

SUMMARY
BMK1 is activated by mitogens and oncogenic signals and, thus, is strongly implicated in tumorigenesis. We found that BMK1 interacted with promyelocytic leukemia protein (PML), and inhibited its tumor-suppressor function through phosphorylation. Furthermore, activated BMK1 notably inhibited PML-dependent activation of p21. To further investigate the BMK-mediated inhibition of the tumor suppressor activity of PML in tumor cells, we developed a small-molecule inhibitor of the kinase activity of BMK1, XMD8-92. Inhibition of BMK1 by XMD8-92 blocked tumor cell proliferation in vitro and significantly inhibited tumor growth in vivo by 95%, demonstrating the efficacy and tolerability of BMK1-targeted cancer treatment in animals.
INTRODUCTION
Four MAP kinase pathways exist in mammalian cells: ERK1/2, JNK, p38 and BMK1 (Chang and Karin, 2001; Johnson and Lapadat, 2002; Pearson et al., 2001; Raman et al., 2007). BMK1 is most similar to ERK1/2 since both contain the Thr-Glu-Tyr dual phosphorylation motif. However, unlike ERK1/2, BMK1 has a unique activating loop structure and an uncommonly large C-terminal non-kinase domain. The C-terminal half of BMK1 contains a nuclear localization signal (NLS) which is critical for nuclear localization of BMK1 (Lee et al., 1995). The ERK1/2 and BMK1 cascades are both activated by mitogens and by oncogenic signals and, thus, are strongly implicated in tumorigenesis (Chang and Karin, 2001; Johnson and Lapadat, 2002; Kato et al., 1997; Kato et al., 1998; Pearson et al., 2001). Specifically, deregulated BMK1 signaling has been associated with properties of human malignancies including chemoresistance of breast tumor cells (Weldon et al., 2002), uncontrolled proliferation of ErbB2-overexpressing carcinomas (Esparis-Ogando et al., 2002), metastatic potential of prostate tumor cells (Mehta et al., 2003), and tumor-associated angiogenesis (Hayashi et al., 2005).
Three sequentially activated kinases make up the central core of the MAP kinase module: a MAP kinase kinase kinase, or MEKK; a MAP kinase kinase, or MEK; and a MAP kinase. The signaling core in the BMK1 pathway consists of the kinases, MEKK2/MEKK3, MEK5 and BMK1 (Hayashi and Lee, 2004). MEK5 is the only known direct upstream regulatory kinase of BMK1 (Kato et al., 1997). However, MEK5 can be inhibited by inhibitors of MEK1/2 (Kamakura et al., 1999; Mody et al., 2001), such as PD98059 and U0126, which have been considered as specific inhibitors of the ERK1/2 pathway. As such, experimental results produced using these two inhibitors need to be re-evaluated using more specific inhibitors of the BMK1 and the ERK1/2 cascades. So far, there is no specific small-molecule inhibitor of BMK1 that is effective both in cells and animals. More importantly, the lack of this kind of BMK1 inhibitor has hampered the understanding of the physiological/pathological roles of BMK1 through cellular and animal studies.
RESULTS
Development of Pharmacological Inhibitors of BMK1
During the course of developing isoform-selective polo kinase inhibitors, we synthesized a library of analogs of the highly selective, ATP-competitive polo kinase inhibitor, BI-2536 (Steegmaier et al., 2007). By screening a subset of the library against a diverse panel of 402 kinases, we were able to explore the full range of potential kinase targets of this class of compounds (Goldstein et al., 2008). We discovered that expansion of the 6-membered aliphatic ring of BI-2536 to a 7-membered ring containing an anthranilic acid resulted in loss of polo kinase inhibition activity but serendipitously resulted in compounds that exhibited high selectivity towards BMK1. Structure-activity guided optimization based on the ability of the compounds to inhibit cellular BMK1 autophosphorylation stimulated by EGF (Kato et al., 1998) in conjunction with kinase selectivity analysis resulted in the synthesis of XMD8-92 (Figure 1A). The kinase selectivity of XMD8-92 was determined by profiling the inhibitor at a concentration of 10 μM against a panel of 402 diverse kinases using an in vitro ATP-site competition binding assay (Fabian et al., 2005; Karaman et al., 2008). Kinases that exhibited greater than 90% displacement by XMD8-92 were determined to be BMK1, DCAMKL1, DCAMKL2, TNK1 and PLK4. XMD8-92 exhibited the greatest affinity towards BMK1 with a measured dissociation constant (Kd) of 80 nM, while DCAMKL2, TNK1 and PLK4 exhibited Kd’s of 190, 890 and 600 nM, respectively (Table S1). This represents a remarkable level of selectivity considering the very large number of kinases that have been assayed. Moreover, XMD8-92 was profiled against all detectable kinases in HeLa cell lysates using the KiNativ method (Patricelli et al., 2007) and was found to be about 10-fold more selective for BMK1 with a IC50 of 1.5 μM than the most potent off-targets, TNK1 (IC50 = 10 μM) and ACK1 (aka TNK2, IC50 = 18 μM). Other weak off-targets included the kinase domain 2 of RSK1 and RSK2, PIK4A and PIK4B, and FAK (Table S2). Notably, MEK5 was not significantly inhibited by XMD8-92 at up to 50 μM. There is also no significant inhibitory effect of XMD8-92 on TNK1 and PLK4 detected in vitro and in vivo (Figure S1). The BMK1 potency and selectivity determined by the KiNativ method support the conclusion that the anti-cancer effects of XMD8-92 detailed herein are due to inhibition of BMK1.
Figure 1. Development of a Pharmacological Inhibitor of BMK1.

(A) Chemical structure of XMD8-92.
(B) HeLa cells were serum starved overnight followed by treatment with 1 or 5 μM XMD8-92 or 1 μM PD184352 as indicated for one hour. Cells were then stimulated with EGF for 17 min and BMK1 activation was detected by mobility retardation (Abe et al., 1996). ERK1/2 activation was detected by an anti-phospho-ERK1/2 (T202/Y204) antibody.
(C) HEK293 cells were co-transfected with expression plasmids of MEK5D and BMK1. After 48 hr, BMK1 was immunoprecipitated and in vitro kinase assays were performed in the presence of the indicated amount of XMD8-92. The ATP concentration was measured by the Kinase-Glo® Luminescent Kinase Assay Platform. Kinase activity is expressed relative to the kinase activity in cells without XMD8-92 treatment, which was taken as 1. n = 3, ± SEM.
(D) Expression plasmids of MEK5D and BMK1 were transfected into HEK293 as indicated. After 36 hr, these cells were co-transfected with vectors for pCMV-β and the reporter plasmid pG5ElbLuc along with a GAL4 fusion expression vector containing MEF2C for 3 hr and then treated with 5 μM XMD8-92 for a further 16 hr. The luciferase activities were normalized against cells transfected with pG5ElbLuc and pGAL4 reporter plasmid alone, whose value was taken as 1. n = 3, ± SEM, *p < 0.01.
