

Abstract
The intermolecular hydroamination of allenes occurs readily with hydrazide nucleophiles, in the presence of 3–12% Ph3PAuNTf2. Mechanistic studies have been conducted to establish the resting state of the gold catalyst, the kinetic order of the reaction, the effect of ligand electronics on the overall rate, and the reversibility of the last steps in the catalytic cycle. We have found the overall reaction to be first order in gold and allene and zero order in nucleophile. Our studies suggest that the rate-limiting transition state for the reaction does not involve the nucleophile and that the active catalyst is monomeric in gold(I). Computational studies support an “outersphere” mechanism and predicts that a two-step, no intermediate mechanism may be operative. In accord with this mechanistic proposal, the reaction can be accelerated with the use of more electron deficient phosphine ligands on the gold(I) catalyst.
Keywords: hydroamination, mechanistic studies, gold(I) catalysis, allene activation, DFT calculations
Introduction
Gold(I)-catalyzed addition of carbon, oxygen, and nitrogen based nucleophiles to allenes has emerged as a powerful synthetic transformation in recent years.1 The stability of gold(I) complexes to air and moisture renders gold catalysis a practical and mild method for bench top synthesis.2 However, the mechanism of this important class of reactions is only beginning to be explored. While multiple mechanisms for the hydroamination, hydroalkoxylation and hydroarylation of allenes have been proposed, few studies regarding the nature of the active catalyst, the rate limiting step of the reaction, and the role of allene and nucleophile in the transformation have been performed.3 Part of the challenge of performing detailed kinetic studies of gold(I) catalyzed hydroamination reactions is that many of the most active catalysts employed are generated in situ from gold(I) trimers or the gold chloride and the corresponding silver salt, making it difficult to determine the precise concentration of catalyst employed.4 In 2009, we reported the intramolecular hydroamination reaction of allenes with hydrazine nucleophiles catalyzed by an isolable gold(I) complexes.5 We hypothesized that an intermolecular variant of this transformation could provide a valuable platform for a systematic study of the mechanism of the reaction.
While experimental observations in gold(III)-catalyzed hydroamination and hydroalkoxylation reactions suggest that these reactions proceed through an innersphere mechanism,6 both innersphere and outersphere mechanisms for nucleophilic addition to allenes promoted by gold(I) complexes have been proposed (Scheme 1).7 In the outersphere mechanism the cationic gold(I) complex acts as a π-acid to induce addition of the nucleophile across the C-C π-bond. Protodemetalation of the vinyl gold intermediate then regenerates the gold(I) catalyst. In the innersphere mechanism a tricoordinate gold complex is formed by complexation of both the allene and the nucleophile. Addition of the nucleophile across the allene then proceeds directly from this complex. In both mechanisms, a monomeric cationic gold(I) complexes are proposed to be the catalytically relevant species. However, the “aurophilicity” of gold(I)8 and the isolation of a vinyl-gold dimer in a gold-catalyzed hydroarylation reaction by Gagné3d, suggest that dinuclear gold(I) complexes should also be considered as potential intermediates in gold(I) mediated processes. Thus, a kinetic study of this class of reactions is needed to determine the role of each reagent in the catalytic cycle and the composition of the rate limiting transition state.
Scheme 1.

