

Abstract
We report the synthesis, structure, and pairing properties in DNA of an isostere for deoxyadenosine which lacks all hydrogen-bonding functionality on the Watson–Crick pairing edge. A deoxyribo-nucleoside derivative of 4-methylbenzimidazole (1), which was recently shown to be inserted into DNA by Klenow DNA polymerase (Morales, J. C.; Kool, E. T. Nature Struct. Biol. 1998, 5, 950), is prepared from 1-chloro-2-deoxy-3,5-bis-O-p-toluoyl-α-D-erythro-pentofuranose. The X-ray crystal structure of the nucleoside confirms that the compound is a close steric match for deoxyadenosine (2), although the methylbenzimidazole base is in the syn glycosidic orientation in the crystal. In D2O solution, 1H NMR studies show that 1 and 2 have similar (60% vs 70% S) sugar conformations and anti glycosidic orientations. Compound 1 is incorporated into a 12mer oligodeoxynucleotide and its base pairing properties in duplexes assessed by thermal denaturation. The results show that 1 has low affinity for the four natural bases but displays a stronger preference for being situated opposite a nonpolar difluorotoluene nucleoside analogue of thymine (3). The structural similarities of 1 and 2, combined with recent polymerase studies, add support to the hypothesis that steric complementarity plays an important role in base pair replication by polymerase enzymes and that Watson–Crick hydrogen bonds are not absolute requirements. Compound 1 should have significant utility as a probe of the importance of electrostatic effects in protein–DNA and protein–nucleotide binding as well as in DNA replication.
Introduction
Nonnatural analogues of nucleosides have been widely useful in probing important noncovalent interactions with other molecules, including metals, small organic compounds, proteins, and other nucleic acids. One of the most useful families of analogues has been derivatives of adenine in which hydrogen-bonding groups have been blocked or deleted.1–5 For example, a number of deaza analogues of deoxyadenosine have been reported recently: 7-Deazadeoxyadenosine has shown significant utility in probing DNA major groove interactions,1 and 1-deaza and 3-deaza derivatives have also been synthesized and utilized as probes.2,3 Interactions with the 6-amino group have been probed by use of a methylated derivative4 and a derivative in which the entire amino group is absent.5 In addition, indole derivatives which may have utility as probes have been reported.6–9
Along its Watson–Crick base pairing edge, adenine carries both a hydrogen bond acceptor (N1) and a donor (NH2), and so testing the importance of these two polar groups together requires an analogue in which both are replaced with nonpolar functionality, ideally maintaining steric size and shape as closely as possible.8,10 Rather than deleting or adding to the amino group, we have used replacement by a methyl group as a strategy; this replacement is isoelectronic and is one of the closest possible steric matches to this polar group.8 Thus, 4-methylbenzimidazole would appear to be a simple but useful structure to act as a nonpolar replacement for adenine. We chose this over a previously reported methlindole derivative9 because the greater reactivity of indoles made that compound less stable during DNA synthesis (C. Shiels and E. Kool, unpublished data).
One important use for nonpolar isosteres of natural DNA bases has been the probing of the hydrogen bonding and steric requirements for DNA replication by polymerase enzymes. Previous work with a difluorotoluene nucleoside (3, F), a nonpolar isostere of thymidine that lacks strong H-bond donors or acceptors, showed that it can be replicated with surprising efficiency and selectivity by the Klenow DNA polymerase.11,12 It was found to direct the highly selective incorporation of adenine, and conversely, it is selectively incorporated as a free nucleotide opposite adenine. Two competing explanations for this behavior are (1) that hydrogen bonds are not strictly required for efficient replication or (2) that the difluorotoluene forms strong hydrogen bonds with adenine.13 We have argued that the first explanation better fits the existing data12 and calculations;14,15 however, a reasonable approach to testing these possibilities in a new context is to construct a nonpolar isostere for adenine. This would allow testing in the absence of electronegative fluorine atoms, and so such a new analogue would have little possibility of forming Watson–Crick hydrogen bonds with natural DNA bases and even less with the thymidine analogue F.