(E) HeLa cells were infected with Ad-EV or Ad-BMK1(AEF), as indicated, 24 hr prior to treatment with or without XMD8-92 (5 μM) for 48 hr, followed by MTT assays. % growth inhibition = (1- MTT value of cells in each experimental group/MTT value of cells treated with Ad-EV only) × 100. n = 3, ± SEM.
(F) Immunofluorescent analysis of CD31 (red) and TUNEL (green) in heart sections from control and XMD8-92 treated. DNase treatments were used as positive controls for TUNEL staining. Scale bar, 100 μm. n = 6 mice, n = 10 slices. (see also Figure S1, Table S1, S2, S3, S4 and S5)
We tested the effect of XMD8-92 on the cellular activity of BMK1. Growth factor-induced activation of cellular BMK1, but not the structurally similar ERK1/2, was effectively blocked by 1 μM XMD8-92 (Figure 1B), while PD184352, an ERK1/2 inhibitor, only blocked ERK1/2 but not BMK1 activation by EGF (Figure 1B). XMD8-92 also significantly reduced BMK1 activity in in vitro kinase assays (Figure 1C). In addition, Trans-reporter assays showed that XMD8-92 dramatically reduced the BMK1-dependent transactivating activity of MEF2C, a known substrate for BMK1 (Kato et al., 1997) (Figure 1D).
As the BMK1 pathway is critical for cell proliferation (Kato et al., 1998), we next tested whether XMD8-92, through its specific ability to block BMK1 activity, has an anti-proliferative effect on cells. The extent of inhibition by XMD8-92 in HeLa cells is indistinguishable from that by dominant negative BMK1, BMK1(Ala-Glu-Phe [AEF]), administered through infection with recombinant adenovirus encoding BMK1(AEF), Ad-BMK1(AEF) (Figure 1E). The inhibitory effect of XMD8-92 on proliferation was observed in a wide variety of cancer cell lines (Figure S2). Moreover, additional treatment of Ad-BMK1(AEF)-infected HeLa cells with XMD8-92 did not have a further inhibitory effect when compared to cancer cells treated with XMD8-92 alone (Figure 1E), indicating that at least some of the anti-proliferative capacity of XMD8-92 is through inhibition of the activity of BMK1.
Subsequently, to assess the utility of XMD8-92 in animal experiments, we analyzed the pharmacokinetics and tolerability of XMD8-92. The pharmacokinetics of XMD8-92 was evaluated in Sprague-Dawley rats given a single intravenous or oral dose. XMD8-92 was found to have a 2.0 hr half life clearance of 26 mL/min/kg. The compound had moderate tissue distribution with a calculated volume of distribution of 3.4 L/kg. XMD8-92 had high oral bioavailability with 69% of the dose absorbed. After a single oral dose of 2 mg/kg, maximal plasma concentrations of approximately 500 nM were observed by 30 minutes, with 34 nM remaining 8 hr post dose. (Figure S1). In tolerability experiments (Table S3, S4 and S5), high plasma concentrations of drug (approximately 10 μM following IP dosing of 50 mg/kg) were maintained throughout the 14 days. The drug appeared to be well tolerated and the mice appeared healthy with no sign of distress. No vasculature instability was observed in the XMD8-92-treated mice (Table S4, S5 and Figure 1F). Together, these results demonstrated that XMD8-92 is an effective and specific inhibitor of BMK1 in vitro and in vivo.
BMK1 Is in Complex with PML and Suppresses Its Anti-Cancer Activity
To study the molecular mechanism by which BMK1 regulates cell proliferation, we performed mass spectrometry (MS) analysis of cellular BMK1-interacting proteins and revealed that BMK1 interacted with promyelocytic leukemia protein (PML) (Table S6). Western blot analysis using two different anti-PML antibodies showed that PML co-precipitated with BMK1 (Figure 2A) and that reciprocal immunoprecipitation of PML using anti-PML antibody brought down cellular BMK1 (Figure 2B). These data indicate that endogenous BMK1 is in complex with PML.
Figure 2. BMK1 is in Complex with PML and Suppresses the Anti-Cancer Function of PML in Tumor Cells.
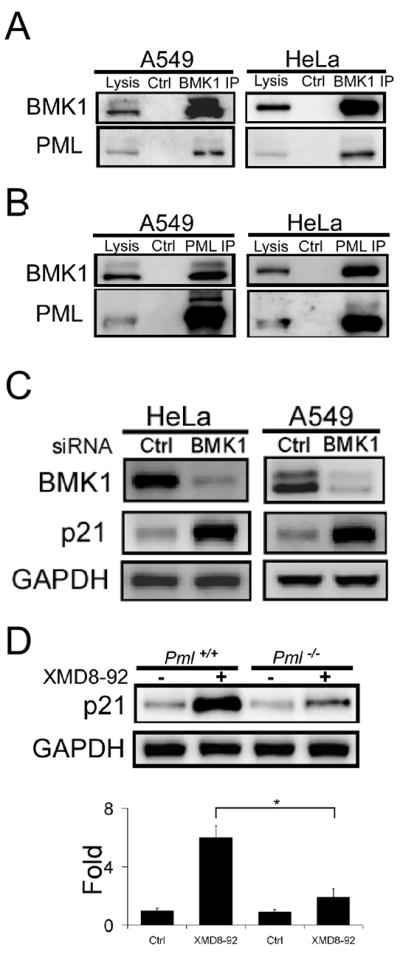
(A) Cellular BMK1 was immunoprecipitated from cell lysates from A549 and HeLa cells with anti-BMK1 antibody or rabbit IgG, as control. The resultant immunoprecipitates and total cell lysates were then tested for the presence of PML using anti-PML antibody.
(B) Endogenous PML was immunoprecipitated from cell lysates of A549 and HeLa cells with anti-PML antibody or rabbit IgG, as control. The existence of BMK1 in these immunoprecipitates was analyzed using western blots as described in (A).
(C) HeLa and A549 cells were transfected with control or BMK1-specific siRNA. Cell lysates were analyzed by western blot using anti-p21 or anti-GAPDH antibodies as noted. n = 3, ± SEM, *p < 0.01.