Outersphere and innersphere mechanisms proposed for the hydroamination of allenes with gold(I) catalysts.
Toward this goal, we have found an intermolecular gold(I)-catalyzed hydroamination reaction of allenes by hydrazide nucleophiles and investigated the mechanism of this transformation. To distinguish between the inner- and outersphere mechanisms, and to discover the stoichiometry of gold catalyst in the rate-determining transition state, a detailed study was performed to determine: (1) the order of the reaction in each reagent (2) the resting state of the catalyst, the nature of other intermediates within the catalytic cycle (3) the effect of electronics of the catalyst on the rate of reaction and (4) the reversibility of the reaction. These investigations provide evidence that the rate-limiting transition state involves an activated allene complex. Trapping of this complex by the nucleophile and protodemetalation to regenerate the gold catalyst are rapid and likely irreversible.
Results and Discussion
We began our studies with 1,7-diphenylhepta-3,4-diene (1), because the symmetry of the substrate greatly simplified the number of chemically distinct environments in which the gold(I) catalyst could coordinate. Recently, Ph3PAuNTf2 has been effectively used in the activation of carbon-carbon multiple bonds9 and we hypothesized that this isolable complex would be a reliable Au+ source for mechanistic analysis. Gratifyingly, we found that 1 underwent the desired reaction with 2.5 equiv of methyl carbazate and 9 mol% Ph3PAuNTf2 in MeNO2 at 45 °C, providing the hydroamination products in a 5:1 trans:cis ratio with >95% conversion after 12 h (eq 1). Monitoring the first 3.25 h of the reaction by 1H NMR at 45 °C showed that the reaction was remarkably clean and that the total concentration of 1 and 2 during the course of the reaction was consistent with the concentration of 2 at t = 0 (Figure 1). In addition, the hydroamination does not proceed appreciably in the presence of 20 mol% HNTf2 or 10 mol% AgNTf2. We were encouraged by these results to further examine the precise role of the Ph3PAuNTf2 in the catalytic cycle.
Figure 1.

Reaction of 1 with methyl carbazate where □ = [ 1], △ = [2], and ○ = [1] + [2].
 |
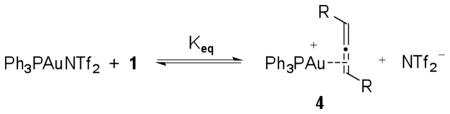
(1) |
Catalyst Resting State
Phosphorous NMR methods have been previously used to study gold(I) intermediates and the 31P signal of the phosphine ligand was found to be very sensitive to the electronic environment around the gold center10. Upon addition of the catalyst to a reaction mixture containing 1 and methyl carbazate, the signal corresponding to the Ph3PAuNTf2 (δ = 30.3 ppm) vanished and a new signal at 45.2 ppm was observed. As the reaction progressed to higher conversion and the concentration of 1 remaining in solution was comparable to that of the catalyst, a second broad peak at 30 ppm also appeared (Figure 2).
Figure 2.

The hydroamination reaction was monitored by 31P NMR spectroscopy.
Both gold-amine11 and gold-allene12 complexes have been previously postulated as intermediates in gold catalyzed reactions. To determine the nature of the compounds comprising the observed peaks at 30 and 45 ppm, we independently prepared two samples containing (Ph3PAuNTf2 and methyl carbazate, in a 1:1 ratio) and (Ph3PAuNTf2 and 1, in a 1:10 ratio) in CD3NO2. The sample containing Ph3PAuNTf2 and methyl carbazate showed a broadened 31P NMR signal at δ 29.7 ppm (eq 2), in the same region as the second peak that appeared as catalytic reaction progressed. The second sample containing Ph3AuNTf2 and 1, however, showed the same 31P NMR signal at 45.2 ppm as was observed in the catalytic reaction (eq 3).
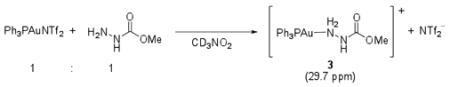 |
(2) |
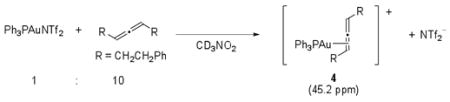 |
(3) |
The signal at 45.2 ppm is consistent with the 31P NMR shifts of other alkyl- and vinyl-AuPPh3 species reported in the literature13. In addition, the same species can be independently generated from a solution of Ph3PAuCl and AgBF4 in the presence of 1 without additional NTf2−. Thus, we hypothesized that this peak corresponded to a Ph3PAu+-allene complex. In the catalytic reaction, this complex is in equilibrium with other cationic gold species, such as Ph3PAu+ and Ph3PAu+-carbazate. When the concentration of 1 was relatively high at the beginning of the reaction, the equilibrium favored the gold-allene complex. As the reaction progressed, the concentration of 1 decreased and the equilibrium shifted such that the other cationic gold species were also observable by NMR.14
Due to the C2 symmetry of the allene, two diastereotopic π-gold complexes could theoretically be formed; yet we could not resolve the two complexes by either 31P or 1H NMR at low temperature (0 to −84 °C). However, computational studies have predicted the barrier for exchange between the two diastereomeric faces of a simple disubstituted alkyl allene to be very low15. In addition, the energy difference between the two π-complexes that could form is calculated to be only 1.0 kcal/mol. Thus, it is likely that under catalytic reaction conditions, this species is two-coordinate cationic gold-allene complex with a fully dissociated counterion, where the gold center is rapidly exchanging between the two faces of the allene.
Kinetic Order of the Reaction
Order in Nucleophile
In order to determine the rate-determining transition state for the catalytic reaction, we sought to establish kinetic order of the reaction in Ph3PAuNTf2, 1, and methyl carbazate. We began our studies by examining the order of the Ph3PAuNTf2 catalyzed hydroamination reaction in nucleophile by the method of initial rates. The allene (1) (90.9 mM) and Ph3PAuNTf2 (5.45 mM) was combined with varying concentrations of methyl carbazate from 90.0 to 295.5 mM in CD3NO2 at 45 °C. The reactions were monitored by 1H NMR and a plot of kobs,16 (as determined by (Δ[2]/Δt)/[1]0, where [1]0 = 90.91 mM) at each concentration of methyl carbazate provided a straight line with R2 = 0.9923 and a slope of −0.0001 h−1 (Figure 3). The order in methyl carbazate was verified by a least squares fit to f(x) = axn, which provided n = −0.108 (Supporting Information, Figure S3). These results indicate that methyl carbazate is not involved in the rate-determining transition state. In contrast, previously proposed mechanisms have implied nucleophilic addition or protodemetalation to be the rate-limiting step in gold(I) catalyzed hydroamination17.
Figure 3.