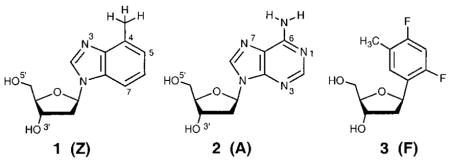
To test the importance of hydrogen bonds and steric effects in the fidelity of enzymatic DNA synthesis, as well as in other systems, we undertook the study of 4-methylbenzimidazole nucleoside 1.16 We now report the structure and pairing properties in DNA of this nonpolar analogue of deoxyadenosine (2). Compound 1 is found to act as a very close steric and conformational mimic of 2 in solution, and consistent with its nonpolar nature, it displays low selectivity and stability with natural DNA bases. However, opposite the difluorotoluene derivative F, compound 1 shows higher stability than natural adenine does.
Results
The synthesis of the nucleoside 1 (Z) (Scheme 1) has been recently described elsewhere.16 Details involving the coupling of 4-methylbenzimidazole17 with 1-chloro-2-deoxy-3,5-di-O-p-toluoyl-α-D-erythro-pentofuranose18 are given in the Supporting Information. Deprotection of the coupled bis(toluoyl) nucleoside with sodium methoxide, 5′-dimethoxytritylation, and 3′-O-phosphitylation proceeded in a 50% yield overall to give the previously unknown β-cyanoethylphosphoramidite derivative (8). Incorporation into oligodeoxynucleotides was carried out with standard coupling cycles, and stepwise yields for coupling of this compound were >98% by trityl monitoring. To test for intact incorporation, we synthesized a trinucleotide having the sequence d(TZT) (9) and examined it by 1H NMR. The spectrum of the crude unpurified oligonucleotide confirmed both the presence of the intact 4-methylbenzimidazole nucleoside and the high coupling yields (see Supporting Information). Incorporation of 1 into a longer oligodeoxynucleotide (10, see Table 2) was confirmed by electrospray mass spectrometry.
Scheme 1.

Table 2.
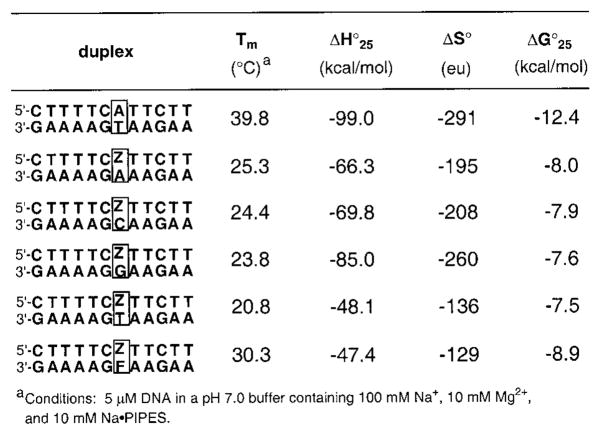
Melting Temperatures (Tm (°C)) and Thermodynamic Parameters for Duplexes Containing Base Pairs of Z (1) with the Natural Bases and Nonpolar Nucleoside F (3)
 |
Nucleoside analogue 1 was crystallized from ethanol. Comparison of the X-ray crystal structures of nucleoside 1 and deoxyadenosine19 indicates very similar bond lengths for the two. All analogous bonds except two are within 0.05 Å of each other in length (Figure 1). The six-membered ring of 1 is slightly larger than that of deoxyadenosine due to the replacement of carbon–nitrogen bonds with slightly longer carbon–carbon bonds within the ring. The C6–N6 bond in deoxyadenosine is 0.18 Å shorter than the analogous C–C bond in nucleoside 1. The addition of hydrogens at C1 and C3 and the replacement of the amino group with a methyl group also increase the steric bulk somewhat relative to adenine. Semiempirical calculations indicate that the most significant difference from a base pairing standpoint is the C3 proton, which adds approximately 0.73 Å of length to the van der Waals surface at that position relative to a nitrogen atom with its lone pair.20
Figure 1.
