

Abstract
We investigated the link between cell movement and plasma membrane recycling using a fast-acting, temperature-sensitive mutant of the Dictyostelium SecA exocytic protein. Strikingly, most mutant cells become almost paralysed within minutes at the restrictive temperature. However, they can still sense cyclic-AMP (cAMP) gradients and polymerise actin up-gradient, but form only abortive pseudopodia, which cannot expand. They also relay a cAMP signal normally, suggesting that cAMP is released by a non-exocytic mechanism. To investigate why SecA is required for motility, we examined membrane trafficking in the mutant. Plasma membrane circulation is rapidly inhibited at the restrictive temperature and the cells acquire a prominent vesicle. Organelle-specific markers show that this is an undischarged contractile vacuole, and we found the cells are correspondingly osmo-sensitive. Electron microscopy shows that many smaller vesicles, probably originating from the plasma membrane, also accumulate at the restrictive temperature. Consistent with this, the surface area of mutant cells shrinks. We suggest that SecA mutant cells cannot move at the restrictive temperature because their block in exocytosis results in a net uptake of plasma membrane, reducing its area, and so restricting pseudopodial expansion. This demonstrates the importance of proper surface area regulation in cell movement.
Keywords: Cell motility, SecA, Exocytosis, Contractile vacuole, Dictyostelium, cAMP relay
Introduction
The plasma membrane, in cooperation with the cytoskeleton, has a vital role in cell movement (Bretscher, 1996). It receives and processes guidance signals from the environment (Van Haastert and Devreotes, 2004; King and Insall, 2009; Swaney et al., 2010) and also supplies attachments to the substratum. In neutrophils, these attachments (integrins) are essential for migration on 2D surfaces (Lammermann et al., 2008), and their endocytosis and return to the cell surface at the leading edge might create membrane flow (Lawson and Maxfield, 1995), which in turn could provide motive force for the cell or assist in its polarisation (Bretscher, 1984; Valdez-Taubas and Pelham, 2003).
The plasma membrane also represents a physical constraint to cell movement because, although fully flexible, it is barely extensible (Mohandas and Evans, 1994). The surface area of moving amoeboid cells is unlikely to remain constant, as such cells often progress by cycles of expansion and rounding up (Wessels et al., 1998), and can extend a pseudopod without first withdrawing their tail (Weber et al., 1995). Measurement from confocal reconstructions confirms that the surface area of moving Dictyostelium cells continuously fluctuates, with increases of 30% sometimes occurring over a few minutes (Traynor and Kay, 2007); even greater increases are found in spreading mammalian cells (Gauthier et al., 2009). As these changes are much greater than membrane stretching allows, it seems likely that cells have evolved mechanisms to increase their surface area on demand, such as membrane ‘unwrinkling’ (Hallett and Dewitt, 2007), or by regulation of their endocytic cycle (Kay et al., 2008).
Dictyostelium cells maintain an active endocytic cycle, taking up their surface area once every 5–10 minutes, even when starving (Aguado-Velasco and Bretscher, 1999; Traynor and Kay, 2007). To test the importance of the endocytic cycle in cell movement, we previously created temperature-sensitive mutants of the essential nsfA gene, and found that they become paralysed within 30 minutes at the restrictive temperature (Thompson and Bretscher, 2002; Traynor and Kay, 2007). This is in stark contrast to most mutations of the actin cytoskeleton, which have only a minimal impact on cell motility (Noegel and Schleicher, 2000). NSF is required for resolving SNARE complexes after vesicle fusion, and impairment of its function is expected to inhibit all membrane fusion events in the cell, resulting in severe perturbation in organelles such the Golgi and endoplasmic reticulum, as well as blocking exocytosis (Novick et al., 1980; Malhotra et al., 1988; Sudhof and Rothman, 2009). Since these perturbations might indirectly affect cell motility, we sought a more specific tool to better dissect the role of exocytosis in cell movement.
SecA is the Dictyostelium homologue of the yeast Sec1p and mammalian Munc18 proteins. These proteins interact with exocytic SNARE proteins during vesicle docking and fusion and so are essential for exocytosis, but they do not appear to function at other points in the secretary pathway (Carr et al., 1999; Grote et al., 2000; Shen et al., 2007; Sudhof and Rothman, 2009). A library of temperature-sensitive secA mutants was previously created by replacing the endogenous gene with randomly mutated variants using homologous recombination, and screening for lack of growth at the restrictive temperature (Bretscher and Clotworthy, 2007). Many of these mutants develop poorly at the permissive temperature, but here we have identified a mutant that does develop and becomes chemotactic to cyclic-AMP (cAMP). This mutant shows a striking movement defect at the restrictive temperature and has a second defect in contractile vacuole discharge. However, cells can still polarise in response to cAMP, and relay a cAMP signal, showing that many other cellular functions continue unimpaired; we argue that the movement defect is due to surface area constriction.
Results
A temperature-sensitive secA mutant that is developmentally competent
The Dictyostelium homologue of the Munc18/Sec1p proteins was identified bioinformatically (Bretscher and Clotworthy, 2007). The SecA primary structure is very similar to its homologues in higher eukaryotes and is devoid of the low complexity repeats found in many Dictyostelium proteins. Temperature-sensitive mutants of SecA were obtained previously by mutagenic replacement of the endogenous gene using homologous recombination (Bretscher and Clotworthy, 2007). We found that that most of these are impaired in early development even at the permissive temperature, making them unsuitable for investigating chemotaxis to cAMP, which is a developmentally acquired property. By re-screening this pool, we found one strain, which carried the secA2 allele (resulting in seven amino acid substitutions: M250L, A296V, M427L, S435R, K505N, V552D, H570Q), where development was relatively normal at the permissive temperature. The secA2 allele is fast acting, with visible effects on cell morphology within 30 minutes at the restrictive temperature (see later) and was reconstituted in the Ax2 wild-type background by homologous recombination.
Cells of the resulting strain (HM1325) grew normally on suspensions of bacteria at the permissive temperature of 18°C (mean generation times: wild type=5.7±0.5 hours; mutant=5.3±0.5 hours; n=3, ± s.e.m.), though they did not grow in shaken axenic culture. However, at the restrictive temperature of 27.5°C, growth completely stopped, both on bacterial lawns and in suspension (mean generation times: wild type=4.1±0.1 hours; mutant cells die, halving every 18.2±3.4 hours; n=3, ± s.e.m.), thereby demonstrating that the gene is essential (Fig. 1A). This growth defect was completely rescued in parasexual diploid strains harbouring one copy of the wild-type gene (Fig. 1A), showing that the secA2 allele is recessive. This indicates that the effect of the mutations is through loss of SecA function, rather than through an indirect dominant inhibitory effect of the mutant protein on other proteins.
