic approach merits a closer scrutiny, considering the potential implications on the primary and secondary prevention strategies for CVD.
Pathogenesis of Type 2 Diabetes
T2DM, previously referred to as adult-onset or non–insulin-dependent diabetes, is characterized by a triad of metabolic derangements: 1) insulin resistance, 2) relative impairment in insulin secretion that is not autoimmune-mediated, and 3) hyperglycemia from increased hepatic gluconeogenesis. The etiogenesis of these metabolic abnormalities is poorly understood, but both genetic and environmental factors play important roles [2]. A family history of diabetes is a strong risk factor for the subsequent development of the disease, exerting a profound genetic influence on the phenotype of patients with T2DM [3]. Ethnicity-related differences in susceptibility exist, with minority ethnic groups such as the American Indians, Pacific Islanders, Asians, Hispanics, and blacks at a particularly higher risk for developing the disease compared with the Caucasian population [4]. Several acquired factors also play a role in the pathogenesis of the disease, including unhealthy dietary patterns, hypertension, obesity [5], smoking [6], and a sedentary lifestyle [7].
Of the three underlying metabolic defects involved in diabetes pathogenesis, insulin resistance is the generally accepted primary mechanism responsible for T2DM and is perhaps the best predictor for it [8]. It is characterized by a subnormal response to a given concentration of insulin, blunting the normal glucose uptake by skeletal muscles and fat cells. A higher fasting plasma insulin level usually indicates a higher degree of insulin resistance. Insulin resistance is thought to be principally genetically linked, although other factors such as obesity, hypertension, or advancing age may play a role [9].
Impaired insulin secretion, the other crucial metabolic event in T2DM, determines the progression and ultimately directs the course of the disease. It is caused by pancreatic β-cell dysfunction, the exact mechanisms for which remain not entirely clear. Derangement in insulin secretion has been demonstrated with abnormalities of the GLUT protein (which partly mediates glucose transport into the pancreatic β cell to stimulate insulin production) [10], as well as deficiency of the ABCA1 protein (an adenosine triphosphate–binding cholesterol transporter in the β cell) [11]. More recently, experimental evidence has highlighted the possible role of oxidative stress in causing β-cell damage and reduced insulin secretory function [12].
The impairment in insulin secretion results in hyperglycemia that, in chronic states, can be toxic to the β cell, leading to further reduction of insulin secretion. Also, hyperglycemia in itself exacerbates insulin resistance [13], perpetuating the vicious metabolic cycle. Diabetic patients present with a combination of varying degrees of insulin resistance and eventually relative insulin deficiency, and whatever the underlying mechanisms, the full phenotypic expression of T2DM requires both insulin resistance and β-cell dysfunction.
The Natural History of Type 2 Diabetes
Insulin resistance is the initial metabolic event that eventually leads to T2DM (Fig. 1), and it precedes the development of hyperglycemia by several years [14]. The pancreatic β cell, for a period of time, compensates by increasing insulin levels (hyperinsulinemia), which keeps the individual normoglycemic during the initial phase of the disease. These initial metabolic events manifest clinically as mild postprandial hyperglycemia (impaired glucose tolerance [IGT]) and/or mild elevation of fasting plasma glucose levels (impaired fasting glucose [IFG]) [15].
Fig. 1.
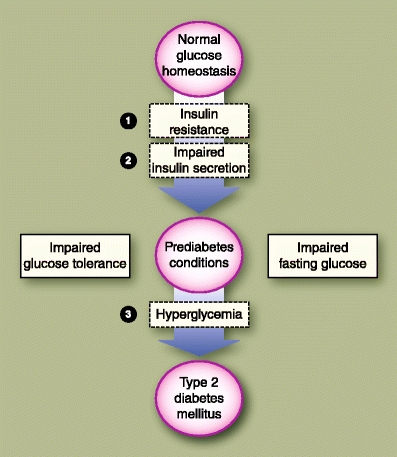
The postulated natural progression from normoglycemic state to frank type 2 diabetes is shown. The numbered items indicate the hypothetical sequence of adverse metabolic events that are associated with the development of the disease
Both IGT and IFG serve as markers for individuals at greatest risk for developing T2DM. It was previously thought that they represented the asymptomatic stages along the continuum between normal glucose tolerance and frank diabetes. Recent data, however, suggest that IGT and IFG are distinct phenotypic pathways that separately lead to the development of diabetes [16, 17]. Although these prediabetes conditions feature dysfunctions in both insulin sensitivity and secretion, they differ in the degree, sites, and patterns by which these metabolic defects manifest. Patients with isolated IGT have severe skeletal muscle insulin resistance with normal (or only mildly impaired) hepatic sensitivity, whereas those with isolated IFG predominantly have hepatic insulin resistance with normal muscle insulin sensitivity. In patients with IGT, both early and late phases of insulin secretory responses are abnormal, leading to prolonged hyperglycemia after a glucose load. In those with isolated IFG, there is reduction in the early phase of insulin secretory response, which causes fasting hyperglycemia; however, the late phase response is preserved, which allows the normalization of blood sugar levels after a glucose load.