(D) Pml+/+ MEF and Pml−/− MEF cells were treated with or without XMD8-92 (5 μM) for 16 hours as indicated. Cell lysates were analyzed by western blot using anti-p21 antibody. The levels of p21 protein expression were quantified by densitometry and normalized against GAPDH. The amount of the p21 protein in the Pml+/+ MEF without XMD8-92 treatment was taken as 1. n = 5, ± SEM, *p < 0.01. (see also Figure S2 and Table S6)
As PML is a tumor suppressor and known to regulate cell proliferation, we next tested whether BMK1 controls the anti-tumor function of PML. We found that p21, one of the downstream effectors of PML and a key modulator of cell proliferation (Bernardi and Pandolfi, 2007; Salomoni and Pandolfi, 2002), was significantly induced when the expression level of BMK1 was suppressed by siRNA (Figure 2C). Inhibition of BMK1 activation by XMD8-92 also significantly induced p21 expression in cells (Figure 2D), and its induction was substantially reduced in PML null cells (Figure 2D). Using PML-knockdown cell lines, we also demonstrated that PML and p21 are involved in XMD8-92-mediated suppression of cancer cell proliferation (Figure S2). These results indicated that BMK1 not only is in complex with PML but also suppresses the expression of the cell-cycle regulator p21, through inhibiting the anti-tumor function of PML.
BMK1 Suppress PML Function through Phosphorylation
Since BMK1 is known to regulate the activity of cellular proteins through phosphorylation (Hayashi et al., 2001; Kato et al., 1997; Kato et al., 2000), we performed an in vitro kinase assay using activated BMK1 kinase and recombinant PML as substrate, followed by mass spectrometry analysis (details in Experimental Procedures) to map the potential phosphorylation sites on PML by BMK1. We identified that S403 and T409 of PML as potential phosphorylation sites (Figure 3A). Mutation of these two sites to Alanine significantly inhibited the phosphorylation of PML by BMK1 in an in vitro kinase assay (Figure 3B) indicating these two sites in PML are major phosphorylation sites by the BMK1 kinase. PML phosphorylation by BMK1 in vivo was also demonstrated (Figure S3). We next mutated S403 and T409 to Aspartic acid (PML-2D) to mimic the phosphorylation of PML by BMK1. We found that recombinant adenovirus-mediated expression of PML2D (S403D/T409D) in PML-deficient cells did not significantly restore their responsiveness to XMD8-92-dependent p21 induction unlike wild type PML, which did restore responsiveness (Figure 3C). As PML is known to modulate p21 through p53 (Guo et al., 2000), the role of BMK1 on p53 regulation is still not clear (Figure S4). These results indicate that BMK1 suppresses PML function through phosphorylation of S403 and T409 of PML (Figure 3C).
Figure 3. BMK1 Regulates PML Function through Phosphorylation.

(A) A schematic representation of wild-type and mutant PMLs. The amino acid sequences of potential BMK1 phosphorylation sites in PML are indicated.
(B) Mutation of potential BMK1 sites in full-length PML reduces phosphorylation by BMK1. Equal amounts of full-length wild-type or mutant recombinant PMLs were subjected to in vitro BMK1-mediated phosphorylation assay of PML in the presence of activated recombinant BMK1. The levels of relative [γ32P]-incorporation in PML or PML mutants were quantified by densitometry and normalized against the loading amount of PML or PML mutants in each reaction mix, respectively. The value of relative [γ32P]-incorporation of the wild-type PML by BMK1 was taken as 100%. n = 3, ± SEM, *p < 0.01.
(C) Pml−/− MEF cells were infected with Ad-EV, Ad-PML or Ad-PML2D as indicated. Three days later, these cells were treated with or without XMD8-92 (5 μM) for 16 hours as noted. The expression levels of p21 were quantified as described in Figure 2D except that the amount of the p21 protein in the Ad-EV-infected Pml−/− cells without XMD8-92 treatment was taken as 1. n = 3, ± SEM, *p < 0.01. (See also Figure S3)
Activated BMK1 Translocates from the Cytosol to Colocalize with PML in the Nucleus
It has been shown that, upon activation, BMK1 translocates from the cytoplasm to the nucleus (Kondoh et al., 2006; Yan et al., 2001). As PML is known to localize to the PML-nuclear body (PML-NB), through which it mediates diverse cellular functions, we wondered whether BMK1, after activation, could move to the nucleus and colocalize with PML in PML-NBs. We expressed a dominant negative form of MEK5, MEK5A (Kato et al., 1997), to inhibit the activation of BMK1 in cells (Figure 4A, first-row panels). Conversely, we expressed a dominant active form of MEK5, MEK5D, to keep BMK1 activated in cells (Figure 4A, second-row panels). With increased expression of PML in cells, activated BMK1 is colocalized with PML in the PML-NBs (Figure 4A, fourth-row panels) while non-activated BMK1 stays in the cytoplasm (Figure 4A, third-row panels). BMK1 contains a kinase domain in its N-terminal half and a non-kinase domain in its C-terminal half (Figure 4B). We next tested the involvement of these two domains in the colocalization of activated BMK1 and PML in PML-NBs. We demonstrate that the kinase region, but not the non-kinase region, of BMK1 is sufficient for the colocalization of BMK1 and PML in PML-NBs (Figure 4B). This result indicates that BMK1, upon activation, translocates from the cytosol to the nucleus and, through its kinase domain, colocalizes with PML in PML-NBs (Figure 5).
Figure 4. Activated BMK1 Translocates from the Cytosol to Colocalize with PML in the Nucleus.

(A) Fluorescent microscopy images of HeLa cells transfected with expression vectors encoding BMK1, PML, MEK5A or MEK5D, as indicated. These cells were stained with anti-BMK1 (green, first-column panels) or with anti-PML (PG-M3) antibody (red, second-column panels) as noted. Nuclei were shown by DAPI staining (blue, third-column panels). Merged images (yellow, fourth-column panels). n = 5. Scale bar, 10 μm.
(B) Fluorescent microscopy images of HeLa cells transfected with expression vectors encoding the N-terminal kinase domain of BMK1 (BMK1-KD), the C-terminal Non-Kinase domain of BMK1 (BMK1-NKD) and/or PML, as indicated. These cells were stained with anti-BMK1 (green, first-column panels) or with anti-PML (PG-M3) antibody (red, second-column panels) as noted. Nuclei were shown by DAPI staining (blue, third-column panels). Merged images (yellow, fourth-column panels). n = 5. Scale bar, 10 μm. (see also Figure S4)
Figure 5. Scheme for PML regulation by BMK1.

BMK1 Is a Potential Drug Target for Treating Cancer
To evaluate the effectiveness of XMD8-92 in inhibiting the activity of BMK1 in tumors, we tested the compound in mice xenografted with human tumors. XMD8-92 was found to effectively inhibit BMK1 activation as well as induce PML’s downstream effector, p21 (Figure 6A). More importantly, treatment of the mice with XMD8-92 one day, several days, or weeks after inoculation of the tumor cells all significantly inhibit the growth of the xenografted human or syngeneic mouse tumors (Figure 6B), without obvious side effects to the animals. Immuno-staining of tumor sections showed that XMD8-92 effectively inhibited the incorporation of BrdU during tumor cell division (Figure 6C) indicating XMD8-92 blocks tumor cell proliferation, one of the anti-cancer effects of PML. As both BMK1 and PML are involved in angiogenesis (Bernardi et al., 2006; Borden and Culjkovic, 2009; Hayashi et al., 2005; Hayashi et al., 2004), we tested whether XMD8-92 blocks angiogenesis in vivo and found that XMD8-92 significantly inhibits basic fibroblast growth factor (bFGF) induced angiogenesis in Matrigel plugs (Figure 6D). These results indicated that XMD8-92 exerts its anti-tumor effect by blocking tumor cell proliferation and tumor-associated angiogenesis, and, possibly, through other BMK1-dependent mechanisms yet to be tested or discovered.