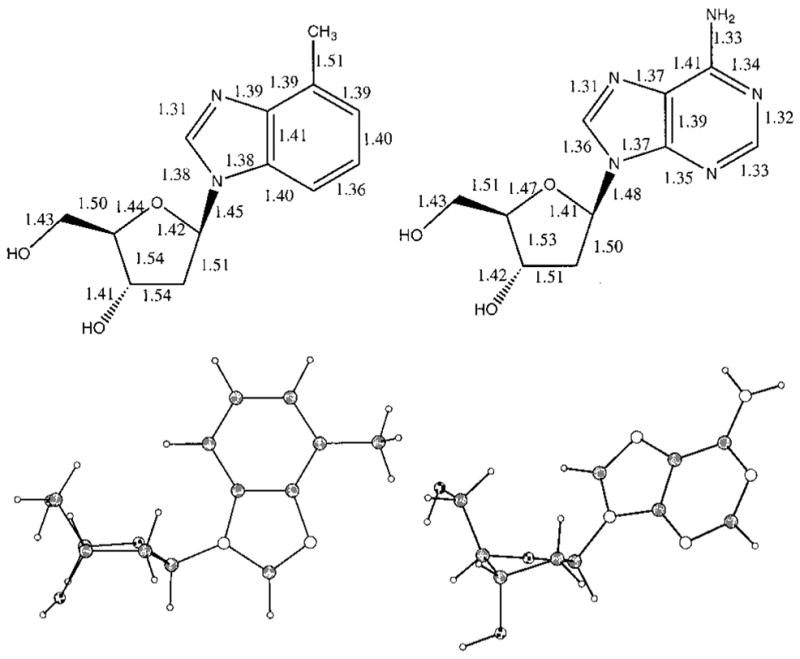
Solid-state structural data for 4-methylbenzimidazole nucleoside analogue 1 (left) and deoxyadenosine (2).19 Bond lengths are given in Å (top), and actual structures are shown below.
Further comparison of the X-ray structures of 1 and 2 shows that they adopt different conformations in the crystalline state (Figure 1). Despite the fact that the atomic differences between the two are remote from the sugar, the sugar puckers and the glycosidic torsion angles are different. Deoxyadenosine adopts a C2′-endo (S) sugar pucker and an anti glycosidic torsion angle, while nucleoside 1 adopts a C1′-exo (also S family) sugar pucker and a syn glycosidic conformation.
Examination of the intermolecular interactions for 1 and 2 in the solid state also shows significant differences. The natural nucleoside 2 exhibits stacking between the aromatic bases of adjacent nucleosides with a base–base distance of 3.67 Å. Nucleoside 1, however, is not base-stacked in the crystal. Both deoxyadenosine and nucleoside 1 show intermolecular hydrogen bonding in their packing diagrams. Deoxyadenosine shows intermolecular hydrogen bonds (heavy atom to heavy atom) from N1 to O5′, O5′ to water, O3′ to water, NH2 to N3, and NH2 to N7.19 Nucleoside 1 shows hydrogen bonds from N3 to O5′, O5′ to O3′, and O5′ to water, but no close contacts are found for the protons on the Watson–Crick pairing edge.
The conformations of deoxyadenosine and nucleoside analogue 1 in D2O solution were examined through a series of NOE and decoupling experiments. The measured coupling constants (Table 1) were treated as described by Rinkel and Altona21 to estimate the percent S conformer in solution. Through this analysis nucleoside 1 was found to be 60% S while adenosine was classified as 70% S. Qualitative classification of the glycosidic torsion angle as syn or anti was determined by 1-D NOE experiments. Irradiation of the H1′ of nucleoside 1 was found to induce a greater enhancement at H7 than H2. Models show that in the anti conformation, assuming an S type sugar pucker, the H2 to H1′ distance is approximately 1.0 Å longer than the H7 to H1′ distance. When rotated to the syn conformation, the H2 to H1′ distance is ca. 0.7 Å shorter than the H7 to H1′ distance. Thus, the NOE enhancements observed for nucleoside 1 are consistent with a primarily anti conformation in solution. Deoxyadenosine, which is known to adopt primarily an anti conformation in solution,22 exhibited NOE’s similar to those seen for 1. Thus the results show that despite their differences in the solid state, compound 1 and deoxyadenosine have nearly indistinguishable conformations in solution.
Table 1.
