

Noncovalent interactions between aromatic nucleobases are the major stabilizing forces which contribute to the structural integrity of duplex DNA and RNA.1–3 However, base pairing in nucleic acids is a consequence of more than just hydrogen bonding alone. The predictive success of nearest neighbor analysis,4 along with “dangling base” measurements which show duplex stabilization in the absence of pairing,5 points out the importance of stacking interactions to duplex integrity. Despite the existing work on nucleic acid stability, however, it is not yet known whether hydrogen bonds are absolutely required for stabilization of a base pair (bp). To test this question, we have designed and synthesized a series of non-hydrogen-bonding nucleoside analogues to probe these fundamental interactions.6 We report herein the finding of the first stable non-hydrogen-bonded base pair, and its selective formation relative to mismatched pairs.
Prior studies on the base pairing properties of nonpolar nucleoside analogues have almost invariably shown that pairs with natural DNA bases are strongly destabilizing.7 When both members of a base pair are nonpolar, the destabilization is lessened somewhat;7a however, one study found that a nonpolar–nonpolar pair still resulted in net destabilization of a 12-bp duplex by 2.9–4.0 kcal mol−1 (9–14 °C in Tm) relative to an A–T base pair.7a Possible reasons for this are the lack of hydrogen bonds between the bases or the imperfect steric fit of the specific pairs studied in the context of duplex B-form DNA. We hypothesized that optimization of the steric fit of the nonpolar base pair within the duplex, combined with the use of strongly stacking groups, might enable selective pairing without compromising duplex stability.
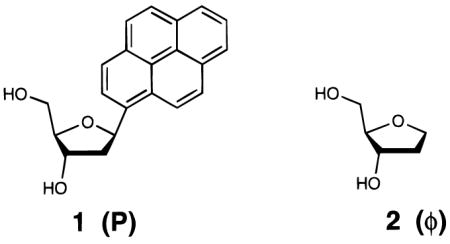
Models of B-form DNA suggested that a recently described pyrene nucleoside analogue (1)8 is sterically large enough to fit well against model abasic site9 2 (also denoted φ) (Figure 1). This combination yields a single contiguous π-system spanning nearly the entire distance between strands that would normally be occupied by two nucleobases. We synthesized 12mer duplexes containing 1 and 2 and measured their thermodynamic stabilities in aqueous buffer (Table 1) with thermal denaturation studies. To evaluate pairing selectivity, 1 was also paired against the four natural bases as well as itself, and abasic nucleoside 2 was paired against each of the four natural bases.
Figure 1.

(A) Space-filling models of base pairs A–T and P–φ in the geometry of B-form duplex DNA, illustrating how the added size of pyrene (P) may compensate sterically for the lack of a base in its partner (φ). (B) Illustration showing the positioning of various pyrene–X pairs in the center of a DNA duplex, with the potential for stacking on neighboring C–G and T–A pairs.
Table 1.
Free Energies (−ΔG°25 (kcal/mol) and Melting Temperatures (Tm (°C) for DNA Duplexes Containing P–X Pairs (P = Pyrene (1); φ = Abasic (2); X = A, C, G, T, φ))
 |
Thermodynamic parameters (25 °C) were determined from van’t Hoff plots using at least five different concentrations for each duplex. Under the conditions described above, control duplex 3, containing an A–T pair at the central position, has a Tm (5 μM) of 43.2 °C with a corresponding free energy of −12.3 kcal mol−1. When A is paired with the abasic site, the result is a strong destabilization of 5.3 kcal/mol (21.2 °C in Tm), which has been ascribed to the disruption of continuous stacking in the helix.10 However, when pyrene nucleoside 1 is paired opposite the abasic site, the duplexes formed (8 and 9) are only slightly less stable than the control containing an A–T pair (by 0.6–0.7 kcal mol−1 and 1.6–2.2 °C in Tm). Comparison to an 11mer duplex in which this pyrene–abasic pair is deleted (11, Table 1) shows that the non-H-bonded pair is thermally (and perhaps thermodynamically) stabilizing to the duplex despite its lack of hydrogen bonds.
Studies were then carried out to investigate the pairing selectivity of the pyrene nucleoside and its abasic partner. Replacing a central pyrene–abasic nucleoside pair with P–X (P = pyrene; X = A, C, G, T) destabilizes the duplex (relative to P–φ) by 0.5–1.8 kcal mol−1 and 2.3–4.6 °C in Tm (Table 1); thus, pyrene shows significant selectivity for the abasic site over natural bases. The pairing preferences of the abasic nucleoside were examined by placing it opposite each of the four natural bases. Thermal denaturation measurements indicated that, confirming previous findings,10 each of the resulting duplexes was severely destabilized relative to the pyrene–abasic pair, with an 18–23 °C decrease in Tm and a loss of ca. 4.0–5.0 kcal mol−1 in stability. Thus, the abasic nucleoside 2 shows a large preference for pairing with pyrene 1. Interestingly, when two pyrenes are paired against one another, a duplex (10) is formed which is only 0.5 kcal mol−1 less stable than the control having an A–T pair.