Figure 6. BMK1 is a Potential Drug Target for Treating Cancer.

(A) A549 cells were injected subcutaneously into the flanks of Nod/Scid mice. After three weeks, these tumor-bearing mice were injected intraperitoneally with XMD8-92 or vehicle for two days. The XMD8-92 concentration in these tumors were analyzed and shown in the supplemental information (See also Figure S5). The tumor homogenates were used to detect both BMK1 activity and p21 expression using western blot as described in Figure 2D.
(B) Mouse xenograft models were established as described in Experimental Procedures. (Top panels) Starting one day or at the indicated time after injection of the human (HeLa) or mouse (LL/2) tumor cells, XMD8-92 was administered twice daily and the tumor growth in these mice was measured every two or three days. (Lower panels) Representative mice from each group (control mice on the left) showing the growth of xenograft tumors at the end of an experiment. n = 6 mice, ± SEM, *p < 0.01.
(C) Fluorescence microscopy images of HeLa or LL/2 tumor sections from XMD8-92 treated or control mice as indicated. The sections were stained with anti-BrdU antibody or DAPI as noted. n = 6 mice, n = 9 slices. Scale bar, 10 μm.
(D) Mice were implanted with Matrigel containing 400 ng/mL bFGF. The next day, mice were treated with XMD8-92 or vehicle for 6 days. The Matrigel plugs were then harvested, sectioned and stained with Masson’s trichrome (n = 6 mice, n = 9 slices.). Hemoglobin content in the Matrigel plugs was assessed as described (Hayashi et al., 2005). n = 6 mice, ± SEM, *p < 0.01. Scale bar, 100 μm.
We further examined whether the anti-tumor effect of XMD8-92 is specific to its anti-BMK1 capacity. The extent of tumor growth inhibition by XMD8-92 is identical to that elicited using dominant negative BMK1 administered through intratumoral injection of Ad-BMK1(AEF) (Figure 7A). Importantly, the additional treatment of Ad-BMK1(AEF) did not have a further inhibitory effect on the tumor-bearing mice treated with XMD8-92 alone (Figure 7A) indicating the anti-tumor capacity of XMD8-92 is, at least partly, through blocking the activity of BMK1.
Figure 7. Pharmacological inhibition of BMK1 suppresses tumor growth through PML.
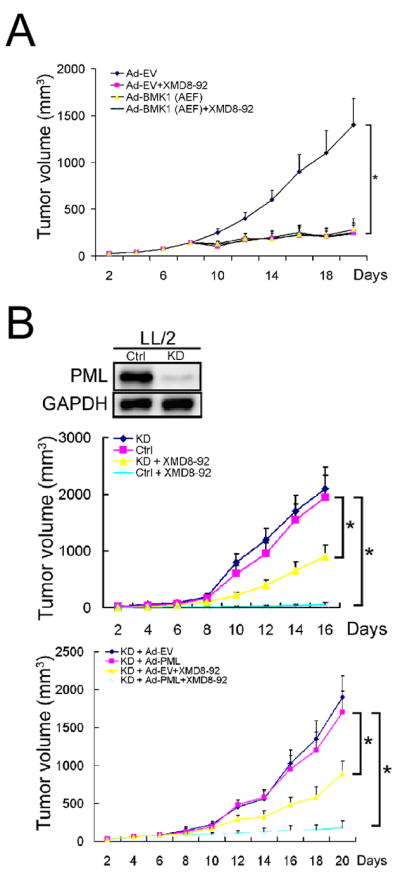
(A) Mouse tumor model with syngeneic LL/2 carcinoma was established as described in Experimental Procedures. Starting seven days after injection of LL/2 tumor cells into mice, the mice were treated with intratumoral injection of recombinant adenovirus encoding BMK1(AEF) [Ad-BMK1(AEF)] or empty vector control (Ad-EV) as indicated. One day after starting the injection of recombinant adenoviruses, XMD8-92 was administered to the indicated mice. n = 6 mice, ± SEM, *p < 0.01.
(B) Mouse tumor models with PML-knockdown (PML-KD) and control LL/2 carcinoma (Ctrl) were established (middle panel) similar to syngeneic LL/2 tumor model. Starting seven days after injection of the LL/2 tumor cells, the mice were treated with XMD8-92 or vehicle for another nine days as described in Figure 7A. (Bottom panel) Mice bearing PML-KD LL/2 tumors for seven days were injected with recombinant adenovirus encoding wild type PML [Ad-PML], mutant PML [Ad-PML2D] or empty vector control [Ad-EV] intratumorally. One day after starting the injection of recombinant adenoviruses, XMD8-92 or vehicle was administered to the indicated mice. n = 6 mice, ± SEM, *p < 0.01.
We next evaluate the role of PML in XMD8-92-dependent inhibition of tumor growth. We found that depleting PML in tumor cells significantly lowered, but did not completely abrogate, the inhibitory effect of XMD8-92 on their growth in mice (Figure 7B). As XMD8-92 inhibits tumor cell proliferation and neovascularization (Figure 6C and 6D), the reason why PML depletion in tumor cells can only partly reverse the anti-tumor effect of XMD8-92 may be that PML in tumor cells is responsible only for mediating XMD8-92-mediated growth arrest of cancer cells and not for XMD8-92-dependent angiogenesis inhibition. Moreover, increasing the expression level of wild type PML in these PML-knockdown (KD) tumor cells restored their sensitivity to XMD8-92-mediated inhibition of tumor growth (Figure 7B). In contrast, expression of PML2D in the PML-KD cells did not re-establish their responsiveness to XMD8-92-mediated suppression of tumor proliferation (Figure 7B). These data indicate that PML plays a role in XMD8-92-mediated suppression of tumor growth.
Taken together, these results demonstrated that the BMK1 pathway in animals can be blocked effectively by a small-molecule inhibitor without apparent adverse effects and, more importantly, BMK1 inhibition is a very effective way to prevent cancer development in animals.