Data Obtained by 1H NMR in D2O for Nucleoside 1 and Deoxyadenosine (2)
| chemical shift | H1′ | H2′ | H2″ | H3′ | H4′ | H5′ |
|---|---|---|---|---|---|---|
| nucleoside 1 (ppm) | 6.70 | 3.06 | 2.80 | 4.85 | 4.37 | 4.00 |
| deoxyadenosine | 6.45 | 2.83 | 2.55 | 4.64 | 4.18 | 3.80 |
| coupling constants | H1′-H2′ | H1′-H2″ | H2′-H3′ | H2″-H3′ | H3′-H4′ | H2′-H2″ |
| nucleoside 1 J (Hz) | 6.84 | 5.86 | 6.35 | 3.90 | 3.70 | 13.52 |
| deoxyadenosine | 7.69 | 6.23 | 6.22 | 3.30 | 3.00 | 13.52 |
| summed J values | Σ(2′3′+2″3′) | Σ(1′) | Σ(2′) | Σ(2″) | Σ(3′) | |
| nucleoside 1 (Hz) | 10.25 | 13.12 | 27.84 | 25.00 | 14.00 | |
| deoxyadenosine | 9.52 | 13.92 | 27.83 | 23.89 | 12.09 | |
We next examined the effects of nucleoside 1 (Z) when placed in DNA opposite each of the four natural bases as well as 3 (F) in the center of a 12-base pair duplex (Table 2). Thermodynamic stabilities of the duplexes were measured by UV-monitored thermal denaturation studies at pH 7.0 in the presence of 100 mM Na+ and 10 mM Mg2+. Results show that all four duplexes containing Z opposite the natural bases are considerably less stable than the one with a natural A–T pair, with Tm values 14–19 °C lower and free energies (25 °C) 4.4–4.9 kcal·mol−1 less favorable. These stabilities are somewhat lower than those seen for mismatches such as T–C at the same position in this sequence context.23 Thus the nucleoside analogue 1 shows little if any preference for being situated opposite any of the natural bases, which is significant in light of its enzymatic properties (see Discussion). Interestingly, the placement of nonpolar deoxyadenosine analogue 1 (Z) opposite a nonpolar thymidine analogue 3 (F) results in an increase in stability relative to the above cases. The duplex containing a single Z–F pair is more stable by 5–9 °C in Tm and 0.9–1.5 kcal·mol−1 in free energy than the cases in which Z is paired with a natural base and approaches the stability of a G–T mismatch in this same context.
Discussion
The structural data show that the 4-methylbenzimidazole nucleoside analogue 1 is a very successful conformational mimic for deoxyadenosine in solution, with few observable differences between the two. This is perhaps not surprising, considering that the atomic replacements (three isoelectronic C–H replacements for N) are conservative in nature and are located at positions remote to the sugar ring. We surmise that the conformational differences between the two in the solid state reflect the influence of different intermolecular interactions in the crystal packing arrangements. In this respect it is worthwhile noting that the polar groups of adenine that are normally involved in hydrogen bonding in duplex DNA also undergo hydrogen bonding in the crystal structure of 2. By contrast, the analogous groups in isostere 1 are not involved in any close contacts with other groups, consistent with the molecule’s low polarity.
The sugar pucker and glycosidic torsion angle adopted by nucleoside 1 in the solid state are different than those in solution, which can be attributed to crystal packing forces that are relieved when 1 is free in solution. Interestingly, a benzimidazole deoxynucleoside (identical to 1 but lacking the methyl group) recently crystallized in our laboratory adopts an anti glycosidic torsion angle with a C2′-endo sugar pucker (D. Tahmassebi, E. Kool, unpublished data). This indicates the importance of crystal packing forces on conformation for deoxyadenosine and these analogues, as well as the low energetic barriers involved in these conformational changes.
In terms of shape mimicry by the base alone, nucleoside 1 is likely to be the closest possible isostere to deoxyadenosine which lacks Watson–Crick hydrogen-bonding ability. Replacement of N1 with CH and NH2 with CH3 does, however, increase the overall steric bulk relative to adenine. The CH replacing N1 adds ca. 0.8 Å to the center of the pairing edge of the molecule, which may be a factor in its base pairing geometry and, therefore, replication efficiency. As for the methyl group, the added bulk resides largely outside the plane of the base, which may be less important to base pairing considerations.