Combined IGT-IFG states are common. Roughly 25% of patients with prediabetes develop frank T2DM, about 25% revert to normoglycemia, and the remaining half remain in this impaired glycemic state [18]. Although asymptomatic, these “prediabetes” conditions are potentially pathologic, and in themselves could induce vascular damage and CVD. Nevertheless, both disorders, when diagnosed early, can be reversible and provide an important window in which appropriate intervention may halt the progression to full-blown diabetes [19].
During the transition from prediabetes to T2DM, insulin resistance becomes more severe, a progression that may be due not only to full expression of genetic defects, but also to acquired factors such as obesity, decreased physical activity, and aging. This is accompanied by an increase in basal hepatic glucose production. β-Cell exhaustion eventually occurs, resulting in inadequate insulin secretion, and ultimately hyperglycemia. This failure of the β cell over time to compensate for insulin resistance with hyperinsulinemia marks the beginning of T2DM. As β-cell function progressively declines an absolute insulin deficiency develops, which could lead to a stage in which exogenous insulin is required to maintain adequate glycemic control [2].
Microvascular and Microvascular Complications of Diabetes
Morbidity from T2DM is generally grouped into the microvascular and macrovascular complications. The microvascular disorders include diabetic retinopathy, neuropathy, and nephropathy. The macrovascular complications include CAD, stroke, and peripheral vascular disease.
Retinopathy is generally considered the most common microvascular complication of diabetes, accounting for an estimated 10,000 new cases of blindness annually in the United States [20]. Diabetic nephropathy is the leading cause of renal failure, and afflicts roughly 40% of patients with diabetes [21]. Approximately 30% of patients with diabetes also suffer from some form of neuropathy [22], which is responsible for a large proportion of limb amputations.
The risk for developing microvascular complications of diabetes is thought to be related more directly to glucose, and depends on both the duration and the severity of hyperglycemia. The exact pathologic mechanisms are unclear, but one of those postulated includes osmotic stress from sorbitol accumulation. High glucose levels increase the flux of sugar molecules through the polyol pathway, which converts glucose into glucose alcohol (sorbitol) inside the cell. This pathway, mediated by the aldose reductase enzyme, causes intracellular sorbitol accumulation and subsequent cell damage. Furthermore, hyperglycemia is thought to promote the nonenzymatic formation of advanced glycosylated end products, substances that have been demonstrated to cause cellular injury. High glucose levels can also stimulate free radical production and induce oxidative stress, as well as incite the pathologic release of different growth factors [23].
Diabetes is a strong independent risk factor for CVD, and is in fact considered a CAD-equivalent. At least 65% of patients with diabetes die from heart disease or stroke. Mortality rates from CVD in diabetic patients are two to four times higher compared with nondiabetic patients [24]. The risk of CVD events is disproportionately higher in women with T2DM, with increases in rates of myocardial infarction (MI) up to 4.5-fold and stroke up to sixfold, compared with twofold and 1.5-fold increases in MI and stroke risks, respectively, in diabetic men [25]. The Framingham study showed that diabetes takes away the entire gender protection for macrovascular disease afforded to women [26].
Although the association between diabetes and CVD is unquestioned, the underlying mechanisms for the macrovascular complications are not as well defined. Atherosclerosis is the central pathophysiologic process involved, and is thought to result from chronic inflammation and injury to the coronary, cerebral, and peripheral arterial walls, stimulating atheromatous plaque formation. Progressive plaque deposition leads to chronic vessel stenosis, whereas rupture of this atherosclerotic lesion results in acute vascular syndromes. T2DM is associated with atherogenic dyslipidemia, which consists of high triglyceride, low high-density lipoprotein, and preponderance of small dense low-density lipoprotein particles, all of which can contribute to the development of atherosclerosis. Additionally, diabetes is associated with increased platelet adhesion (from free radical formation, impaired nitric oxide (NO) generation, and calcium dysregulation) as well as hypercoagulability (from elevated levels of plasminogen activator inhibitor). This combination of increased coagulability and impaired fibrinolysis further predisposes to CVD events [27].
RAS Agents and Prevention of Diabetic Complications
The beneficial effects of RAS agents in the prevention of microvascular and macrovascular complications of T2DM are well established, making them mainstay therapies in the management of diabetic patients, as recommended by current primary and secondary prevention guidelines [28]. In a substudy of patients with T2DM in the HOPE trial, ACEI therapy using ramipril resulted in reductions in MI by 22%, stroke by 33%, cardiovascular mortality by 37%, and nephropathy by 24% compared with placebo [29]. Treatment with ARB has also been shown to have vasculoprotective effects, as shown in the LIFE study in which losartan was associated with reductions in MI by 19%, stroke by 22%, cardiovascular mortality by 38%, and albuminuria by 46% compared with atenolol in diabetic patients [30].