These findings suggested that multiple substitution of natural base pairs with the pyrene–abasic site (P–φ) pairs might be possible while still retaining reasonable duplex stability. To test this, oligonucleotides were designed to form duplexes 17 (20% P–φ pairs) and 18 (40% P–φ), and controls containing A–T pairs were studied for comparison. Although the melting behavior of putative duplex 17 made exact Tm determination difficult due to a highly sloped upper baseline, nonlinear least-squares fitting8 approximated the Tm as 67 °C. In comparison, the A–T substituted duplex had a Tm 8 °C lower. Since melting of single-stranded oligomers containing pyrene also showed sloping baselines, we hypothesize that the sloped upper baseline with 17 is the result of single strand melting at elevated temperatures, which may be the result of strong stacking by the pyrene residues. This possibility is also supported by CD spectra of single strands containing pyrene residues, which show evidence of stacked secondary structure (data not shown). A nondenaturing gel shift assay with duplex 17 was used to generate a Jobs plot, which clearly showed formation of a 1:1 duplex (full complexation at 0.48 mole fraction) (see Supporting Information). CD studies with the duplex in the same buffer revealed spectra characteristic of B-form DNA,12 similar to those for control duplex 16. Increasing the fraction of P–φ pairs to 40% (duplex 18) resulted in no helix formation being observed by either UV or gel shift experiments; it is possible that the added hydrophobicity causes intramolecular aggregation, preventing the desired complexation, or that the additional, relatively flexible abasic sites contribute an overly large entropic cost to helix formation.
These results establish that both pairing selectivity and stability in DNA can be achieved without hydrogen bonds. Pyrene was chosen to be paired against the abasic site because it may occupy (220 Å2) the area normally covered by two natural bases (269 Å2).13 The pairing selectivity between pyrene and the abasic nucleoside (compared to the natural bases) can be rationalized through a best fit (least steric hindrance) argument, since pyrene is far too large to fit in B-form DNA when placed edge-to-edge with a natural base. The strong preference of the abasic site in pairing with pyrene over natural bases is most easily explained by the latter’s robust stacking ability (nearly twice that of adenine).5e The notable lack of destabilization with the pyrene–abasic pair is most likely a result of the strong stacking and of the large size of pyrene, which may be able to maintain the continuity of stacking in both strands of the helix. Structural studies will, of course, be important in evaluating these hypotheses.
It was initially surprising that pairing of pyrene against a natural base only moderately destabilized the duplex relative to the case with previous nonpolar nucleosides.7 We believe this is best explained by the pyrene being accommodated within the helix, partially intercalated between the opposing residue and an adjoining base pair. This intercalation would allow the pyrene to bury its hydrophobic surface, compensating for most of the steric perturbation caused by its bulk being present in the duplex. This hypothesis is supported by the unexpected stability of duplex 10, which has two pyrenes paired against each other.
The stable pairing of pyrene and the abasic nucleoside represents a marked improvement in stability and selectivity over previous base pairs having no hydrogen-bonding functionality.7 As such, the new findings suggest that base stacking and geometric fit alone can stabilize duplex DNA.
Supplementary Material
Acknowledgments
We thank the National Institutes of Health (GM52956) for support.
Footnotes
Supporting Information Available: Experimental details, sample melting curves, plots of thermodynamic data, Jobs plot, and CD spectra (5 pages, print/PDF). See any current masthead page for ordering information and Web access instructions.