Fig. 1.

A temperature-sensitive mutant of SecA. (A) secA is an essential gene. The temperature-sensitive mutant strain HM1325 (ts-mutant) carrying the secA2 allele grows normally at the permissive temperature of 18°C, but not at the restrictive temperature of 27.5°C. A diploid strain harbouring the mutant and wild-type alleles grows normally at both temperatures, showing that the mutant allele is recessive. Cells were picked onto bacterial lawns and photographed after 4 days. Scale bar: 5 mm. (B) Cell morphology at permissive and restrictive temperatures. Mutant and wild type are similar at 18°C. After 30 minutes at 27.5°C, mutant cells become rounded and heavily vacuolated (arrowheads). Freshly starved cells were incubated at 18°C or 27.5°C for 30 minutes before imaging (Normarski). Scale bar: 10 μm. (C) Cell survival at the restrictive temperature. The wild-type (solid line) and temperature-sensitive (HM1325; dashed line) strains were incubated at the restrictive temperature for up to 4 hours (over twice the duration of any assay in this study) and viability determined clonal plating on bacterial lawns at the permissive temperature. No significant change in viability was observed (n=3, error bars represent s.e.m.).
The mutant cells remained intact at the restrictive temperature, but rapidly rounded up and acquired prominent vacuoles (Fig. 1B), which we show later to be undischarged contractile vacuoles. Crucially, they did not lose viability for at least 4 hours at 27.5°C (Fig. 1C), which is longer than used in subsequent experiments, indicating that the acute effects on motility are not due to cell death, and are reversible.
Cell motility requires SecA function
We compared the motility of mutant and wild-type cells at the permissive and restrictive temperatures (18–20°C and 27.5°C, respectively, with a 30 minute pre-incubation). Vegetative mutant and wild-type cells move similarly at the permissive temperature (not shown), but movement of the mutant is strongly suppressed at the restrictive temperature (Fig. 2A). Only around 20% of the mutant cells move more than 20 μm from their initial positions (Fig. 2B) and, because the average diameter of a Dictyostelium cell is approximately 10 μm, this means that most mutant cells still overlapped their original position at the end of the experiment, contrasting starkly with the highly motile wild type. This is reflected in the average speed distribution, which peaked at 1.8 μm/minute in the mutant, whereas the mean for the much broader wild-type distribution was 8.5 μm/minute (Fig. 2C).
Fig. 2.

Random motility of vegetative SecA mutant cells is almost abolished at the restrictive temperature. (A) Tracks of wild-type and SecA mutant cells (HM1325; ts-mutant) at 27.5°C. The tracks of 100 randomly sampled cells are shown centred at the origin. (B) Horizon plot. The fraction of the population moving a given distance from the origin is plotted. More than 75% of mutant cells (lower curve) remain within 2 cell diameters (~ 20 μm) and less than 5% reach a distance of 55 μm from their origin. By contrast, over 50% of the wild-type cells (upper curve) travel at least 85 μm from the origin. (C) Speed distribution. The average mean speed distribution for wild-type cells, white bars, and for the mutant, black bars, is shown. The mean speeds are 8.5±1.6 μm/minute and 1.8±0.4 μm/minute for the wild type and mutant, respectively. Error bars represent s.e.m., n=3; wild type=212 cells, temperature sensitive mutant=348 cells. Cells were washed free of bacteria and plated in axenic medium and incubated for 30 minutes at 27.5°C before imaging for 30 minutes at 3 frames per minute.
Similarly, chemotaxis to cAMP was strongly impaired in the mutant at the restrictive temperature. In these experiments, mutant and wild-type aggregation-competent cells were placed on opposite sides of a Dunn chamber and filmed in parallel using a motorised stage, so that they experienced the same gradient and temperature conditions. It was apparent that the mutant was barely motile in comparison with the wild type (Fig. 3A,B; supplementary material Movie 1).
Fig. 3.

Cells of the SecA mutant can sense chemotactic gradients at the restrictive temperature, but barely move. (A) Tracks of wild-type and SecA mutant cells in a cAMP gradient at the restrictive temperature of 27.5°C. Tracks of aggregation-competent cells in a Dunn chamber (35 wild-type and 45 mutant cells) from paired samples are shown, centred at the origin; filmed for 30 minutes at 3 frames per minute. Representative of four independent experiments; see supplementary material Movie S1. (B) Speed distributions of wild-type and SecA mutant cells. Taken from four independent data sets (124 wild-type cells, white bars; 158 mutant cells, black bars). Mean speeds are wild type, 13.4±1.4 μm/minute; mutant, 2.0±0.3 μm/minute; error bars represent s.e.m. (C) Actin polymerisation produced by uniform cAMP stimulus. All cells display a robust peak 5–10 seconds after stimulation and a more variable second peak about 60 seconds thereafter, which is similar to bulk assays. Aggregation-competent cells were pre-incubated at 27.5°C for 30 minutes and then bath-stimulated with 10 μM cAMP at t0. Actin polymerisation at the cortex of individual cells was monitored using ABD120-GFP as reporter for F-actin, and fluorescence quantified using QuimP. Average from 22 wild-type and 23 mutant cells from three independent experiments. (D) Polarised actin polymerisation induced by a cAMP gradient. Wild-type and mutant cells respond to cAMP gradients by polarised F-actin accumulation up the gradient. This drives expansion of a pseudopod and cell movement in the wild type at either temperature, but the SecA mutant can only extend a pseudopod at the permissive temperature, producing a bulge or even remaining rounded at the restrictive temperature (the extreme cell is from the same population). Cells expressing ABD120-GFP were made aggregation competent and either used immediately or shifted to the restrictive temperature for 30 minutes before imaging. They were stimulated using a needle filled with cAMP (white dot). Scale bar: 10 μm; see supplementary material Movies 2 and 3.
Conceivably, the poor motility of mutant cells at the restrictive temperature might be due to a failure to deploy their actin cytoskeletons effectively. We first tested their ability to polymerise actin in response to a uniform cAMP stimulus, using the F-actin reporter ABP120-GFP (Pang et al., 1998). The initial actin response was very similar in mutant and wild-type cells, with F-actin levels robustly peaking at 5–10 seconds and then falling off. F-actin levels peaked for a second time in the wild type at 1–2 minutes, but this peak was much reduced in the mutant (Fig. 3C). The second peak correlates with pseudopod projection, but in our hands it was very variable and often barely discernable even in the wild type (Langridge and Kay, 2006; Hoeller and Kay, 2007).