RAS Agents in the Prevention of Diabetes Development and Progression
In addition to the mitigation of diabetic complications, there is also mounting evidence, mostly from post hoc analyses of early large randomized trials, on the role of RAS agents in modifying the natural course of diabetes itself. The HOPE study, for example, showed that compared with placebo, the ACEI ramipril was associated with a 34% reduction in self-reported incident diabetes in patients at high risk for CVD events [31]. In the ALLHAT trial, nondiabetic patients who were treated with the ACEI lisinopril had a significantly lower rate of development of new diabetes after 2 years compared with those given amlodipine and chlorthalidone [32]. Similarly, the ACEI trandolapril, when added to other antihypertensive agents, was found to significantly reduce the risk of incident diabetes, and appeared to neutralize the dysglycemic effects associated with thiazides in the INVEST study involving patients without diabetes at baseline [33].
Treatment with ARBs appeared to have the same effect as ACEIs in preventing the development of diabetes. In hypertensive nondiabetic patients, the risk of new-onset diabetes was reduced by 25% with losartan therapy in the LIFE study [34], and by 20% with candesartan in the SCOPE [35]. In patients with heart failure, analysis of data from the CHARM program showed that ARB therapy was associated with a 29% reduction in incident diabetes [36].
A systematic review of 22 trials of antihypertensive drugs involving over 143,000 nondiabetic patients found that among the different classes of antihypertensive agents, the RAS blockers were associated with the highest reduction in the risk of development of diabetes (reductions of 33% with ACEI and 43% with ARB) [37]. Data from the ONTARGET trial [38], which studied over 25,000 patients with CVD, demonstrated similar rates of incident diabetes with ACEI or ARB treatment, and that combination therapy with ACEI and ARB did not lead to further reduction in the incidence of new-onset diabetes or CVD events compared to treatment with either agent alone. It was estimated that an average of 45 patients would need to be treated with RAS agents over 4 to 5 years to avoid one new case of T2DM [39].
Mechanisms for Diabetes-Modifying Properties of RAS Agents
The glycometabolic effects of ACEIs are purported to be modulated by their actions on bradykinin, NO, and angiotensin II (AT II). Bradykinin has been shown to increase insulin sensitivity and enhance skeletal muscle glucose uptake by activating B2 kinin receptors in the muscle cells [40] and by upregulating the GLUT glucose transport system [41]. Bradykinin-potentiated glucose uptake has been demonstrated in cardiac tissue as well [42]. NO is purported to mediate insulin-related glucose uptake through the same mechanisms as bradykinin (Fig. 2). In addition, NO also appears to be the primary mediator for the non–insulin-dependent transport and utilization of glucose in the skeletal muscle [43]. AT II, a powerful vasoconstrictor and growth factor, can interfere with the normal insulin signaling pathway through serine phosphorylation of specific insulin receptors in the muscle cell (Fig. 3), contributing to the insulin resistance observed in essential hypertension [44]. AT II is also a strong mediator of oxidative stress via the NADPH pathway [45].
Fig. 2.
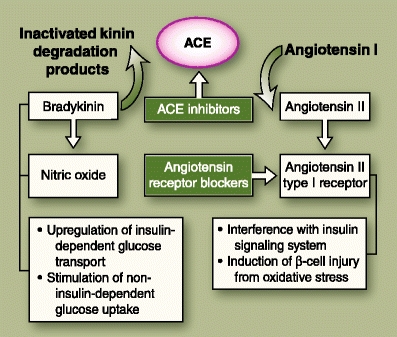
Shows the sites of action of the renin-angiotensin system blockers in relation to the different mediators that are thought to have favorable (bradykinin, nitric oxide) or deleterious (angiotensin II) effects on insulin sensitivity and/or secretion. ACE—angiotensin-converting enzyme
Fig. 3.
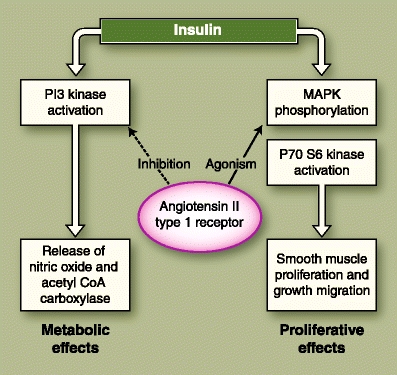
Shows the cross-talk between the insulin signaling pathway and the angiotensin II type 1 receptor, in which the metabolic effects of insulin are antagonized, but the proliferative effects are not. CoA—coenzyme A; MAPK—mitogen-activated protein kinase; PI3—phosphatidylinositol-3
ACE catalyzes the conversion of angiotensin I to AT II, increasing the levels of circulating AT II. Being biochemically identical to the kininase II enzyme, ACE also promotes the degradation of bradykinin [46], leading to reductions in both bradykinin and NO levels. Treatment with ACEI agents, by blocking the actions of ACE, causes reduction in the levels of AT II, as well as increase in the levels of bradykinin and NO, which results not only in vasodilatation but also enhanced insulin insensitivity and improved cellular glucose uptake.