DISCUSSION
The promyelocytic leukemia protein PML (also called MYL, RNF71, PP8675 or TRIM19) has been implicated in the regulation of a range of cellular functions, such as inhibition of proliferation, induction of cellular senescence and apoptosis, as well as maintenance of genomic stability (Bernardi and Pandolfi, 2007). It is well known that compromising PML anti-cancer function by gene translocation (to produce the PML-RAR fusion protein) leads to acute promyelocytic leukemia. PML-RAR fusion protein not only represses PML function but also represses transcriptional activity mediated by RAR-RXR, thereby disrupting retinoid signaling (Altucci and Gronemeyer, 2001). Although the physiological roles of PML are not yet fully understood, the tumor-suppressive function of PML is generally recognized (Bernardi et al., 2006; Salomoni and Pandolfi, 2002; Trotman et al., 2006). One of the critical anti-cancer/anti-proliferative functions of PML is through activation of the tumor suppressor, p21, through transcriptional regulation of p53 (Bernardi and Pandolfi, 2007; Bernardi et al., 2004; Fogal et al., 2000; Guo et al., 2000; Pearson et al., 2000; Pearson and Pelicci, 2001). Previous reports have described the regulation of PML through phosphorylation by kinases such as ERK1/2 and CK2 (Hayakawa and Privalsky, 2004; Scaglioni et al., 2006). Herein, we report that the mitogenic MAP kinase, BMK1, interacts with PML and suppresses its anti-tumor actions such as p21 activation. However, the effect of BMK1 on p53 regulation is not clear (Figure S4) and needs further investigation. Since BMK1 negatively regulates PML tumor suppressor function and BMK1 is activated by a variety of deregulated oncogenes (e.g., Her2, Ras and STAT3), it is likely that the oncogene-induced BMK1 activation increases the proliferation/survival/chemo-resistance potential of tumor cells by dampening the anti-cancer capacity of PML.
BMK1 promotes tumor development not only by inhibiting the PML tumor suppressor described herein but also by supporting tumor angiogenesis (Hayashi et al., 2005; Hayashi and Lee, 2004; Pi et al., 2005), tumor metastasis (Sawhney et al., 2009; Sticht et al., 2008; Zhou et al., 2008) and chemo-resistance of tumor cells (Weldon et al., 2002). In addition, no significant feed-back compensatory activation of any other signaling pathway was detected, so far, by blocking the BMK1 pathway using XMD8-92 (Figure S4). Conditional knockout of BMK1 in various tissues of mice (e.g., in cardiomyocytes, neurons, hepatocytes, and T and B cells) has no obvious effect on the development, behavior, reproduction and aging of the animals (Hayashi and Lee, 2004), suggesting that BMK1 should be an attractive target for pharmaceutical intervention in cancer therapy. However, the instability of the vasculature observed in mice depleted of endothelial BMK1 (Hayashi et al., 2004) has discouraged the therapeutic development of a BMK1 inhibitor. Compared to BMK1 knockout, which is much more severe as it leads to complete and irreversible annihilation of BMK1 gene product in the tissues targeted, pharmaceutical inhibition of BMK1 only targets the kinase activity of the enzyme. Therefore, we believe that applying the BMK1 inhibitor in animals should have no or little unfavorable effect on vascular integrity. Indeed, we have demonstrated that XMD8-92 is a very effective ATP-competitive inhibitor that inhibits BMK1 activity in animals and reduces tumor growth by 95% without inducing vasculature abnormalities (Table S4, S5 and Figure 1F). These results strengthen the notion that blocking the BMK1 pathway may be an effective approach for treating human cancer.
EXPERIMENTAL PROCEDURES
KiNativ Profiling Method
KiNativ profiling of XMD8-92 was carried out with both an ATP and ADP acylphosphate-desthiobiotin as described previously (Patricelli et al., 2007) with the following modifications. HeLa cell lysates (5 mg/mL total protein) were incubated in the presence of XMD8-92 at 50 μM, 10 μM, 2 μM, 0.8 μM, and 0 μM for 15 minutes prior to addition of the ATP or ADP acylphosphate probe (5 μM final probe concentration). All reactions were performed in duplicate. Probe reactions proceeded for 10 minutes and the reaction stopped by the addition of urea and processed for MS analysis as described (Patricelli et al., 2007). Samples were analyzed by LC-MS/MS on a linear ion trap mass spectrometer using a time segmented “target list” designed to collect MS/MS spectra from all kinase peptide-probe conjugates that can be detected in HeLa cell lysates. This target list was generated and validated by prior exhaustive analysis of HeLa lysates. Up to four characteristic fragment ions for each kinase peptide-probe conjugate were used to extract signals for each kinase, and a comparison of inhibitor treated to control (untreated) lysates allowed for precise determination of % inhibition at each point. A manuscript describing the details of this targeted mass spectrometry approach is in preparation.
Cell Culture, Stable Isotope Labeling with Amino Acid in Cell Culture (SILAC)-Labeling and Sample Preparation for Mass Spectrometry
Gibco SILAC DMEM basal cell culture medium (Invitrogen, Carlsbad, CA) containing 2 mM L-Glutamine, 10% dialyzed fetal bovine serum (FBS) (Invitrogen, Carlsbad, CA) and 100 U/ml penicillin and streptomycin was supplemented with 100 mg/L L-Lysine and 20 mg/L L-Arginine or 100 mg/L [U-13C6]-L-Lysine and 20 mg/L [U-13C6, 15N4]-L-Arginine (Invitrogen, Carlsbad, CA) to make the “Light” SILAC or “Heavy” SILAC culture media, respectively. HeLa cells were obtained from ATCC and were propagated in SILAC medium for more than nine generations to ensure nearly 100% incorporation of labeled amino acids before the experiment was performed. After being washed with precooled PBS buffer, HeLa cells were lysed in E1A (250 mM NaCl, 50 mM HEPES (pH 7.5), 0.1% NP40, 5 mM EDTA, protease inhibitor cocktail (Roche, Indianapolis, IN) and phosphatase inhibitor cocktail (Roche, Indianapolis, IN)) or RIPA (1X PBS, 1% NP40, 0.5% sodium deoxycholate, 0.1% SDS, 0.1 mg/ml PMSF, 1mM sodium orthovanadate, protease inhibitor cocktail (Roche, Indianapolis, IN) and phosphatase inhibitor cocktail (Roche, Indianapolis, IN)) lysis buffer respectively. “Light” cell lysate and “Heavy” lysate were mixed at a 1:1 ratio, and the mixed lysates were incubated with the precipitating antibody for 8 hr, followed by 16 hr incubation with protein G beads (Invitrogen, Carlsbad, CA). Immune complexes were washed three times in lysis buffer (E1A or RIPA respectively) and once in sterile water, and then incubated in 8 M urea for 30 min at room temperature. The supernatant was reduced with DTT, and then alkylated with iodoacetamide. The resulting samples from E1A or RIPA lysis buffer were dialyzed against 2 M urea/100 mM NH4HCO3 at 37°C for 5 hr and then analyzed by Multidimensional Liquid Chromatography Coupled with Tandem Mass Spectrometry (MudPIT) twice. Since the existence of the stable (heavy) isotope in nature is less than 1%, any “light” peptide detected by MudPIT with no similar amount of “heavy” peptide detected was considered as a contaminant.
In Vivo Tumorigenesis Experiments
The following animal handling and procedures were approved by the Scripps Institutional Animal Care and Use Committee and followed the National Institutes of Health guidelines.