Similarly, both mutant and wild type responded to a cAMP gradient by polymerising actin on the up-gradient side of the cell. In the wild type, this resulted in extension of a pseudopod and movement up the gradient, whereas the mutant generally could not produce a distinct pseudopod or move (Fig. 3D; supplementary material Movies 2 and 3); in the extreme example, the mutant cell remained rounded and could do no more than slightly bulge towards the needle, despite appropriate actin polymerisation.
We also considered two more trivial explanations for the movement defect: that it might be due to the presence of the undischarged contractile vacuole (see later), or loss of adhesion to the substratum, as a result of cell rounding. We found that placing cells in solutions of increasing osmolarity (KK2 supplemented with 100–200 mM sorbitol) reduced the size of the contractile vacuole, but did not restore wild-type motility (result not shown). Similarly, pressing cells against the substratum with an agarose overlay (Laevsky and Knecht, 2001), which can restore motility to at least one poorly adhesive mutant (Langridge and Kay, 2007), also did not rescue motility of the SecA mutant (not shown).
cAMP relay does not require SecA function
As well as performing chemotaxis, aggregation-competent cells respond to cAMP by producing and releasing more cAMP (Shaffer, 1975). This relay response depends on many of the same signal transduction components as chemotaxis and we therefore used it as a more global test for impairment of mutant cell function. Since the mechanism of cAMP release from the cell is not known, we also asked whether release depends on SecA function.
It is clear from the measurements at the restrictive temperature that SecA was not required for cAMP production. More surprisingly, it was also dispensable for cAMP release, which might reasonably have been assumed to be by vesicle exocytosis (Fig. 4A). We microscopically checked the integrity of the cells at the end of the experiments and found no difference from the wild type. To confirm these findings, the same experiment was repeated with the NSF temperature-sensitive mutant (Thompson and Bretscher, 2002), with the same result (Fig. 4B).
Fig. 4.

cAMP relay by aggregation-competent cells. Cells were stimulated with 15 μM 2′-deoxy cAMP and total cAMP produced over 5 minutes (extracellular + cytosolic) and the proportion released determined. (A) SecA mutant (means of five independent experiments; error bars represent s.e.m.). (B) NsfA mutant (means of three independent experiments; error bars represent s.e.m.). No significant differences were observed.
Thus, SecA is required for efficient cell motility, but is not required to transduce a chemotactic gradient into appropriate actin polymerisation, or for the closely related signal transduction events required for cAMP relay.
SecA localisation
To understand the connection between SecA and cell motility, we first attempted to localise SecA protein. An affinity-purified serum against full-length, recombinant SecA, which specifically recognises SecA in western blots (supplementary material Fig. S1), stained the plasma membrane of vegetative cells and puncta within them. Cells chemotaxing to cAMP stained in patches at the front and rear, often in the same cell, but also independently (supplementary material Fig. S2). Cells with a knocked in SecA C-terminal GFP fusion were viable, but did not grow in axenic medium, showing that the fusion was only partially functional, and unfortunately the GFP was too faint to detect. Overexpressed N- or C-terminal fusion proteins showed similar localisation patterns, often strongly staining the plasma membrane at the rear of moving cells (supplementary material Fig. S2, Movie 4). These results reveal a complex SecA localisation pattern in vegetative cells and a variable one in chemotaxing cells, where SecA can also be at the front or back. Neither pattern is especially informative. We next investigated membrane trafficking.
SecA is required for normal plasma membrane circulation
Plasma membrane circulation was measured in a continuous assay using the lipophilic dye FM1-43, which becomes intensely fluorescent when it partitions into the membrane (Betz et al., 1996; Traynor and Kay, 2007). The initial fluorescence produced upon mixing dye and cells gives a measure of collective cell surface area, and the subsequent increase a measure of exocytosis, because previously internal membrane becomes available for binding (endocytosed membrane retains its dye and continues to fluoresce). From Table 1 it is apparent that at the permissive temperature, the wild-type and SecA mutant cells circulate their plasma membrane at similar rates, taking approximately 10 minutes to exocytose the equivalent of their surface area, with no statistically significant difference between them. At the restrictive temperature, the mutant circulates its plasma membrane more slowly than the wild type: if no corrections are made, the rate is approximately half (P<0.05) that of the wild type; but if allowance is made for the increase in fluorescence that occurs even in azide-poisoned cells, then it appears that the endocytic cycle is almost completely suppressed in the mutant. This indicates that normal plasma membrane circulation requires the function of SecA.
Table 1.
Exocytic rate of wild-type and SecA temperature-sensitive cellsa

SecA is required for discharge of the contractile vacuole
The most visible perturbation of the mutant membrane system at the restrictive temperature is the presence of one, or more, large translucent vesicles in each cell (Fig. 1B). Cells of the temperature-sensitive NSF mutant similarly round up at the restrictive temperature, but do not produce these characteristic vesicles (Traynor and Kay, 2007).
We investigated the origin of these vesicles using standard markers for subcellular compartments. They were not stained by markers for ER, Golgi, late endosome or plasma membrane, nor was the distribution of these markers visibly disturbed in the mutant. The markers used were: calnexin-GFP and calreticulin-GFP for the ER (Muller-Taubenberger et al., 2001); GFP-golvesin, GFP-Δ(1–75)golvesin, Δ(1–75,119–579)golvesin-GFP for the Golgi (Schneider et al., 2000; Gerisch et al., 2004); vacuolinB-GFP for late endosomes (Jenne et al., 1998); and ACA-YFP and cAR1-GFP for the plasma membrane (Kriebel et al., 2003; Xiao et al., 1997); although the latter did faintly stain the vesicle (not shown).
The only exception was the Dajumin-GFP construct, an integral membrane protein that strictly defines the contractile vacuole system (Gabriel et al., 1999); this was found to strongly stain all the large vesicles and virtually no other structure (Fig. 5A).
Fig. 5.