ARBs, conversely, are able to attenuate the negative effects of AT II on cellular insulin signaling by specifically inhibiting the AT II type 1 receptors. The ARB-related improvement in insulin-stimulated glucose uptake has been documented not only in muscle and hepatic cells, but in adipose tissue as well [47]. ARBs have also been shown to have beneficial outcomes on glucose metabolism, which are independent of their effects on AT II receptors, and are thought to be related to their partial agonism of peroxisome proliferator-activated receptors [48]. Thus, the protection against type 2 diabetes afforded by some ARBs may be partially linked to a thiazolidinedione-like effect.
It is speculated that the hemodynamic action of RAS agents on the microcirculation leads to increased blood flows to the skeletal muscle (resulting in increased insulin sensitivity) and to the pancreatic islet cells (resulting in increased insulin secretion). RAS agents have also been shown to reduce sympathetic activity, modulate changes in muscle fiber composition, and improve the ionic homeostasis of potassium and magnesium, all of which may contribute to favorable changes in insulin release and sensitivity [49].
Recently, attention has focused on the role of ARBs in the preservation of pancreatic β-cell function. Experimental models have shown that ARB treatment resulted in significant reductions in reactive oxygen species, protein kinase C, and NADPH oxidase activities in pancreatic islet cells, leading to enhanced β-cell survival [50, 51]. The protection afforded by ARBs against the deleterious effects of inflammatory and oxidative stress was associated with increased β-cell mass, augmented insulin secretion, and improved glucose tolerance. Furthermore, ARB therapy was also shown to prevent the loss of endothelial cells and reduce fibrosis in and around the islets [52]. Despite these multiple postulated mechanisms, however, the precise processes by which the RAS blockers exert their favorable effects on glucose metabolism in humans are still not established.
Recent Clinical Evidence on the Role of RAS Blockers in Diabetes Prevention
Although retrospective studies pointed to a consistent beneficial effect of RAS agents on the development and progression of diabetes, recent randomized trials that prospectively evaluated this effect showed somewhat less convincing results. In the DREAM [53] study, which enrolled nearly 5300 patients with prediabetes, treatment with the ACEI ramipril was associated with a 16% higher likelihood of regression of IGT and/or IFG to normoglycemia. There was a trend toward a reduced incidence of new-onset diabetes compared with placebo after 3 years of follow-up; however, this did not reach statistical significance.
The recently published results of the NAVIGATOR [54•] trial showed more positive findings in favor of the ARB valsartan. This international, multicenter, double-blind, randomized study enrolled over 9300 subjects with IGT and/or IFG having or known CVD who were treated with valsartan or nateglinide for an average of 5 years. Although there was no beneficial effect of the antidiabetic medication nateglinide, treatment with valsartan was associated with a 14% reduction in the incidence of new-onset diabetes. However, there was no significant difference in the rates of CVD events compared with placebo after 5 years of follow-up.
One possible reason that could explain the observed differences in results between the ACEI and ARB trials was the shorter follow-up period in DREAM (3 years) compared with NAVIGATOR (5 years). Also, although both studies enrolled patients with prediabetes, the subjects of the DREAM trial appeared to have lesser risk profiles. Hypertension, a major risk factor for diabetes development, was present in only 43% of DREAM patients, as opposed to 77% in NAVIGATOR. All subjects of the NAVIGATOR trial had pre-existing CVD (if 50 years of age or older), or one or more CVD risk factors (if 55 years of age or older). Younger patients (30 years minimum age) were recruited in the DREAM study, and those with pre-existing CVD were excluded. Because of the disparity in subject selection, it is thought that NAVIGATOR likely enrolled significantly more patients with overactive RAS compared with DREAM, which could partially account for the disproportionate results seen with RAS blockade.
Although the prospective studies showed that both ACEI and ARB do have favorable glycometabolic effects, similar to those suggested by the earlier retrospective analyses, they failed to demonstrate the beneficial effects of RAS agents on “harder” end points in patients with prediabetes and/or the presence of CVD risk factors. The lack of favorable effect on harder end points could potentially be related to the relatively short follow-up periods. These results do corroborate the assertion that RAS blockers have the potential to prevent the progression of prediabetes conditions to frank T2DM.