HeLa Xenograft Model
5 × 105 HeLa cells were resuspended in DMEM and injected subcutaneously into the right flank of 6-week-old Nod/Scid mice (day 0). On the second day (day 1) after tumor cell injection, mice were randomized into 2 groups {6 animals [XMD8-92 (1–28 day)] and 18 animals [control]}. The XMD8-92 (1–28 day) group was treated with XMD8-92 at the dose of 50 mg/kg twice a day intraperitoneally. The control group received daily injections of the carrier solution as control. On the day 7, the control group was randomized into 2 groups {6 animals [XMD8-92 (7–28 day)] and 12 animals [control]}. And on the day 14, the remaining control group was randomized into 2 groups {6 animals [XMD8-92 (14–28 day)] and 6 animals [control]}. Treatment with XMD8-92 in XMD8-92 (7–28 day) and XMD8-92 (14–28 day) groups was initiated on day 7 and day 14, respectively. Tumor size was measured using a caliper, and tumor volume was determined by using the formula: L × W2 × 0.52, where L is the longest diameter and W is the shortest diameter.
LL/2 Xenograft Model
1 × 106 LL/2 cells were resuspended in DMEM and injected subcutaneously into the right flank of 6-week-old C57Bl/6 mice (day 0). On the second day (day 1) after tumor cell injection, mice were randomized into 2 groups {6 animals [XMD8-92 (1–17 day)] and 18 animals [control]}. The XMD8-92 (1–17 day) group was treated with XMD8-92 at the dose of 50 mg/kg twice a day intraperitoneally. The control group received daily injections of the carrier solution as control. On the day 7, the control group was randomized into 2 groups {6 animals [XMD8-92 (7–17 day)] and 12 animals [control]}. And on the day 14, the remaining control group was randomized into 2 groups {6 animals [XMD8-92 (10–17 day)] and 6 animals [control]}. Treatment with XMD8-92 in XMD8-92 (7–17 day) and XMD8-92 (10–17 day) groups was initiated on day 7 and day 10, respectively.
Recombinant Adenovirus Treatment
1 × 106 LL/2 cells were injected subcutaneously into C57Bl/6 mice (day 0). Recombinant adenoviral particles were generated as described in our previous study (Hayashi et al., 2001). On the day 7, mice were randomized into 4 groups [Ad-EV, Ad-BMK1(AEF), Ad-EV+XMD8-92 and Ad-BMK1(AEF) +XMD8-92] (6 animals per group). Mice were injected intratumorally with either empty adenovirus (Ad-EV) or recombinant adenovirus encoding BMK1(AEF) [Ad-BMK1(AEF)] on day 7, day 11, day 15, day 19, using the procedure previously described (Kim et al., 2004). Ad-EV+XMD8-92 and Ad-BMK1 (AEF)+XMD8-92 groups were treated with XMD8-92 from day 8 to day 20 using the procedure described in the LL/2 xenograft model. The other two groups [Ad-EV and Ad-BMK1(AEF)] received injections of the carrier solution instead as control.
PML Reconstitution
PML shRNAi knockdown and control cell lines were built using pGIPZ-shRNAmir-PML (Lentiviral) and pGIPZ (Lentiviral) plasmids from Openbiosystems (Huntsville, AL). 1 × 106 knockdown (48 animals) or control (12 animals) LL/2 cells were injected subcutaneously into C57Bl/6 mice (day 0). On the second day (day 1) after tumor cell injection, mice were randomized into 10 groups (6 animals per group) [KD, Ctrl, KD+XMD8-92 and Ctrl+XMD8-92; KD+Ad-EV, KD+Ad-PML, KD+Ad-PML2D(S403D/T409D), KD+Ad-EV+XMD8-92, KD+Ad-PML+XMD8-92 and KD+Ad-PML2D(S403D/T409D)+XMD8-92]. The KD+XMD8-92 and Ctrl+XMD8-92 groups were treated with XMD8-92 at the dose of 50 mg/kg twice a day intraperitoneally for 16 days. The KD and Ctrl groups received daily injections of the carrier solution as control for 16 days. The KD+Ad-EV, KD+Ad-PML, KD+Ad-PML2D, KD+Ad-EV+XMD8-92, KD+Ad-PML+XMD8-92 and KD+Ad-PML2D+XMD8-92 groups were injected intratumorally with either empty adenovirus (Ad-EV), Ad-PML or Ad-PML2D recombinant adenovirus encoding PML or PML2D on day 7, day 11, day 15, day 19, using the procedure previously described (Kim et al., 2004). KD+Ad-EV+XMD8-92, KD+Ad-PML+XMD8-92 and KD+Ad-PML2D+XMD8-92 groups were treated with XMD8-92 from day 8 to day 20 using the procedure described in the LL/2 xenograft model. The KD+Ad-EV and KD+Ad-PML and KD+Ad-PML2D groups received injections of the carrier solution instead as control.
A549 Xenograft Model
To ascertain that XMD8-92 can block BMK1 in vivo, 1 × 106 A549 cells, whose endogenous BMK1 autophosphorylation was detectable by western blotting, were resuspended in DMEM and injected subcutaneously into the right flank of 6-week-old Nod/Scid mice. On the 21st day after the injection, mice were randomized into 2 groups (2 animals per group). One group was treated with XMD8-92 at the dose of 50 mg/kg twice a day. The other group was treated with the carrier solution as control. After 2 days, the A549 tumor was homogenized in E1A buffer followed by western blot analysis.
The effectiveness of BMK1 inhibition in xenograft tumors was shown in the supplemental information (Figure S5).
Statistical Analysis
p values were calculated with the Student’s t test.
Significance.
The BMK1/ERK5 pathway is the last discovered and the least studied mammalian MAP kinase cascade. Herein, we describe the development of the small-molecule inhibitor for BMK1 that is effective not only in cells but also in animals. Using this inhibitor, we determined that BMK1 inhibits the tumor suppressor activity of cellular PML, and more importantly, we demonstrated the efficacy and tolerability of BMK1-targeted cancer treatment in animals. As BMK1 is expressed in most, if not all, tumor cells, our results suggest that cancer therapies targeting BMK1 will have broad application for treating diverse types of human tumors.
HIGHLIGHTS.
Development of a potent inhibitor, XMD8-92, for BMK1 kinase.
XMD8-92 significantly inhibits tumor growth in animal.
BMK1 inhibits p21 expression through PML.