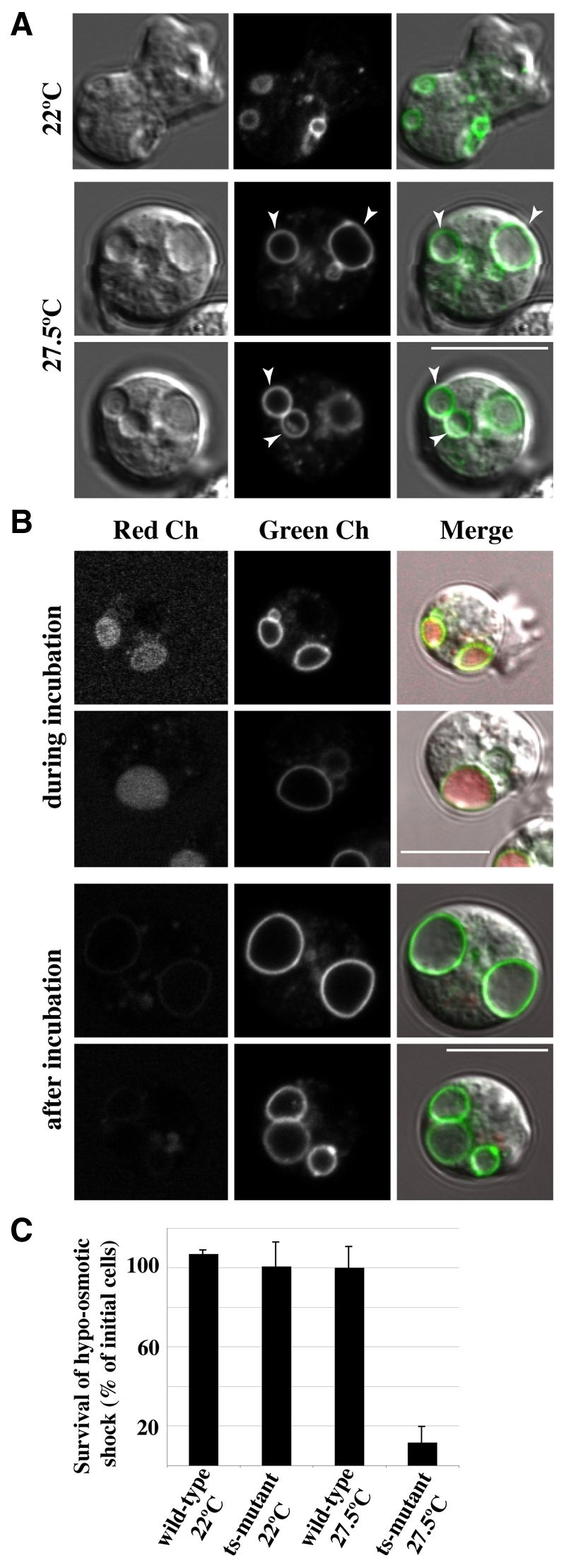
Contractile vacuole discharge requires SecA activity. (A) The large vesicles formed at the restrictive temperature are undischarged contractile vacuoles. SecA mutant cells, expressing Dajumin-GFP as a contractile vacuole marker, were incubated for 30 minutes at the restrictive temperature of 27.5°C. All large vesicles visible by DIC display the marker. Scale bar: 10 μm. (B) The contractile vacuole ceases discharge at the restrictive temperature. When the contractile vacuole (Dajumin-GFP, green channel) discharges, it becomes accessible to back-filling by small bulk-phase tracers such as Alexa Fluor 594 (red channel). Cells were incubated with dye, either for the first 30 minutes at the restrictive temperature (27.5°C), when it has access to the contractile vacuole (top panels), or for the next 30 minutes, when it does not (lower panels). In both cases, dye in the medium was washed away before observing the cells. Scale bars: 10 μm. (C) The SecA mutant is osmo-sensitive at the restrictive temperature. Cells were transferred from growth medium to water, and after incubation for 30 minutes, their viability was determined by clonal plating on bacterial lawns. Viability of the SecA mutant (ts-mutant 27.5°C) was reduced to less than 10% at the restrictive temperature.
The contractile vacuole fills with fluid and periodically discharges by fusion with the plasma membrane. At this point it becomes accessible to the external medium and can be back-filled with a small dye, such as Alexa Fluor 594. When dye was added to mutant cells at the start of their incubation at the restrictive temperature, the contractile vacuole was accessible to the label; but if addition was delayed for 30 minutes, the vesicle remained unstained (Fig. 5B), indicating that SecA activity is required for fusion of the contractile vacuole with the plasma membrane.
As the contractile vacuole is the organelle of osmoregulation, we tested whether the SecA mutant is osmosensitive. Cells were transferred to distilled water before or after incubation at 27.5°C. Wild-type cells, or the SecA mutant at the permissive temperature were able to effectively resist this osmotic shock, but not the mutant at the restrictive temperature, where approximately 90% of the cells died after 30 minutes (P=0.003, n=3; Fig. 5C). Thus we conclude that the large vesicles are undischarged contractile vacuoles.
Small vesicles accumulate, which might originate from the plasma membrane
Electron microscopy reveals a second perturbation of the SecA mutant membrane system at the restrictive temperature: an increased number of small vesicles in the cytoplasm (Fig. 6A; the vesicles are taken as those less than 0.5 μm diameter). In central sections of the cell – those containing at least part of the nucleus – their number nearly doubles from 0.68±0.42 per μm2 of cytoplasm in the wild type to 1.22±0.40 μm2 of cytoplasm in the mutant (n=8 sections, ± s.d.; the area of the nucleus and of large vesicles is excluded from the calculation), accompanied by a marginal increase in average diameter (193±48 nm to 266±24 nm). The smaller vesicles are often found in the proximity of the plasma membrane and sometimes in close juxtaposition with it. Apart from these vesicles, other cellular compartments were not visibly changed in the mutant.
Fig. 6.

Ultrastructure of SecA mutant cells. (A) Electron micrographs of representative wild-type and mutant aggregation-competent cells at the restrictive temperature (27.5°C). (a–c) wild-type cell morphology. This is unchanged by the temperature shift. (d–i) Mutant cell morphology at low power, with selected regions at higher power. Two changes in the endomembrane system are apparent: large vacuoles (Va) – undischarged contractile vacuoles – appear, and the number of small vacuoles increases. Often these cluster near the plasma membrane (g and h), sometimes in tight juxtaposition (i). Scale bars: 1 μm (f, for a–f; h, for g,h), 0.2 μm (i). (B) Redistribution of plasma membrane components in the SecA mutant at the restrictive temperature detected using phosphotungstic acid and chromic acid stain. (a–c) Permissive temperature. The plasma membrane stains more strongly than any other membrane and the contractile vacuole does not stain. (d–f) After incubation for 30 minutes at the restrictive temperature. Staining of the plasma membrane is greatly reduced, but a subset of large multi-vesiculated compartments become strongly stained (d and e) and the small vesicles are finely stained, suggesting that they also receive plasma membrane material (f). Scale bars: 1 μm (d, for a and d; c, for b and c; f, for e and f). Aph, autophagosome; MVC, multi-vesciculated compartments; N, nucleus; PM, plasma membrane; Va, vacuole; Ve, vesicle.