Implications to Clinical Practice
The value of RAS agents in preventing CVD events and other complications related to diabetes is established. That they provide additional, albeit modest, benefit in terms of modifying the natural history of T2DM only strengthens their position as a viable first-line therapeutic option in diabetes as well as prediabetes conditions. For the primary prevention of CVD in T2DM, current standards of medical care recommend that ACEI or ARB be administered to all diabetic patients with hypertension [28]. Furthermore, the guidelines also advocate the use of RAS blockers for the prevention of diabetic nephropathy, especially in those with microalbuminuria [55].
For the prevention of T2DM in high-risk individuals, the American Diabetes Association has approved only one drug (metformin) so far for this purpose, citing cost, safety, and efficacy issues related to the other potential agents [18]. Given the preponderance of evidence pointing to the potential beneficial effects of ACEIs and ARBs on glycometabolic parameters, it is reasonable to consider these agents to prevent progression to diabetes in high-risk individuals with IGT or IFG. It is conceivable that future updates to the management guidelines might include the use of RAS agents in patients with prediabetes or metabolic syndrome.
Conclusions
As a major risk factor for CVD morbidity and mortality worldwide, the T2DM pandemic is a major global public health concern. RAS blockers have been indispensable in diminishing the macrovascular and microvascular complications of diabetes. Cumulative evidence from retrospective studies pointed toward a beneficial effect of RAS agents in preventing the development and progression of the disease, and this has been substantiated by recent prospective trials. Contemporary concepts regarding the natural history of T2DM and the pathophysiologic processes involved have increased our understanding of the mechanisms underlying the therapeutic potential of these agents in diabetes management. In addition to their established roles in the primary prevention of CVD in patients with diabetes, RAS blockers might be considered a suitable therapeutic choice for the prevention of diabetes development in high-risk patients with IGT or IFG.
Acknowledgments
Disclosure
No potential conflicts of interest relevant to this article were reported.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Clinical Trial Acronyms
- ALLHAT
Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial
- CHARM
Candesartan in Heart Failure-Assessment of Reduction in Mortality and Morbidity
- DREAM
Diabetes Reduction Assessment with Ramipril and Rosiglitazone Medication
- HOPE
Heart Outcomes Prevention Evaluation
- INVEST
International Verapamil SR-Trandolapril Study
- LIFE
Losartan Intervention for Endpoint Reduction in Hypertension
- NAVIGATOR
Nateglinide and Valsartan in Impaired Glucose Tolerance Outcomes Research
- ONTARGET
Ongoing Telmisartan Alone and in Combination with Ramipril Global Endpoint Trial
- SCOPE
Study on Cognition and Prognosis in the Elderly
Contributor Information
Joel A. Lardizabal, Email: dr_jolar@yahoo.com
Prakash C. Deedwania, Email: deed@fresno.ucsf.edu
References
Papers of particular interest, published recently, have been highlighted as: • of importance
- 1.International Diabetes Federation . The Diabetes Atlas. 4. Brussels: International Diabetes Federation; 2009. [Google Scholar]
- 2.Ramlo-Halsted BA, Edelman SV. The natural history of type 2 diabetes: practical points to consider in developing prevention and treatment strategies. Clin Diabetes. 2000;18:80–85. [Google Scholar]
- 3.Li H, Isomaa B, Taskinen MR, et al. Consequences of a family history of type 1 and type 2 diabetes on the phenotype of patients with type 2 diabetes. Diabetes Care. 2000;23:589–594. doi: 10.2337/diacare.23.5.589. [DOI] [PubMed] [Google Scholar]
- 4.Shai I, Jiang R, Manson JE, et al. Ethnicity, obesity, and risk of type 2 diabetes in women: a 20-year follow-up study. Diabetes Care. 2006;29:1585–1590. doi: 10.2337/dc06-0057. [DOI] [PubMed] [Google Scholar]
- 5.Mokdad AH, Ford ES, Bowman BA, et al. Prevalence of obesity, diabetes, and obesity-related health risk factors, 2001. JAMA. 2003;289:76–79. doi: 10.1001/jama.289.1.76. [DOI] [PubMed] [Google Scholar]