References
- 1.(a) Cantor CR, Schimmel PR. In: Biophysical Chemistry. Part I. Freeman WH, editor. New York: 1980. pp. 318–341. [Google Scholar]; (b) Serra MJ, Turner DH. Methods Enzymol. 1995;259:242. doi: 10.1016/0076-6879(95)59047-1. [DOI] [PubMed] [Google Scholar]; (c) Marky LA, Breslauer KJ. Biopolymers. 1982;21:2185. doi: 10.1002/bip.360211107. [DOI] [PubMed] [Google Scholar]
- 2.(a) Kawase Y, Iwai S, Inoue H, Miura K, Ohtsuka E. Nucleic Acids Res. 1986;14:7727. doi: 10.1093/nar/14.19.7727. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Howard FB, Miles HT. Biochemistry. 1984;23:6723. doi: 10.1021/bi00321a068. [DOI] [PubMed] [Google Scholar]; (c) Freier SM, Sugimoto N, Sinclair A, Alkema D, Meilson T, Kierzek R, Caruthers MH, Turner DH. Biochemistry. 1986;25:3214. doi: 10.1021/bi00359a020. [DOI] [PubMed] [Google Scholar]
- 3.(a) SantaLucia J, Kierzek R, Turner DH. Science. 1992;256:217. doi: 10.1126/science.1373521. [DOI] [PubMed] [Google Scholar]; (b) SantaLucia J, Kierzek R, Turner DH. J Am Chem Soc. 1991;113:4313. [Google Scholar]
- 4.(a) Freier SM, Kierzek R, Jaeger JA, Sugimoto N, Caruthers MH, Neilson T, Turner DH. Proc Natl Acad Sci USA. 1986;83:9373. doi: 10.1073/pnas.83.24.9373. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Breslauer KJ, Frank R, Blocker H, Marky LA. Proc Natl Acad Sci USA. 1986;83:3746. doi: 10.1073/pnas.83.11.3746. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) SantaLucia J, Allawi HT, Senevirante PA. Biochemistry. 1996;35:3555. doi: 10.1021/bi951907q. [DOI] [PubMed] [Google Scholar]; (d) Sugimoto N, Nakano S, Yoneyama M, Honda K. Nucleic Acids Res. 1996;24:4501. doi: 10.1093/nar/24.22.4501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.(a) Freier SM, Alkema D, Sinclair A, Neilson T, Turner DH. Biochemistry. 1985;24:4533. doi: 10.1021/bi00338a008. [DOI] [PubMed] [Google Scholar]; (b) Sugimoto N, Kierzek R, Turner DH. Biochemistry. 1987;26:4554. doi: 10.1021/bi00388a058. [DOI] [PubMed] [Google Scholar]; (c) Senior M, Jones RA, Breslauer KJ. Biochemistry. 1988;27:3879. doi: 10.1021/bi00410a053. [DOI] [PubMed] [Google Scholar]; (d) Doktycz MJ, Paner TM, Amaratunga M, Benight AS. Biopolymers. 1990;30:829. doi: 10.1002/bip.360300718. [DOI] [PubMed] [Google Scholar]; (e) Guckian KM, Schweitzer BA, Ren RX-F, Sheils CJ, Paris PL, Tahmassebi DC, Kool ET. J Am Chem Soc. 1996;118:8182. doi: 10.1021/ja961733f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.(a) Schweitzer BA, Kool ET. J Org Chem. 1994;59:7238. doi: 10.1021/jo00103a013. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Chaudhuri NC, Ren RXF, Kool ET. Synlett. 1997:341. doi: 10.1055/s-1997-788. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Guckian KM, Kool ET. Angew Chem. 1997;109:2942. [Google Scholar]; Angew Chem, Int Ed Engl. 1998;36:22825. [Google Scholar]
- 7.(a) Schweitzer BA, Kool ET. J Am Chem Soc. 1995;117:1863. doi: 10.1021/ja00112a001. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Millican TA, Mock GA, Chauncey MA, Patel TP, Eaton MAW, Gunning J, Cutbush SD, Neidle S, Mann J. Nucleic Acids Res. 1984;12:7435. doi: 10.1093/nar/12.19.7435. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Moran S, Ren RXF, Kool ET. Proc Natl Acad Sci USA. 1997;94:10506. doi: 10.1073/pnas.94.20.10506. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Nichols R, Andrews PC, Zhang P, Bergstrom DE. Nature. 1994;369:492. doi: 10.1038/369492a0. [DOI] [PubMed] [Google Scholar]; (e) Loakes D, Brown DM. Nucleic Acids Res. 1994;22:4039. doi: 10.1093/nar/22.20.4039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ren RXF, Chaudhuri NC, Paris P, Rumney S, Kool ET. J Am Chem Soc. 1996;118:7671. doi: 10.1021/ja9612763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.(a) Millican TA, Mock GA, Chauncey MA, Patel TP, Eaton MA, Gunning J, Cutbush SD, Neidle S, Mann J. Nucleic Acids Res. 1984;12:7435. doi: 10.1093/nar/12.19.7435. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Takeshita M, Chang C, Johnson F, Will S, Grollman AP. J Biol Chem. 1987;262:10171. [PubMed] [Google Scholar]
- 10.Vesnaver G, Chang CN, Eisenberg M, Grollman AP, Breslauer KJ. Proc Natl Acad Sci USA. 1989;86:3614. doi: 10.1073/pnas.86.10.3614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Petersheim M, Turner DH. Biochemistry. 1983;22:256. doi: 10.1021/bi00271a004. [DOI] [PubMed] [Google Scholar]; (b) Breslauer KJ. Methods Enzymol. 1995;259:221. doi: 10.1016/0076-6879(95)59046-3. [DOI] [PubMed] [Google Scholar]
- 12.Gray DM, Ratliff RL, Vaughan MR. Methods Enzymol. 1991;112:389. doi: 10.1016/0076-6879(92)11021-a. [DOI] [PubMed] [Google Scholar]
- 13.Surface areas were calculated using MacroModel v.6.0 (W. C. Still, Columbia University) using a probe radius of 1 Å.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.