To investigate the origin of the small vesicles, we used a phosphotungstic acid and chromic acid stain (Fig. 6B). Although the basis for the selectivity of this stain is not known, it strongly stains the plasma membrane of the wild type and other membranes to a lesser extent, but the contractile vacuole system is not stained at all (Quiviger et al., 1978). Staining of the mutant plasma membrane was reduced at the restrictive temperature, whereas a number of internal vesicles became intensely stained. The simplest explanation for this is that components from the plasma membrane are sequestered into these vesicles when SecA is inactivated.
Loss of SecA function results in reduced plasma membrane area
Two features of the mutant phenotype seem particularly relevant to understanding its movement defect: the cells round up at the restrictive temperature, and the number of small vesicles, apparently originating from the plasma membrane, increases. Both features suggest that the plasma membrane area decreases, and we supposed that such a constriction might physically restrict the ability of a cell to extend pseudopodia and therefore to move. We therefore measured the surface area of cells after transfer to the restrictive temperature, using cAR1-GFP as a surface marker, and reconstructing the surface from confocal stacks.
As is apparent from Fig. 7, the surface area of mutant cells shrunk by 20–30% during incubation for 30 minutes at the restrictive temperature, whereas wild-type cells showed a transient increase, before returning gradually to their initial value. Mutant cells also decreased in volume, although to a lesser extent. Since the surface to volume ratio of a sphere increases as it shrinks (3/r), these measurements imply that the mutant cells shrink from a shape with a higher surface to volume ratio than a sphere towards a more sphere-like ratio.
Fig. 7.

Changes in surface area and volume of mutant cells at the restrictive temperature. Vegetative cells expressing the membrane marker cAR1-GFP were imaged at 27.5°C, taking confocal stacks every 5 minutes. The volume and surface area measurements of wild-type (open circles) and mutant cells (closed circles) are normalised to the initial values (t=0, room temperature). Error bars represent s.e.m. A significant decrease in surface area could be detected in the mutant after 20 minutes at 27.5°C, resulting on average in a ~25% reduction in surface area at 30 minutes. A small decrease in the volume of the mutant is also noticeable, but this is not statistically significant. *P<0.05, **P<0.01, ***P<0.001, two-tailed sample student's t-test (n=6 for the wild-type sample, n=7 for the mutant sample, acquired over 6 different days).
Discussion
There is now a powerful genetic case that a functional endocytic cycle is required for Dictyostelium cell motility. Cells of both the NSF mutant studied previously (Thompson and Bretscher, 2002; Traynor and Kay, 2007), and the SecA mutant studied here, rapidly become essentially immotile at the restrictive temperature. Since the appearance of the movement defect is so rapid – it is apparent within minutes and complete in around 30 minutes – it cannot be due to long-term changes in gene expression, nor is it probably due to defects in trafficking of newly synthesised proteins. Rather, it seems likely that the endocytic cycle is directly required for movement.
In both the SecA and NSF mutants, chemotactic signalling to the cytoskeleton appears to be normal, and both can polymerise actin at the up-gradient side of the cell. Therefore it is unlikely that their movement defects are caused by a failure of signal transduction, or of cytoskeletal mobilisation.
By contrast, the endocytic cycle of both mutants is substantially blocked and they rapidly round up at the restrictive temperature, resulting in the significant reduction in surface area shown here for the SecA mutant. Although NsfA and SecA have very different roles membrane trafficking – NsfA is expected to be essential for all membrane-fusion events, whereas SecA is specifically required in exocytosis – neither is directly required for endocytosis, at least in yeast (Hicke et al., 1997). We therefore suggest the following interpretation of the NsfA and SecA phenotypes. At the restrictive temperature, exocytosis is substantially blocked, but endocytosis runs on for a while, resulting in a net uptake of plasma membrane and a decrease in surface area. As the surface area decreases, membrane tension increases, so physically restricting the extension of pseudopodia and inhibiting cell movement. It is notable that the few mutant cells that do move at the restrictive temperature seem to ‘shuffle’ with greatly reduced pseudopodia, presumably reflecting the limited slack in the plasma membrane. In the converse situation, where membrane tension is artificially decreased, cell extension is correspondingly stimulated (Raucher and Sheetz, 2000).
Our attempts to understand how SecA is required for cell movement revealed two other aspects of SecA biology. First, neither SecA nor NSF is required for cAMP release into the medium during cAMP relay. Relay is fundamental to aggregation, because it allows cAMP waves to propagate from signalling centres through a field of responsive cells, and guide their inward movement. Electron microscopy reveals that signalling cells accumulate and release small vesicles (Maeda and Gerisch, 1977), suggesting that cAMP is released by a conventional exocytic mechanism. Our work contradicts this idea and is consistent with other work showing that cAMP is completely released from cells by lysis in conditions not expected to break small vesicles (Schoen et al., 1989) and that there is a build-up of free cAMP in the cytoplasm during cAMP relay (Bagorda et al., 2009). It seems likely that cAMP is released by a non-exocytic mechanism, possibly through a membrane transporter. The small vesicles (Maeda and Gerisch, 1977) might have some other function, perhaps serving as a membrane reservoir for the regulation of surface area.
Second, SecA appears to be essential for discharge of the contractile vacuole. The contractile vacuole cycle involves filling, fusion with the plasma membrane, and emptying (Gerisch et al., 2002; Heuser, 2006; Du et al., 2008). When SecA function is impaired, the cells carry a prominent, engorged contractile vacuole, which does not empty, suggesting that fusion with the plasma membrane requires SecA and thus resembles other exocytic processes. Fusion is believed to be ‘kiss and run’ and is not thought to involve intermingling of the contractile vacuole and plasma membranes, so it is unlikely that the contractile vacuole normally contributes to surface area regulation, except perhaps in specific cases (Yoshida and Inouye, 2001).
This work confirms the importance of a functional endocytic cycle for cell movement. At the very minimum, the cycle must be regulated to provide the cell with sufficient plasma membrane area to project pseudopodia. The nature of this regulation remains open, but we favour a model that involves membrane tension: if tension becomes too high, extra membrane is supplied by exocytosis, increasing the surface area and decreasing the tension; if it is too low, membrane is withdrawn by endocytosis. The link between membrane tension and the endocytic cycle might involve stretch-operated receptors in the plasma membrane, or perhaps direct inhibition of endocytosis by increasing tension. Finally, the availability of a fast-acting temperature-sensitive mutant in the essential secA gene will be invaluable for future studies of exocytosis and contractile vacuole discharge.