The duplexes containing analogue 1 opposite the natural bases show a sizeable destabilization compared to the case with a natural A–T pair. This is similar to results for nonpolar nucleoside 312 and may be explained by the substantial energetic cost of desolvation of the Watson–Crick pairing edges of the polar natural bases. It is worth noting that there is as yet no structural information available regarding the conformation of 1 in DNA opposite natural bases. As a result, we cannot rule out the possibility that 1 may adopt a syn orientation in DNA and form a Hoogsteen hydrogen bond with a hydrogen bond donor or that 1 or its partner may even be flipped out of the helix. However, to the extent that the nonpolar edge of 1 tends to be near the pairing edge of a natural base, one would expect a significant desolvation penalty. Consistent with this, the nucleoside 1 does display a significant preference for being placed opposite nonpolar thymidine isostere 3, probably because in this case the cost of desolvation of both bases is lower than for the polar natural bases.24 As for the geometry of Z opposite F in DNA, preliminary NMR evidence in a different sequence suggests that both prefer to be stacked in the helix with anti conformations (K. Guckian, T. R. Krugh, E. T. Kool, unpublished data). The possible lack of H-bonded interactions between 1 and the natural bases may explain why it shows little or no inherent pairing selectivity among them in the absence of enzymes.
Significantly, the 1–3 (Z–F) pair is still considerably less stable than the natural A–T pair in this sequence context. This could be explained by two possibilities: First, the lack of hydrogen bonds may be important, especially at a site near the center of this duplex sequence, and this apparently cannot be compensated for by the fact that F shows stacking abilities superior to those of T.25 A second contributing factor is the added size of Z relative to A, which would tend to force the Z–F pair to be wider than an A–T pair by at least 0.8 Å, thus causing the DNA backbone to be distorted signficantly.
Finally, it is important to examine the relationship of the steric similarities of 1 and deoxyadenosine to the properties of the two as substrates in enzymatic DNA replication. Recent studies have shown that when situated in a DNA template strand, analogue 1 acts in a number of respects similar to its natural analogue deoxyadenosine.26 For example, of the four natural nucleotides, dTTP is preferentially inserted opposite 1 by the Klenow fragment of DNA polymerase I. In addition, the nucleoside triphosphate derivative of thymidine shape mimic 3 is inserted with surprising efficiency opposite 1. Thus the enzymatic data establish that hydrogen bonding between the bases is not an absolute requirement for base pair synthesis by this enzyme. Previous findings showing efficient replication opposite deoxyadenosine by thymidine analogue 3 raised the question of whether hydrogen bonds by the fluorine and C-3 hydrogen, rather than steric mimicry alone, might explain those results.13 However, base–base hydrogen bonds are even less possible for the new Z–F pair. The present data for deoxyadenosine analogue 1 show that it is a good shape mimic for the natural compound and should thus act as a good steric complement to thymidine analogue 3. The efficient replication of that pair adds support to our conclusion that steric size and shape complementarity, even in the absence of hydrogen bonding, are likely to be important in the efficiency and fidelity of DNA replication.
Experimental Section
General Remarks
1H and 13C NMR spectra were obtained on a 300 MHz instrument, chemical shifts are reported in δ(ppm) using solvent as internal reference, and the coupling constants are in hertz (Hz). NOE difference spectra were measured on a 500 MHz instrument. Column chromatography was performed with EM Science silica gel 60 (230–400 mesh). High-resolution mass spectral analyses were performed by the University of California Mass Spectrometry Facility, Riverside, CA. Pyridine was dried by distillation from barium oxide. Methylene chloride and acetonitrile were distilled from calcium hydride. 4,4′-dimethoxytrityl chloride and 2-cyanoethyl diiso-propylchlorophosphoramidite were purchased from Aldrich.
1-[2-Deoxy-5-O-(4,4′-dimethoxytriphenylmethyl)-β-D-erythro-pentofuranosyl]-4-methyl-1H-benzimidazole (7)
1-[2-Deoxy-β-D-erythro-pentofuranosyl]-4-methyl-1H-benzimidazole (1)16 (300 mg, 1.21 mmol) was coevaporated with dry pyridine (2 × 10 mL) and dissolved in pyridine (9 mL) and dichloromethane (9 mL). To this mixture was added diisopropylethylamine (315 mL) in one portion. A solution of 4,4′-dimethoxytrityl (DMT) chloride (737 mg, 2.17 mmol) in dichloromethane (9 mL) was added slowly during 1 h. The mixture was stirred for 30 min more and quenched by adding methanol (25 mL). The mixture was concentrated and purified by silica column chromatography (dichloromethane–methanol–triethylamine, 20:2:1) to obtain 595 mg (90%) of 7 as a yellow foam: 1H NMR (CDCl3, ppm) δ 8.02 (1H, s), 7.39–7.15 (12H, m), 6.75–6.68 (4H, m), 6.34 (1H, m), 4.52 (1H, m), 4.40 (1H, m), 3.72 (6H, s) 3.30 (2H, m), 2.63 (3H, s), 2.68–2.47 (2H, m); 13C NMR (CDCl3, ppm) δ 158.46, 144.51, 139.71, 135.64, 135.59, 132.46, 130.08, 130.00, 129.00, 128.09, 127.84, 126.84, 123.15, 122.94, 113.13, 108.50, 86.45, 85.81, 85.11, 72.15, 63.83, 55.16, 40.28, 16.67; HRMS calcd for C34H34O5N2 m/e 551.2546, m/e found 551.2572.