- 6.Foy CG, Bell RA, Farmer DF, et al. Smoking and incidence of diabetes among U.S. adults: findings from the Insulin Resistance Atherosclerosis Study. Diabetes Care. 2005;28:2501–2507. doi: 10.2337/diacare.28.10.2501. [DOI] [PubMed] [Google Scholar]
- 7.Helmrich SP, Ragland DR, Leung RW, Paffenbarger RS. Physical activity and reduced occurrence of non-insulin-dependent diabetes mellitus. N Engl J Med. 1991;325:147–152. doi: 10.1056/NEJM199107183250302. [DOI] [PubMed] [Google Scholar]
- 8.Eriksson J, Franssila-Kallunki A, Ekstrand A, et al. Early metabolic defects in persons at increased risk for non-insulin-dependent diabetes mellitus. N Engl J Med. 1989;321:337–343. doi: 10.1056/NEJM198908103210601. [DOI] [PubMed] [Google Scholar]
- 9.Kahn CR. Banting Lecture: Insulin action, diabetogenes, and the cause of type II diabetes. Diabetes. 1994;43:1066–1084. doi: 10.2337/diabetes.43.4.587. [DOI] [PubMed] [Google Scholar]
- 10.Ohtsubo K, Takamatsu S, Minowa MT, et al. Dietary and genetic control of glucose transporter 2 glycosylation promotes insulin secretion in suppressing diabetes. Cell. 2005;123:1307–1321. doi: 10.1016/j.cell.2005.09.041. [DOI] [PubMed] [Google Scholar]
- 11.Brunham LR, Kruit JK, Pape TD, et al. Beta-cell ABCA1 influences insulin secretion, glucose homeostasis and response to thiazolidinedione treatment. Nat Med. 2007;13:340–347. doi: 10.1038/nm1546. [DOI] [PubMed] [Google Scholar]
- 12.Oprescu AI, Bikopoulos G, Naassan A, et al. Free fatty acid-induced reduction in glucose-stimulated insulin secretion: evidence for a role of oxidative stress in vitro and in vivo. Diabetes. 2007;56:2927–2937. doi: 10.2337/db07-0075. [DOI] [PubMed] [Google Scholar]
- 13.Li Y, Xu W, Liao Z, et al. Induction of long-term glycemic control in newly diagnosed type 2 diabetic patients is associated with improvement of beta-cell function. Diabetes Care. 2004;27:2597–2602. doi: 10.2337/diacare.27.11.2597. [DOI] [PubMed] [Google Scholar]
- 14.Tabak AG, Jokela M, Akbaraly TN, et al. Trajectories of glycaemia, insulin sensitivity, and insulin secretion before diagnosis of type 2 diabetes: an analysis from the Whitehall II study. Lancet. 2009;373:2215–2221. doi: 10.1016/S0140-6736(09)60619-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Weyer C, Bogardus C, Mott DM, Pratley RE. The natural history of insulin secretory dysfunction and insulin resistance in the pathogenesis of type 2 diabetes mellitus. J Clin Invest. 1999;104:787–794. doi: 10.1172/JCI7231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Meigs JB, Muller DC, Nathan DM, et al. The natural history of progression from normal glucose tolerance to type 2 diabetes in the Baltimore Longitudinal Study of Aging. Diabetes. 2003;52:1475–1484. doi: 10.2337/diabetes.52.6.1475. [DOI] [PubMed] [Google Scholar]
- 17.Faerch K, Vaag A, Holst JJ, et al. Natural history of insulin sensitivity and insulin secretion in the progression from normal glucose tolerance to impaired fasting glycemia and impaired glucose tolerance: the Inter99 study. Diabetes Care. 2009;32:439–444. doi: 10.2337/dc08-1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nathan DM, Davidson MB, DeFronzo RA, et al. Impaired fasting glucose and impaired glucose tolerance: implications for care. Diabetes Care. 2007;30:753–759. doi: 10.2337/dc07-9920. [DOI] [PubMed] [Google Scholar]
- 19.Twigg SM, Kamp MC, Davis TM, et al. Prediabetes: a position statement from the Australian Diabetes Society and Australian Diabetes Educators Association. Med J Aust. 2007;186:461–465. doi: 10.5694/j.1326-5377.2007.tb00998.x. [DOI] [PubMed] [Google Scholar]
- 20.Fong DS, Aiello LP, Ferris FL, Klein R. Diabetic retinopathy. Diabetes Care. 2004;27:2540–2553. doi: 10.2337/diacare.27.10.2540. [DOI] [PubMed] [Google Scholar]
- 21.Gross JL, de Azevedo MJ, Silveiro SP, et al. Diabetic nephropathy: diagnosis, prevention, and treatment. Diabetes Care. 2005;28:164–176. doi: 10.2337/diacare.28.1.164. [DOI] [PubMed] [Google Scholar]
- 22.Perkins BA, Orszag A, Ngo M, et al. Prediction of incident diabetic neuropathy using the monofilament exam: a 4-year prospective study. Diabetes Care. 2010;33:1549–1554. doi: 10.2337/dc09-1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fowler MJ. Microvascular and macrovascular complications of diabetes. Clin Diabetes. 2008;26:77–82. doi: 10.2337/diaclin.26.2.77. [DOI] [Google Scholar]