Materials and Methods
Cell procedures
Cells were grown with bacteria on SM agar plates at 18°C (Kay, 1987), freed of bacteria by centrifugation in KK2 (16.5 mM KH2PO4, 3.8 mM K2HPO4, 2 mM MgCl2, 0.1 mM CaCl2) and used immediately (vegetative cells) or made aggregation competent by pulsing shaken cells with 50–90 nM cAMP every 6 minutes for 4–5 hours, starting at 1 hour of starvation. The secA2 allele was identified from a library of mutants (Bretscher and Clotworthy, 2007) and recreated by homologous recombination (Knecht and Pang, 1995) in the Kay laboratory stock of Ax2 wild-type cells, which has minimal duplications (Bloomfield et al., 2008).
Three independent strains (HM1323, HM1324, HM1325) all had the same secA2 allele of the secA gene, resulting in 7 amino acid changes: M250L, A296V, M427L, S435R, K505N, V552D, H570Q. All behaved similarly and HM1325 was studied further. It was transformed with ABD120-GFP (Pang et al., 1998) and GFP-SecA [GFP S65T cDNA fused via a GGRGSEFKLLE linker to the N-terminus of the full length secA cDNA and inserted into the pDXA-3C expression vector (Manstein et al., 1995)] using G418 selection.
Diploid strain DM246 was created by crossing HM1325 and HM2068, a G418-resistant strain expressing GFP (Thompson and Kay, 2000), and diploids selected with 10 μg/ml each of blasticidin and G418. Growth was measured in shaken suspension cultures of heat-killed bacteria in KK2 and viability by plating cells clonally on bacterial lawns at 18°C. Osmotic shock was produced by shifting cultures from growth medium to de-ionised water.
Membrane exocytosis was measured from the initial rate of change in fluorescence of cells incubated with 5 μM FM1-43 (Molecular Probes) in a stirred cuvette (470±2.5 nm excitation, 570±5 nm emission), normalised to the initial value on mixing (Traynor and Kay, 2007). cAMP production was measured with 5 mM dithiothreitol to inhibit breakdown (Traynor et al., 2000); total cAMP is that produced in 5 minutes following stimulation with 15 μM 2′-deoxy cAMP minus that present at t0, and released cAMP is the proportion in the supernatant after the cells are rapidly centrifuged (5–10 seconds at 13,000 g).
Live cell imaging and analysis
Cells were imaged at ~2×105 cells/cm2 in Lab-Tek (Nalgene) chambered coverglasses in axenic medium (vegetative) or KK2 (aggregation competent), on a Nikon Eclipse TE300 inverted microscope with a Bio-Rad confocal system, or an Axiovert S100 with a motorised stage. Temperature was controlled by an ASI 400 Air Stream Stage Incubator (Nevtek) plus a heated stage on the confocal system (Linkam MC60); cells were pre-incubated at 27.5°C for 30 minutes as necessary. Randomly moving cells were imaged every 20 seconds for 30 minutes. Cells were stimulated uniformly with 10 μM cAMP or with a microneedle containing 1 μM cAMP. Dunn chambers had 1 μM cAMP in the chemoattractant reservoir and two cell populations on opposite sides of the same coverslip were imaged in parallel (1 frame/20 seconds).
Films were processed by threshold binarisation and centroid tracking with ImageJ (NIH) and MTrack2. Only tracks lasting for the entire movie were analysed for random motility, and only those of at least 30 frames for chemotaxis. Parameters and trajectories were extracted using custom scripts in the R statistical language (http://www.R-project.org). In the actin polymerisation assays, fluorescence was compared using QuimP plugin (Dormann et al., 2002).
Stacks for 3D reconstruction were acquired on an Olympus IX71 inverted microscope equipped with a Perkin Elmer Ultra View RS spinning disk confocal system, using a 60× 1.20 NA water objective (Olympus) and piezoelectric focusing device, taking images at 0.5 μm intervals, and thus allowing a single volume to be imaged in 2–4 seconds. Stage temperature was regulated as above. Images were processed and reconstructed using Volocity (Improvision).
Immunofluorescence
Cells were fixed with formaldehyde and picric acid (Jungbluth et al., 1994), blocked with 0.1% BSA in phosphate saline buffer, 0.05% Tween-20, incubated with affinity-purified rabbit polyclonal antibody against SecA (1:200, 1 hour at room temperature) followed by a secondary anti-rabbit conjugated to Alexa Fluor 488 (Molecular Probes, 1:500, 1 hour at room temperature). Phalloidin-TRITC (Fluka) was used 1:500 (30 minutes at room temperature).
Electron microscopy
Aggregation-competent cells were prepared in NS (20 mM MES, 20 mM KCl, 20 mM NaCl, 1 mM CaCl2, 1 mM MgCl2, pH 6.2) and fixed overnight on ice with 1% glutaraldehyde in the same buffer. Cells were washed, embedded in 2% agar and osmicated (1% in NS, for 1 hour on ice), then washed in deionised water and dehydrated in an ascending graded ethanol series. Ethanol was replaced with propylene oxide before embedding in low viscosity resin (Agar Scientific Ltd.). Sections of 60-80 nm (silver to pale gold iridescence) were post-stained with saturated aqueous uranyl acetate (30 minutes) washed in deionised water, then with Reynolds lead citrate (3-5 minutes) and washed. Alternatively, they were stained with phosphotungstic acid and chromic acid for 30 minutes (Ryter and de Chastellier, 1977). Sections were viewed on a Philips EM200 transmission electron microscope with an 80 kV beam; vesicle quantification was carried out with ImageJ (NIH).
Supplementary Material
Acknowledgments
We would like to acknowledge all members of the Kay group for useful discussions and Gunther Gerisch for the dajumin-GFP construct. Core funding was from the Medical Research Council. Deposited in PMC for release after 6 months.