2-Deoxy-5-O-(4,4′-dimethoxytriphenylmethyl)-3-O-(2-cyanoethyl N, N-diisopropylphosphoramidite)-1-(4-methyl-1H-benzimidazolyl)-β-D-erythro-pentofuranose (8)
The 5′-O-DMT compound 7 (595 mg, 1.086 mmol) was dissolved in dry dichloromethane (15 mL), and to this were added diisopropylethylamine (0.710 mL, 4.07 mmol) and 2-cyanoethyl diisopropylchlorophosphoramidite (0.365 mL, 1.63 mmol). The reaction mixture was stirred at room temperature for 90 min. Hexanes (30 mL) was added, and the concentrated mixture was purified by silica column chromatography (hexanes–ethyl acetate–triethylamine, 10:10:1). The two diastereoisomers were obtained as oils, phosphoramidite 8a (242 mg, 30%) and phosphoramidite 8b (273 mg, 33%). Spectroscopic data for 8a: 1H NMR (CDCl3, ppm) δ 8.12 (1H, s), 7.48–6.86 (12H, m), 6.82–6.78 (4H, m), 6.32 (1H, m), 4.80–4.75 (1H, m), 4.37 (1H, m), 3.78 (6H, s), 3.80–3.60 (3H, m), 3.48–3.30 (2H, m), 2.77–2.25 (5H, m), 2.72 (3H, s), 1.35–1.17 (12 H, m); 13C NMR (CDCl3, ppm) δ 158.42, 144.37, 143.43, 139.59, 135.49, 132.42, 130.29, 130.24, 130.12, 129.96, 128.08, 127.68, 126.72, 122.97, 122.74, 117.17, 113.02, 108.34, 86.37, 85.33, 85.10, 73.45, 73.22, 63.13, 58.38, 58.12, 55.02, 43.28, 43.11, 39.44, 39.40, 24.47, 24.38, 20.08, 19.99, 16.48; HRMS (FAB, 3-NBA matrix) calcd for C43H51O6N4P (M + 1) m/e 751.3624, m/e found 751.3620. Data for phosphoramidite 8b: 1H NMR (CDCl3, ppm) δ 8.10 (1H, s), 7.50–6.95 (12H, m), 6.83–6.77 (4H, m), 6.34 (1H, m), 4.76 (1H, m), 4.36 (1H, m), 3.78 (6H, s), 3.90–3.25 (5H, m), 2.84–2.30 (5H, m), 2.72 (3H, s), 1.28–1.12 (12 H, m); 13C NMR (CDCl3, ppm) δ 158.43, 144.39, 143.46, 139.63, 135.52, 135.48, 132.51, 130.11, 129.95, 128.06, 127.69, 126.71, 122.98, 122.75, 117.29, 113.05, 108.43, 86.39, 85.16, 74.05, 73.81, 63.35, 58.33, 58.07, 55.04, 43.28, 43.11, 39.43, 39.41, 24.44, 22.37, 20.29, 20.20, 16.50; HRMS (FAB, 3-NBA matrix) calcd for C43H51O6N4P (M + 1) m/e 751.3624, m/e found 751.3640.