Supplementary Material
Acknowledgments
We thank Dr. Giovanni Blandino, Dr. Myung Kim and Dr. Pier Paolo Pandolfi for the PML null and control cell lines. We also thank Ambit Biosciences for performing the KinomeScan kinase selectivity profiling. This work was supported by grants from the NIH, CA079871 and CA114059, (to J.-D.L) and by funds from the Tobacco-Related Disease, Research Program of the University of California, 15RT-0104, (to J.-D.L.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abe J, Kusuhara M, Ulevitch RJ, Berk BC, Lee JD. Big mitogen-activated protein kinase 1 (BMK1) is a redox-sensitive kinase. J Biol Chem. 1996;271:16586–16590. doi: 10.1074/jbc.271.28.16586. [DOI] [PubMed] [Google Scholar]
- Altucci L, Gronemeyer H. The promise of retinoids to fight against cancer. Nat Rev Cancer. 2001;1:181–193. doi: 10.1038/35106036. [DOI] [PubMed] [Google Scholar]
- Bernardi R, Guernah I, Jin D, Grisendi S, Alimonti A, Teruya-Feldstein J, Cordon-Cardo C, Simon MC, Rafii S, Pandolfi PP. PML inhibits HIF-1alpha translation and neoangiogenesis through repression of mTOR. Nature. 2006;442:779–785. doi: 10.1038/nature05029. [DOI] [PubMed] [Google Scholar]
- Bernardi R, Pandolfi PP. Structure, dynamics and functions of promyelocytic leukaemia nuclear bodies. Nat Rev Mol Cell Biol. 2007;8:1006–1016. doi: 10.1038/nrm2277. [DOI] [PubMed] [Google Scholar]
- Bernardi R, Scaglioni PP, Bergmann S, Horn HF, Vousden KH, Pandolfi PP. PML regulates p53 stability by sequestering Mdm2 to the nucleolus. Nat Cell Biol. 2004;6:665–672. doi: 10.1038/ncb1147. [DOI] [PubMed] [Google Scholar]
- Borden KL, Culjkovic B. Perspectives in PML: a unifying framework for PML function. Front Biosci. 2009;14:497–509. doi: 10.2741/3258. [DOI] [PubMed] [Google Scholar]
- Chang L, Karin M. Mammalian MAP kinase signalling cascades. Nature. 2001;410:37–40. doi: 10.1038/35065000. [DOI] [PubMed] [Google Scholar]
- Esparis-Ogando A, Diaz-Rodriguez E, Montero JC, Yuste L, Crespo P, Pandiella A. Erk5 participates in neuregulin signal transduction and is constitutively active in breast cancer cells overexpressing ErbB2. Mol Cell Biol. 2002;22:270–285. doi: 10.1128/MCB.22.1.270-285.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabian MA, Biggs WH, 3rd, Treiber DK, Atteridge CE, Azimioara MD, Benedetti MG, Carter TA, Ciceri P, Edeen PT, Floyd M, et al. A small molecule-kinase interaction map for clinical kinase inhibitors. Nat Biotechnol. 2005;23:329–336. doi: 10.1038/nbt1068. [DOI] [PubMed] [Google Scholar]
- Fogal V, Gostissa M, Sandy P, Zacchi P, Sternsdorf T, Jensen K, Pandolfi PP, Will H, Schneider C, Del Sal G. Regulation of p53 activity in nuclear bodies by a specific PML isoform. Embo J. 2000;19:6185–6195. doi: 10.1093/emboj/19.22.6185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein DM, Gray NS, Zarrinkar PP. High-throughput kinase profiling as a platform for drug discovery. Nat Rev Drug Discov. 2008;7:391–397. doi: 10.1038/nrd2541. [DOI] [PubMed] [Google Scholar]
- Guo A, Salomoni P, Luo J, Shih A, Zhong S, Gu W, Pandolfi PP. The function of PML in p53-dependent apoptosis. Nat Cell Biol. 2000;2:730–736. doi: 10.1038/35036365. [DOI] [PubMed] [Google Scholar]
- Hayakawa F, Privalsky ML. Phosphorylation of PML by mitogen-activated protein kinases plays a key role in arsenic trioxide-mediated apoptosis. Cancer Cell. 2004;5:389–401. doi: 10.1016/s1535-6108(04)00082-0. [DOI] [PubMed] [Google Scholar]
- Hayashi M, Fearns C, Eliceiri B, Yang Y, Lee JD. Big mitogen-activated protein kinase 1/extracellular signal-regulated kinase 5 signaling pathway is essential for tumor-associated angiogenesis. Cancer Res. 2005;65:7699–7706. doi: 10.1158/0008-5472.CAN-04-4540. [DOI] [PubMed] [Google Scholar]
- Hayashi M, Kim SW, Imanaka-Yoshida K, Yoshida T, Abel ED, Eliceiri B, Yang Y, Ulevitch RJ, Lee JD. Targeted deletion of BMK1/ERK5 in adult mice perturbs vascular integrity and leads to endothelial failure. J Clin Invest. 2004;113:1138–1148. doi: 10.1172/JCI19890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi M, Lee JD. Role of the BMK1/ERK5 signaling pathway: lessons from knockout mice. J Mol Med. 2004;82:800–808. doi: 10.1007/s00109-004-0602-8. [DOI] [PubMed] [Google Scholar]
- Hayashi M, Tapping RI, Chao TH, Lo JF, King CC, Yang Y, Lee JD. BMK1 mediates growth factor-induced cell proliferation through direct cellular activation of serum and glucocorticoid-inducible kinase. J Biol Chem. 2001;276:8631–8634. doi: 10.1074/jbc.C000838200. [DOI] [PubMed] [Google Scholar]
- Johnson GL, Lapadat R. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science. 2002;298:1911–1912. doi: 10.1126/science.1072682. [DOI] [PubMed] [Google Scholar]
- Kamakura S, Moriguchi T, Nishida E. Activation of the protein kinase ERK5/BMK1 by receptor tyrosine kinases. Identification and characterization of a signaling pathway to the nucleus. J Biol Chem. 1999;274:26563–26571. doi: 10.1074/jbc.274.37.26563. [DOI] [PubMed] [Google Scholar]
- Karaman MW, Herrgard S, Treiber DK, Gallant P, Atteridge CE, Campbell BT, Chan KW, Ciceri P, Davis MI, Edeen PT, et al. A quantitative analysis of kinase inhibitor selectivity. Nat Biotechnol. 2008;26:127–132. doi: 10.1038/nbt1358. [DOI] [PubMed] [Google Scholar]
- Kato Y, Kravchenko VV, Tapping RI, Han J, Ulevitch RJ, Lee JD. BMK1/ERK5 regulates serum-induced early gene expression through transcription factor MEF2C. Embo J. 1997;16:7054–7066. doi: 10.1093/emboj/16.23.7054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato Y, Tapping RI, Huang S, Watson MH, Ulevitch RJ, Lee JD. Bmk1/Erk5 is required for cell proliferation induced by epidermal growth factor. Nature. 1998;395:713–716. doi: 10.1038/27234. [DOI] [PubMed] [Google Scholar]