Footnotes
Supplementary material available online at http://jcs.biologists.org/cgi/content/full/123/19/3226/DC1
References
- Aguado-Velasco C., Bretscher M. S. (1999). Circulation of the plasma membrane in Dictyostelium. Mol. Biol. Cell 10, 4419-4427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagorda A., Das S., Rericha E. C., Chen D., Davidson J., Parent C. A. (2009). Real-time measurements of cAMP production in live Dictyostelium cells. J. Cell Sci. 122, 3907-3914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betz W. J., Mao F., Smith C. B. (1996). Imaging exocytosis and endocytosis. Curr. Opin. Neurobiol. 6, 365-371 [DOI] [PubMed] [Google Scholar]
- Bloomfield G., Tanaka Y., Skelton J., Ivens A., Kay R. R. (2008). Widespread duplications in the genomes of laboratory stocks of Dictyostelium discoideum. Genome Biol. 9, R75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bretscher M. S. (1984). Endocytosis: relation to capping and cell locomotion. Science 224, 681-686 [DOI] [PubMed] [Google Scholar]
- Bretscher M. S. (1996). Getting membrane flow and the cytoskeleton to cooperate in moving cells. Cell 87, 601-606 [DOI] [PubMed] [Google Scholar]
- Bretscher M. S., Clotworthy M. (2007). Using single loxP sites to enhance homologous recombination: ts mutants in Sec1 of Dictyostelium discoideum. PLoS ONE 2, e724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carr C. M., Grote E., Munson M., Hughson F. M., Novick P. J. (1999). Sec1p binds to SNARE complexes and concentrates at sites of secretion. J. Cell Biol. 146, 333-344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dormann D., Libotte T., Weijer C. J., Bretschneider T. (2002). Simultaneous quantification of cell motility and protein-membrane-association using active contours. Cell Motil. Cytoskeleton 52, 221-230 [DOI] [PubMed] [Google Scholar]
- Du F., Edwards K., Shen Z., Sun B., De Lozanne A., Briggs S., Firtel R. A. (2008). Regulation of contractile vacuole formation and activity in Dictyostelium. EMBO J. 27, 2064-2076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabriel D., Hacker U., Kohler J., Muller-Taubenberger A., Schwartz J. M., Westphal M., Gerisch G. (1999). The contractile vacuole network of Dictyostelium as a distinct organelle: its dynamics visualized by a GFP marker protein. J. Cell Sci. 112, 3995-4005 [DOI] [PubMed] [Google Scholar]
- Gauthier N. C., Rossier O. M., Mathur A., Hone J. C., Sheetz M. P. (2009). Plasma membrane area increases with spread area by exocytosis of a GPI-anchored protein compartment. Mol. Biol. Cell 20, 3261-3272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerisch G., Heuser J., Clarke M. (2002). Tubular-vesicular transformation in the contractile vacuole system of Dictyostelium. Cell Biol. Int. 26, 845-852 [DOI] [PubMed] [Google Scholar]
- Gerisch G., Benjak A., Kohler J., Weber I., Schneider N. (2004). GFP-golvesin constructs to study Golgi tubulation and post-Golgi vesicle dynamics in phagocytosis. Eur. J. Cell Biol. 83, 297-303 [DOI] [PubMed] [Google Scholar]
- Grote E., Carr C. M., Novick P. J. (2000). Ordering the final events in yeast exocytosis. J. Cell Biol. 151, 439-452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallett M. B., Dewitt S. (2007). Ironing out the wrinkles of neutrophil phagocytosis: membrane reservoirs for surface area expansion. Trends Cell Biol. 17, 209-214 [DOI] [PubMed] [Google Scholar]
- Heuser J. (2006). Evidence for recycling of contractile vacuole membrane during osmoregulation in Dictyostelium amoebae-a tribute to Gunther Gerisch. Eur. J. Cell Biol. 85, 859-871 [DOI] [PubMed] [Google Scholar]
- Hicke L., Zanolari B., Pypaert M., Rohrer J., Riezman H. (1997). Transport through the yeast endocytic pathway occurs through morphologically distinct compartments and requires an active secretory pathway and Sec18p/N-ethylmaleimide-sensitive fusion protein. Mol. Biol. Cell 8, 13-31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoeller O., Kay R. R. (2007). Chemotaxis in the absence of PIP3 gradients. Curr. Biol. 17, 813-817 [DOI] [PubMed] [Google Scholar]
- Jenne N., Rauchenberger R., Hacker U., Kast T., Maniak M. (1998). Targeted gene disruption reveals a role for vacuolin B in the late endocytic pathway and exocytosis. J. Cell Sci. 111, 61-70 [DOI] [PubMed] [Google Scholar]
- Jungbluth A., Vonarnim V., Biegelmann E., Humbel B., Schweiger A., Gerisch G. (1994). Strong increase in the tyrosine phosphorylation of actin upon inhibition of oxidative phosphorylation-correlation with reversible rearrangements in the actin skeleton of Dictyostelium cells. J. Cell Sci. 107, 117-125 [DOI] [PubMed] [Google Scholar]
- Kay R. R. (1987). Cell differentiation in monolayers and the investigation of slime mold morphogens. Methods Cell Biol. 28, 433-448 [DOI] [PubMed] [Google Scholar]
- Kay R. R., Langridge P., Traynor D., Hoeller O. (2008). Changing directions in the study of chemotaxis. Nat. Rev. Mol. Cell Biol. 9, 455-463 [DOI] [PubMed] [Google Scholar]
- King J. S., Insall R. H. (2009). Chemotaxis: finding the way forward with Dictyostelium. Trends Cell Biol. 19, 523-530 [DOI] [PubMed] [Google Scholar]
- Knecht D., Pang K. M. (1995). Electroporation of Dictyostelium discoideum. Methods Mol. Biol. 47, 321-330 [DOI] [PubMed] [Google Scholar]
- Kriebel P. W., Barr V. A., Parent C. A. (2003). Adenylyl cyclase localization regulates streaming during chemotaxis. Cell 112, 549-560 [DOI] [PubMed] [Google Scholar]
- Laevsky G., Knecht D. A. (2001). Under-agarose folate chemotaxis of Dictyostelium discoideum amoebae in permissive and mechanically inhibited conditions. Biotechniques 31, 1140-1149 [DOI] [PubMed] [Google Scholar]
- Lammermann T., Bader B. L., Monkley S. J., Worbs T., Wedlich-Soldner R., Hirsch K., Keller M., Forster R., Critchley D. R., Fassler R., et al. (2008). Rapid leukocyte migration by integrin-independent flowing and squeezing. Nature 453, 51-55 [DOI] [PubMed] [Google Scholar]
- Langridge P. D., Kay R. R. (2006). Blebbing of Dictyostelium cells in response to chemoattractant. Exp. Cell Res. 312, 2009-2017 [DOI] [PubMed] [Google Scholar]