- 24.American Heart Association Statistics Committee and Stroke Statistics Subcommittee Heart disease and stroke statistics–2010 update: a report from the American Heart Association. Circulation. 2010;121:e46–e215. doi: 10.1161/CIRCULATIONAHA.109.192667. [DOI] [PubMed] [Google Scholar]
- 25.Almdal T, Scharling H, Jensen JS, Vestergaard H. The independent effect of type 2 diabetes mellitus on ischemic heart disease, stroke, and death: a population-based study of 13, 000 men and women with 20 years of follow-up. Arch Intern Med. 2004;164:1422–1426. doi: 10.1001/archinte.164.13.1422. [DOI] [PubMed] [Google Scholar]
- 26.Abbott RD, Donahue RP, Kannel WB, Wilson PW. The impact of diabetes on survival following myocardial infarction in men vs women. The Framingham Study. JAMA. 1988;260:3456–3460. doi: 10.1001/jama.260.23.3456. [DOI] [PubMed] [Google Scholar]
- 27.Boyle PJ. Diabetes mellitus and macrovascular disease: mechanisms and mediators. Am J Med. 2007;120:S12–S17. doi: 10.1016/j.amjmed.2007.07.003. [DOI] [PubMed] [Google Scholar]
- 28.Buse JB, Ginsberg HN, Bakris GL, et al. Primary prevention of cardiovascular diseases in people with diabetes mellitus: a scientific statement from the American Heart Association and the American Diabetes Association. Diabetes Care. 2007;30:162–172. doi: 10.2337/dc07-9917. [DOI] [PubMed] [Google Scholar]
- 29.The Heart Outcomes Prevention Evaluation Study Investigators. Effects of ramipril on cardiovascular and microvascular outcomes in people with diabetes mellitus: results of the HOPE study and MICRO-HOPE substudy. Lancet 2000, 355:253–259. [PubMed]
- 30.Lindholm LH, Ibsen H, Dahlöf B, et al. Cardiovascular morbidity and mortality in patients with diabetes in the Losartan Intervention For Endpoint reduction in hypertension study (LIFE): a randomised trial against atenolol. Lancet. 2002;359:1004–1010. doi: 10.1016/S0140-6736(02)08090-X. [DOI] [PubMed] [Google Scholar]
- 31.Yusuf S, Gerstein H, Hoogwerf B, et al. Ramipril and the development of diabetes. JAMA. 2001;286:1882–1885. doi: 10.1001/jama.286.15.1882. [DOI] [PubMed] [Google Scholar]
- 32.Barzilay JI, Davis BR, Cutler JA, et al. Fasting glucose levels and incident diabetes mellitus in older nondiabetic adults randomized to receive 3 different classes of antihypertensive treatment: a report from the Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial (ALLHAT) Arch Intern Med. 2006;166:2191–2201. doi: 10.1001/archinte.166.20.2191. [DOI] [PubMed] [Google Scholar]
- 33.Cooper-Dehoff R, Cohen JD, Bakris GL, et al. Predictors of development of diabetes mellitus in patients with coronary artery disease taking antihypertensive medications (findings from the INternational VErapamil SR-Trandolapril STudy [INVEST]) Am J Cardiol. 2006;98:890–894. doi: 10.1016/j.amjcard.2006.04.030. [DOI] [PubMed] [Google Scholar]
- 34.Lindholm LH, Ibsen H, Borch-Johnsen K, et al. Risk of new-onset diabetes in the Losartan Intervention For Endpoint reduction in hypertension study. J Hypertens. 2002;20:1879–1886. doi: 10.1097/00004872-200209000-00035. [DOI] [PubMed] [Google Scholar]
- 35.Lithell H, Hansson L, Skoog I, et al. The Study on Cognition and Prognosis in the Elderly (SCOPE): principal results of a randomized double-blind intervention trial. J Hypertens. 2003;21:875–886. doi: 10.1097/00004872-200305000-00011. [DOI] [PubMed] [Google Scholar]
- 36.Yusuf S, Ostergren JB, Gerstein HC, et al. Effects of candesartan on the development of a new diagnosis of diabetes mellitus in patients with heart failure. Circulation. 2005;112:48–53. doi: 10.1161/CIRCULATIONAHA.104.528166. [DOI] [PubMed] [Google Scholar]
- 37.Elliott WJ, Meyer PM. Incident diabetes in clinical trials of antihypertensive drugs: a network meta-analysis. Lancet. 2007;369:201–207. doi: 10.1016/S0140-6736(07)60108-1. [DOI] [PubMed] [Google Scholar]
- 38.ONTARGET Investigators. Yusuf S, Teo KK, et al. Telmisartan, ramipril, or both in patients at high risk for vascular events. N Engl J Med. 2008;358:1547–1559. doi: 10.1056/NEJMoa0801317. [DOI] [PubMed] [Google Scholar]