X-ray Crystallographic Methods
Single crystals of nucleoside 1 were grown from a concentrated ethanol solution. A yellowish crystal of approximate dimensions 0.4 × 0.08 × 0.08 mm was mounted on a glass fiber with epoxy. The X-ray intensity data were collected at −50 °C on a standard Siemens SMART CCD Area Detector System equipped with a normal focus molybdenum-target X-ray tube operated at 1.5 kW (50 kV, 30 mA). A total of 1321 frames of data were collected using a narrow frame method with scan widths of 0.3° in ω and exposure times of 10 s/frame using a detector-to-crystal distance of 5.094 cm (maximum 2θ angle of 56.52°). The total data collection time was approximately 7 h. Frames were integrated with the Siemens SAINT program to yield a total of 8410 reflections, of which 3155 were independent (Rint = 7.55%). A semiempirical absorption correction, included in the SHELXTL program package, was applied to the data using the program SADABS (m = 0.099 mm−1; min/max transmission, 0.928/0.282). The crystal belongs to the orthorhombic crystal system. The unit cell parameters (at −50 °C) of a = 7.104(6) Å, b = 8.716(8) Å, c = 21.67(2) Å, and V = 1342(2) were based upon the least-squares refinement of three-dimensional centroids of >1000 reflections. The space group was assigned as P212121 on the basis of systematic absences and intensity statistics by using the XPREP program (Siemens, SHELXTL 5.04). The structure was solved with direct methods included in the SHELXTL program package and refined by full-matrix least-squares on F2. For a Z value of 4, there is one molecule in the asymmetric unit. All non-hydrogen atoms were refined anisotropically with hydrogens included in idealized positions giving a data:parameter ratio of approximately 17:1. The largest peak in the final difference map was 0.23 e/Å3. The enantiomer was assigned based upon the known stereochemical configuration of the deoxyribose starting material, as the flack parameter was unreliable. The structure refined to a goodness of fit (GOF) of 0.926 and final residuals of R1 = 6.83% (I > 2σ(I)) and wR2 = 16.18% (I > 2σ(I)).
Oligodeoxynucleotide Synthesis
DNA oligonucleotides were synthesized on an Applied Biosystems 392 synthesizer using standard β-cyanoethylphosphoramidite chemistry. Oligomers were purified by preparative 20% denaturing polyacrylamide gel electrophoresis and were quantitated by absorbance at 260 nm. Sequences incorporating 1 (Z) were d(TZT) (9) and d(CTTTTCZTTCTT) (10). Molar extinction coefficients were calculated by the nearest-neighbor method. The value for oligonucleotide 10 was derived from the molar extinction coefficient for 1 (measured to be 5620 at 260 nm) and the calculated values of the adjacent sequences dTTCTT and dCTTTTC. The actual value for 10 was 93,608 M−1·cm−1.
Thermal Denaturation Studies
Solutions for the thermal denaturation studies contained a one-to-one ratio of two complementary oligomers. The buffer solution contained 100 mM NaCl, 10 mM MgCl2, and 10 mM Na(PIPES) (pH 7.0). After the solutions were prepared, they were heated at 90 °C for 5 min and allowed to cool slowly to room temperature prior to the melting experiments. The sequences were monitored at 260 nm. Thermodynamic parameters were determined by plotting 1/Tm vs ln([oligonucleotide]/4) using four to six different concentrations for each duplex. All of the measured complexes generated van’t Hoff plots with good linear fits (see Supporting Information) and displayed apparently two-state melting behavior.
Supplementary Material
Acknowledgments
We thank the National Institutes of Health (Grant GM52956) for support; K.M.G. acknowledges a Merck Fellowship, and J.C.M. acknowledges the Spanish Government for a fellowship from the Ministry of Education and Culture. We thank Mr. D. Vicic, Dr. R. Lachicotte, and Prof. W. D. Jones for assistance with the X-ray data.
Footnotes
Supporting Information Available: Proton NMR spectra for all compounds including oligonucleotide 9, tables of supporting data and on ORTEP diagram for the X-ray structure of compound 1, figures providing mass spectral data for oligonucleotide 10 and thermodynamic data, and text providing synthetic details for the preparation of 1 (16 pages). This material is contained in libraries on microfiche, immediately follows this article in the microfilm version of the journal, and can be ordered from the ACS; see any current masthead page for ordering information.