- Langridge P. D., Kay R. R. (2007). Mutants in the Dictyostelium Arp2/3 complex and chemoattractant-induced actin polymerization. Exp. Cell Res. 313, 2563-2574 [DOI] [PubMed] [Google Scholar]
- Lawson M. A., Maxfield F. R. (1995). Ca(2+)- and calcineurin-dependent recycling of an integrin to the front of migrating neutrophils. Nature 377, 75-79 [DOI] [PubMed] [Google Scholar]
- Maeda Y., Gerisch G. (1977). Vesicle formation in Dictyostelium discoideum cells during oscillations of cAMP synthesis and release. Exp. Cell Res. 110, 119-126 [DOI] [PubMed] [Google Scholar]
- Malhotra V., Orci L., Glick B. S., Block M. R., Rothman J. E. (1988). Role of an N-ethylmaleimide-sensitive transport component in promoting fusion of transport vesicles with cisternae of the Golgi stack. Cell 54, 221-227 [DOI] [PubMed] [Google Scholar]
- Manstein D. J., Schuster H. P., Morandini P., Hunt D. M. (1995). Cloning vectors for the production of proteins in Dictyostelium discoideum. Gene 162, 129-134 [DOI] [PubMed] [Google Scholar]
- Mohandas N., Evans E. (1994). Mechanical properties of the red cell membrane in relation to molecular structure and genetic defects. Annu. Rev. Biophys. Biomol. Struct. 23, 787-818 [DOI] [PubMed] [Google Scholar]
- Muller-Taubenberger A., Lupas A. N., Li H. W., Ecke M., Simmeth E., Gerisch G. (2001). Calreticulin and calnexin in the endoplasmic reticulum are important for phagocytosis. EMBO J. 20, 6772-6782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noegel A. A., Schleicher M. (2000). The actin cytoskeleton of Dictyostelium: a story told by mutants. J. Cell Sci. 113, 759-766 [DOI] [PubMed] [Google Scholar]
- Novick P., Field C., Schekman R. (1980). Identification of 23 complementation groups required for post-translational events in the yeast secretory pathway. Cell 21, 205-215 [DOI] [PubMed] [Google Scholar]
- Pang K. M., Lee E., Knecht D. A. (1998). Use of a fusion protein between GFP and an actin-binding domain to visualize transient filamentous-actin structures. Curr. Biol. 8, 405-408 [DOI] [PubMed] [Google Scholar]
- Quiviger B., de Chastellier C., Ryter A. (1978). Cytochemical demonstration of alkaline phosphatase in the contractile vacuole of Dictyostelium discoideum. J. Ultrastruct. Res. 62, 228-236 [DOI] [PubMed] [Google Scholar]
- Raucher D., Sheetz M. P. (2000). Cell spreading and lamellipodial extension rate is regulated by membrane tension. J. Cell Biol. 148, 127-136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryter A., de Chastellier C. (1977). Morphometric and cytochemical studies of Dictyostelium discoideum in vegetative phase. Digestive system and membrane turnover. J. Cell Biol. 75, 200-217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider N., Schwartz J. M., Kohler J., Becker M., Schwarz H., Gerisch G. (2000). Golvesin-GFP fusions as distinct markers for Golgi and post-Golgi vesicles in Dictyostelium cells. Biol. Cell 92, 495-511 [DOI] [PubMed] [Google Scholar]
- Schoen C. D., Arents J. C., Bruin T., Van Driel R. (1989). Intracellular localization of secretable cAMP in relaying Dictyostelium discoideum cells. Exp. Cell Res. 181, 51-62 [DOI] [PubMed] [Google Scholar]
- Shaffer B. M. (1975). Secretion of cyclic AMP induced by cyclic AMP in the cellular slime mould Dictyostelium discoideum. Nature 255, 549-552 [DOI] [PubMed] [Google Scholar]
- Shen J., Tareste D. C., Paumet F., Rothman J. E., Melia T. J. (2007). Selective activation of cognate SNAREpins by Sec1/Munc18 proteins. Cell 128, 183-195 [DOI] [PubMed] [Google Scholar]
- Sudhof T. C., Rothman J. E. (2009). Membrane fusion: grappling with SNARE and SM proteins. Science 323, 474-477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swaney K. F., Huang C. H., Devreotes P. N. (2010). Eukaryotic chemotaxis: a network of signaling pathways controls motility, directional sensing, and polarity. Annu. Rev. Biophys. 39, 265-289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson C. R. L., Kay R. R. (2000). Cell-fate choice in Dictyostelium: intrinsic biases modulate sensitivity to DIF signaling. Dev. Biol. 227, 56-64 [DOI] [PubMed] [Google Scholar]
- Thompson C. R. L., Bretscher M. S. (2002). Cell polarity and locomotion, as well as endocytosis, depend on NSF. Development 129, 4185-4192 [DOI] [PubMed] [Google Scholar]
- Traynor D., Kay R. R. (2007). Possible roles of the endocytic cycle in cell motility. J. Cell Sci. 120, 2318-2327 [DOI] [PubMed] [Google Scholar]
- Traynor D., Milne J. L., Insall R. H., Kay R. R. (2000). Ca(2+) signalling is not required for chemotaxis in Dictyostelium. EMBO J. 19, 4846-4854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valdez-Taubas J., Pelham H. R. (2003). Slow diffusion of proteins in the yeast plasma membrane allows polarity to be maintained by endocytic cycling. Curr. Biol. 13, 1636-1640 [DOI] [PubMed] [Google Scholar]
- Van Haastert P. J. M., Devreotes P. N. (2004). Chemotaxis: signalling the way forward. Nat. Rev. Mol. Cell Biol. 5, 626-634 [DOI] [PubMed] [Google Scholar]
- Weber I., Wallraff E., Albrecht R., Gerisch G. (1995). Motility and substratum adhesion of Dictyostelium wild-type and cytoskeletal mutant cells: a study by RICM/bright-field double-view image analysis. J. Cell Sci. 108, 1519-1530 [DOI] [PubMed] [Google Scholar]
- Wessels D., Voss E., Von B. N., Burns R., Stites J., Soll D. R. (1998). A computer-assisted system for reconstructing and interpreting the dynamic three-dimensional relationships of the outer surface, nucleus and pseudopods of crawling cells. Cell. Motil. Cytoskeleton 41, 225-246 [DOI] [PubMed] [Google Scholar]
- Xiao Z., Zhang N., Murphy D. B., Devreotes P. N. (1997). Dynamic distribution of chemoattractant receptors in living cells during chemotaxis and persistent stimulation. J. Cell Biol. 139, 365-374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida K., Inouye K. (2001). Myosin II-dependent cylindrical protrusions induced by quinine in Dictyostelium: antagonizing effects of actin polymerization at the leading edge. J. Cell Sci. 114, 2155-2165 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.