A plot of kobs versus concentration of methyl carbazate, where kobs was determined by the method of initial rates.
Order in Allene
The order in 1 was next determined by monitoring the reaction of 1 (150 mM), methyl carbazate (325 mM) and Ph3PAuNTf2 (13.5 mM) in CD3NO2 at 45 °C (Figure 4) by 1H NMR.18 The reaction followed pseudo first order kinetics in 1 and provided linear plots of −ln([1]t/[1]0) vs. time with R2 = 0.9995. The pseudo first order rate constants (kobs) measured reflects the rate of formation of both trans and cis hydroamination products. The first order dependence on 1 implies that the allene is involved in the turnover limiting transition state and that there is an equilibrium between the free and gold-bound allene.
Figure 4.

A representative plot of −ln[1(t)/10] versus time (h) for the reaction of 1 and methyl carbazate shows first order dependence in 1.
Order in Gold
The order in Ph3PAuNTf2 was examined by measuring the pseudo-first order rate constant, kobs, for the reaction at various catalyst loadings. Allene 1 (150 mM) and methyl carbazate (325 mM) were combined with various concentrations of Ph3PAuNTf2 (4.5 to 18 mM) in CD3NO2 at 45 °C. Plotting the measured kobs of the reaction at a range of catalyst loadings provided a straight line with R2 = 0.9993 (Figure 5). This result suggests that the overall reaction is first order in gold and implies that the rate-limiting transition state contains only one gold center.
Figure 5.

A plot of kobs versus [Ph3PAuNTf2], using 2.5 equiv methyl carbazate.
Hammett Analysis
Our group has previously investigated the ability ligand electronics to affect the rate of certain pathways in a gold(I) catalyzed process.19 Thus, a Hammett study of the reaction was performed to probe the sensitivity of the reaction to changes in the electronic properties of the phosphine ligand in the Ar3PAuNTf2 catalyst. Toward this goal, phosphine gold catalysts bearing substitution in the para position were prepared. The reaction of 1 (150 mM) and methyl carbazate (325 mM) in the presence of 6 mol % (p-X-C6H4)3PAuNTf2 (X = CF3, Cl, H, and OMe) in CD3NO2 at 45 °C was monitored by 1H NMR. Plotting log[(kobs,X)/(kobs,H)] against σ provided a straight line (R2 = 0.96) with ϱ = 0.21 (Figure 6). A positive ϱ value is consistent with a decrease in partial positive charge on the phosphine ligand in the transition state. The kinetic order and Hammett analysis of the reaction are consistent with the gold(I) activation of allene as the rate-limiting transition state in the catalytic cycle.
Figure 6.