- 39.Scheen AJ. Renin-angiotensin system inhibition prevents type 2 diabetes mellitus. Part 1. A meta-analysis of randomised clinical trials. Diabetes Metab. 2004;30:487–496. doi: 10.1016/S1262-3636(07)70146-5. [DOI] [PubMed] [Google Scholar]
- 40.Figueroa CD, Dietze G, Muller-Esterl W. Immunolocalization of bradykinin B2 receptors on skeletal muscle cells. Diabetes. 1996;45:S24–S28. doi: 10.2337/diab.45.1.s24. [DOI] [PubMed] [Google Scholar]
- 41.Miyata T, Taguchi T, Uehara M, et al. Bradykinin potentiates insulin-stimulated glucose uptake and enhances insulin signal through the bradykinin B2 receptor in dog skeletal muscle and rat L6 myoblasts. Eur J Endocrinol. 1998;138:344–352. doi: 10.1530/eje.0.1380344. [DOI] [PubMed] [Google Scholar]
- 42.Rett K, Wicklmayr M, Dietze GJ, Haring HU. Insulin-induced glucose transporter (GLUT1 and GLUT4) translocation in cardiac muscle tissue is mimicked by bradykinin. Diabetes. 1996;45:S66–S69. doi: 10.2337/diab.45.1.s66. [DOI] [PubMed] [Google Scholar]
- 43.Roberts CK, Barnard RJ, Scheck SH, Balon TW. Exercise-stimulated glucose transport in skeletal muscle is nitric oxide dependent. Am J Physiol Endocrinol Metab. 1997;273:E220–E225. doi: 10.1152/ajpendo.1997.273.1.E220. [DOI] [PubMed] [Google Scholar]
- 44.Folli F, Saad MJ, Velloso LA, et al. Crosstalk between insulin and angiotensin II signalling systems. Exp Clin Endocrinol Diabetes. 1999;107:133–139. doi: 10.1055/s-0029-1212088. [DOI] [PubMed] [Google Scholar]
- 45.Rueckschloss U, Quinn MT, Holtz J, Morawietz H. Dose-dependent regulation of NADPH oxidase expression by angiotensin II in human endothelial cells: protective effect of angiotensin II type 1 receptor blockade in patients with coronary artery disease. Arterioscler Thromb Vasc Biol. 2002;22:1845–1851. doi: 10.1161/01.ATV.0000035392.38687.65. [DOI] [PubMed] [Google Scholar]
- 46.Dragovic T, Minshall R, Jackman HL, et al. Kininase II-type enzymes. Their putative role in muscle energy metabolism. Diabetes. 1996;45:S34–S37. doi: 10.2337/diab.45.1.s34. [DOI] [PubMed] [Google Scholar]
- 47.Muñoz MC, Giani JF, Dominici FP, et al. Long-term treatment with an angiotensin II receptor blocker decreases adipocyte size and improves insulin signaling in obese Zucker rats. J Hypertens. 2009;27:2409–2420. doi: 10.1097/HJH.0b013e3283310e1b. [DOI] [PubMed] [Google Scholar]
- 48.Rong X, Li Y, Ebihara K, et al.: Angiotensin II type 1 receptor-independent beneficial effects of telmisartan on dietary-induced obesity, insulin resistance and fatty liver in mice. Diabetologia 2010 Apr 15 [Epub ahead of print]. [DOI] [PubMed]
- 49.Scheen AJ. Renin-angiotensin system inhibition prevents type 2 diabetes mellitus. Part 2. Overview of physiological and biochemical mechanisms. Diabetes Metab. 2004;30:498–505. doi: 10.1016/S1262-3636(07)70147-7. [DOI] [PubMed] [Google Scholar]
- 50.Saitoh Y, Hongwei W, Ueno H, et al. Telmisartan attenuates fatty-acid-induced oxidative stress and NAD(P)H oxidase activity in pancreatic beta-cells. Diabetes Metab. 2009;35:392–397. doi: 10.1016/j.diabet.2009.04.005. [DOI] [PubMed] [Google Scholar]
- 51.Cole BK, Keller SR, Wu R, et al. Valsartan protects pancreatic islets and adipose tissue from the inflammatory and metabolic consequences of a high-fat diet in mice. Hypertension. 2010;55:715–721. doi: 10.1161/HYPERTENSIONAHA.109.148049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shao J, Iwashita N, Ikeda F, et al. Beneficial effects of candesartan, an angiotensin II type 1 receptor blocker, on beta-cell function and morphology in db/db mice. Biochem Biophys Res Commun. 2006;344:1224–1233. doi: 10.1016/j.bbrc.2006.04.011. [DOI] [PubMed] [Google Scholar]
- 53.DREAM Trial Investigators. Bosch J, Yusuf S, et al. Effect of ramipril on the incidence of diabetes. N Engl J Med. 2006;355:1551–1562. doi: 10.1056/NEJMoa065061. [DOI] [PubMed] [Google Scholar]
- 54.NAVIGATOR Study Group. McMurray JJ, Holman RR, et al. Effect of valsartan on the incidence of diabetes and cardiovascular events. N Engl J Med. 2010;362:1477–1490. doi: 10.1056/NEJMcp0909392. [DOI] [PubMed] [Google Scholar]
- 55.American Diabetes Association Standards of medical care in diabetes-2008. Diabetes Care. 2008;31(Suppl 1):S12–S54. doi: 10.2337/dc08-S012. [DOI] [PubMed] [Google Scholar]