- Kato Y, Zhao M, Morikawa A, Sugiyama T, Chakravortty D, Koide N, Yoshida T, Tapping RI, Yang Y, Yokochi T, Lee JD. Big mitogen-activated kinase regulates multiple members of the MEF2 protein family. J Biol Chem. 2000;275:18534–18540. doi: 10.1074/jbc.M001573200. [DOI] [PubMed] [Google Scholar]
- Kim SW, Chao TH, Xiang R, Lo JF, Campbell MJ, Fearns C, Lee JD. Tid1, the human homologue of a Drosophila tumor suppressor, reduces the malignant activity of ErbB-2 in carcinoma cells. Cancer Res. 2004;64:7732–7739. doi: 10.1158/0008-5472.CAN-04-1323. [DOI] [PubMed] [Google Scholar]
- Kondoh K, Terasawa K, Morimoto H, Nishida E. Regulation of nuclear translocation of extracellular signal-regulated kinase 5 by active nuclear import and export mechanisms. Mol Cell Biol. 2006;26:1679–1690. doi: 10.1128/MCB.26.5.1679-1690.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JD, Ulevitch RJ, Han J. Primary structure of BMK1: a new mammalian map kinase. Biochem Biophys Res Commun. 1995;213:715–724. doi: 10.1006/bbrc.1995.2189. [DOI] [PubMed] [Google Scholar]
- Mehta PB, Jenkins BL, McCarthy L, Thilak L, Robson CN, Neal DE, Leung HY. MEK5 overexpression is associated with metastatic prostate cancer, and stimulates proliferation, MMP-9 expression and invasion. Oncogene. 2003;22:1381–1389. doi: 10.1038/sj.onc.1206154. [DOI] [PubMed] [Google Scholar]
- Mody N, Leitch J, Armstrong C, Dixon J, Cohen P. Effects of MAP kinase cascade inhibitors on the MKK5/ERK5 pathway. FEBS Lett. 2001;502:21–24. doi: 10.1016/s0014-5793(01)02651-5. [DOI] [PubMed] [Google Scholar]
- Patricelli MP, Szardenings AK, Liyanage M, Nomanbhoy TK, Wu M, Weissig H, Aban A, Chun D, Tanner S, Kozarich JW. Functional interrogation of the kinome using nucleotide acyl phosphates. Biochemistry. 2007;46:350–358. doi: 10.1021/bi062142x. [DOI] [PubMed] [Google Scholar]
- Pearson G, Robinson F, Beers Gibson T, Xu BE, Karandikar M, Berman K, Cobb MH. Mitogen-activated protein (MAP) kinase pathways: regulation and physiological functions. Endocr Rev. 2001;22:153–183. doi: 10.1210/edrv.22.2.0428. [DOI] [PubMed] [Google Scholar]
- Pearson M, Carbone R, Sebastiani C, Cioce M, Fagioli M, Saito S, Higashimoto Y, Appella E, Minucci S, Pandolfi PP, Pelicci PG. PML regulates p53 acetylation and premature senescence induced by oncogenic Ras. Nature. 2000;406:207–210. doi: 10.1038/35018127. [DOI] [PubMed] [Google Scholar]
- Pearson M, Pelicci PG. PML interaction with p53 and its role in apoptosis and replicative senescence. Oncogene. 2001;20:7250–7256. doi: 10.1038/sj.onc.1204856. [DOI] [PubMed] [Google Scholar]
- Pi X, Garin G, Xie L, Zheng Q, Wei H, Abe J, Yan C, Berk BC. BMK1/ERK5 is a novel regulator of angiogenesis by destabilizing hypoxia inducible factor 1alpha. Circ Res. 2005;96:1145–1151. doi: 10.1161/01.RES.0000168802.43528.e1. [DOI] [PubMed] [Google Scholar]
- Raman M, Chen W, Cobb MH. Differential regulation and properties of MAPKs. Oncogene. 2007;26:3100–3112. doi: 10.1038/sj.onc.1210392. [DOI] [PubMed] [Google Scholar]
- Salomoni P, Pandolfi PP. The role of PML in tumor suppression. Cell. 2002;108:165–170. doi: 10.1016/s0092-8674(02)00626-8. [DOI] [PubMed] [Google Scholar]
- Sawhney RS, Liu W, Brattain MG. A novel role of ERK5 in integrin-mediated cell adhesion and motility in cancer cells via Fak signaling. J Cell Physiol. 2009;219:152–161. doi: 10.1002/jcp.21662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scaglioni PP, Yung TM, Cai LF, Erdjument-Bromage H, Kaufman AJ, Singh B, Teruya-Feldstein J, Tempst P, Pandolfi PP. A CK2-dependent mechanism for degradation of the PML tumor suppressor. Cell. 2006;126:269–283. doi: 10.1016/j.cell.2006.05.041. [DOI] [PubMed] [Google Scholar]
- Steegmaier M, Hoffmann M, Baum A, Lenart P, Petronczki M, Krssak M, Gurtler U, Garin-Chesa P, Lieb S, Quant J, et al. BI 2536, a potent and selective inhibitor of polo-like kinase 1, inhibits tumor growth in vivo. Curr Biol. 2007;17:316–322. doi: 10.1016/j.cub.2006.12.037. [DOI] [PubMed] [Google Scholar]
- Sticht C, Freier K, Knopfle K, Flechtenmacher C, Pungs S, Hofele C, Hahn M, Joos S, Lichter P. Activation of MAP kinase signaling through ERK5 but not ERK1 expression is associated with lymph node metastases in oral squamous cell carcinoma (OSCC) Neoplasia. 2008;10:462–470. doi: 10.1593/neo.08164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trotman LC, Alimonti A, Scaglioni PP, Koutcher JA, Cordon-Cardo C, Pandolfi PP. Identification of a tumour suppressor network opposing nuclear Akt function. Nature. 2006;441:523–527. doi: 10.1038/nature04809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weldon CB, Scandurro AB, Rolfe KW, Clayton JL, Elliott S, Butler NN, Melnik LI, Alam J, McLachlan JA, Jaffe BM, et al. Identification of mitogen-activated protein kinase kinase as a chemoresistant pathway in MCF-7 cells by using gene expression microarray. Surgery. 2002;132:293–301. doi: 10.1067/msy.2002.125389. [DOI] [PubMed] [Google Scholar]
- Yan C, Luo H, Lee JD, Abe J, Berk BC. Molecular cloning of mouse ERK5/BMK1 splice variants and characterization of ERK5 functional domains. J Biol Chem. 2001;276:10870–10878. doi: 10.1074/jbc.M009286200. [DOI] [PubMed] [Google Scholar]
- Zhou C, Nitschke AM, Xiong W, Zhang Q, Tang Y, Bloch M, Elliott S, Zhu Y, Bazzone L, Yu D, et al. Proteomic analysis of tumor necrosis factor-alpha resistant human breast cancer cells reveals a MEK5/Erk5-mediated epithelial-mesenchymal transition phenotype. Breast Cancer Res. 2008;10:R105. doi: 10.1186/bcr2210. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.