References
- 1.(a) Robins MJ, Muhs WH. J Chem Soc, Chem Commun. 1976:269. [Google Scholar]; (b) Robins MJ, Wilson JS. J Am Chem Soc. 1981;103:932. [Google Scholar]; (c) Seela F, Kehne A. Liebigs Ann Chem. 1983:876. [Google Scholar]; (d) Ono A, Sato M, Ohtani Y, Ueda T. Nucleic Acids Res. 1984;12:8939. doi: 10.1093/nar/12.23.8939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cristalli G, Franchetti P, Grifantini M, Vittori S, Bordoni T, Geroni C. J Med Chem. 1987;30:1686. doi: 10.1021/jm00392a029. [DOI] [PubMed] [Google Scholar]
- 3.(a) Robins RK, Townsend LB, Rousseau RJ. Biochemistry. 1966;5:756. doi: 10.1021/bi00866a050. [DOI] [PubMed] [Google Scholar]; (b) Ono A, Ueda T. Nucleic Acids Res. 1987;15:3059. doi: 10.1093/nar/15.7.3059. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Cosstick R, Li X, Tuli DK, Williams DM, Connolly BA, Newman PC. Nucleic Acids Res. 1990;18:4771. [PMC free article] [PubMed] [Google Scholar]; (d) Lever C, Li X, Cosstick R, Ebel S, Brown T. Nucleic Acids Res. 1993;21:1743. doi: 10.1093/nar/21.8.1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jones JW, Robins RK. J Am Chem Soc. 1963;85:193. [Google Scholar]
- 5.Nair V, Chamberlain SD. Synthesis. 1984:401. [Google Scholar]
- 6.Coleman RS, Dong Y, Arthur JC. Tetrahedron Lett. 1993;34:6867. [Google Scholar]
- 7.Loakes D, Brown DM. Nucleic Acids Res. 1994;22:4039. doi: 10.1093/nar/22.20.4039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schweitzer BA, Kool ET. J Org Chem. 1994;59:7238. doi: 10.1021/jo00103a013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Moran S, Ren RXF, Sheils CJ, Kool ET. Nucleic Acids Res. 1996;24:2044. doi: 10.1093/nar/24.11.2044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chaudhuri NC, Ren RXF, Kool ET. SYNLETT. 1997:341. doi: 10.1055/s-1997-788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Moran S, Ren RXF, Rumney S, Kool ET. J Am Chem Soc. 1997;119:2056. doi: 10.1021/ja963718g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Moran S, Ren RXF, Kool ET. Proc Natl Acad Sci USA. 1997;94:10506. doi: 10.1073/pnas.94.20.10506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Evans TA, Seddon KR. Chem Commun. 1997:2023. [Google Scholar]
- 14.Meyer M, Sühnel J. J Biomol Struct Dyn. 1997;15:619. doi: 10.1080/07391102.1997.10508972. [DOI] [PubMed] [Google Scholar]
- 15.Barsky D, Colvin M, Kool ET. Manuscript submitted. [Google Scholar]
- 16.Seela F, Bourgeois W, Rosemeyer H, Wenzel T. Helv Chim Acta. 1996;79:488. [Google Scholar]
- 17.Mathias LJ, Overberger CG. J Org Chem. 1978;43:3518. [Google Scholar]
- 18.Hoffer M. Chem Ber. 1960;93:2777. [Google Scholar]
- 19.Watson DG, Sutor DJ, Tollin P. Acta Crystallogr. 1965;19:11. doi: 10.1107/s0365110x65002852. [DOI] [PubMed] [Google Scholar]
- 20.Geometries for the bases of 1 and 2 were optimized using the semiempirical AM1 Hamiltonian in Spartan v.5.0.1 (Wavefunction, Inc.).
- 21.Rinkel LJ, Altona C. J Biomol Struct Dyn. 1987;4:621. doi: 10.1080/07391102.1987.10507665. [DOI] [PubMed] [Google Scholar]
- 22.Davies DB. Prog NMR Spectrosc. 1978;12:135. [Google Scholar]
- 23.Wang S, Friedman AF, Kool ET. Biochemistry. 1995;34:9774. doi: 10.1021/bi00030a015. [DOI] [PubMed] [Google Scholar]
- 24.Schweitzer BA, Kool ET. J Am Chem Soc. 1995;117:1863. doi: 10.1021/ja00112a001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Guckian KM, Schweitzer BA, Ren RX-F, Sheils CJ, Paris PL, Tahmassebi DC, Kool ET. J Am Chem Soc. 1996;118:8182. doi: 10.1021/ja961733f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Morales JC, Kool ET. Nature Struct Biol. 1998;5:950. doi: 10.1038/2925. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.