A Hammett plot of kobs for hydroamination of 1 in the presence of Ar3PAuNTf2.
Chirality Transfer
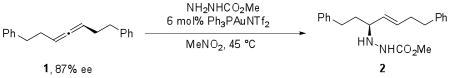
We sought to determine whether chirality transfer was possible from the allene to the hydroamination product to determine whether the reaction went through chiral or planar gold intermediates. While trisubstituted allenes are known to racemize rapidly in the presence of gold(I) catalysts,20 good chirality transfer has been observed with some unfunctionalized disubstituted allenes.21 In addition, chirality transfer would exclude a planar allyl cation as part of the catalytic cycle, as such a species would lead to complete racemization. Enantioenriched 1 (101 nM, 87% ee) was prepared by the method described by Myers22 and was combined with methyl carbazate and Ph3PAuNTf2 at 45 °C. The enantiomeric excess of the major hydroamination product 2trans was determined by chiral HPLC analysis and summarized in Table 1.
Table 1.
Chirality transfer for the hydroamination of chiral 1 in the presence of Ph3PAuNTf2 catalyst at various equiv of methyl carbazate.
 | ||
|---|---|---|
| Equiv of Nuc | yield (%) | ee (%) |
| 1.0 | 81 | 28 |
| 2.0 | 75 | 48 |
| 4.0 | 81 | 56 |
| 8.0 | 79 | 56 |
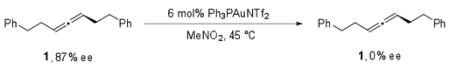
Interestingly, though some chirality transfer was observed at all concentrations of nucleophile, the highest levels of transfer were observed for reactions where greater than 4 equiv of methyl carbazate was used. This indicates that the reaction cannot be proceeding exclusively through a planar intermediate. In addition, the ability of nucleophile concentration to affect chirality transfer suggests that racemization can occur from the chiral allyl-gold intermediate and this process proceeds at a rate competitive with trapping of the intermediate by methyl carbazate. In the absence of any nucleophile, 1 completely racemizes in the presence of the catalyst after 12 h at 45 °C (eq 4).
 |
(4) |
Reversibility of the Reaction
While protodeauration of the gold catalyst is conventionally depicted as an irreversible process in a catalytic reaction, Lee recently reported an example of reversible hydroalkoxylation23 in the presence of Ph3PAuNTf2. Furthermore, an irreversible reaction under catalytic conditions would allow us to exclude the possibility that the incomplete chirality transfer we observed previously was due to racemization of the product. To probe whether the intermolecular hydroamination reaction is reversible, enantioenriched 2 (56% ee) was subjected to 2 equiv of methyl carbazate and 10 mol % achiral Ph3PAuNTf2. After 6 h at 45 °C, 2 was recovered in 56% ee with no apparent erosion of enantiopurity (eq 9), supporting an irreversible protodemetalation. However, conservation of enantiomeric excess could also result from a reversible reaction that regenerates a enantioenriched allene-gold intermediate that can then reform 2 with good chirality transfer.
 |
(5) |
We hypothesized that a competition experiment with a second hydrazide nucleophile would be able to distinguish between these possibilities. If a chiral allene or gold-allene complex was indeed being reversibly formed, hydroamination products from trapping with both hydrazide nucleophiles should be observed. In a competition experiment with 1 equiv methyl carbazate and 3 equiv t-butyl carbazate, both hydroamination products were observed in a 15:85 ratio in >95% conversion, indicating that formation of 5 is kinetically competitive with formation of 2. Thus, if the reaction is reversible and 2 is resubjected to catalytic conditions in the presence of 3 equiv of t-butyl carbazate, a mixture of 2 and 5 should be expected. However, heating the reaction to 45 °C for 8 hr produced 2 as the sole product. The lack of detectible amounts of 5 indicated that chiral allene or gold-allene intermediates could not be forming reversibly under the reaction conditions.
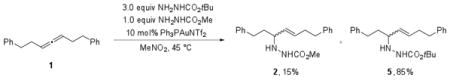 |
(6) |
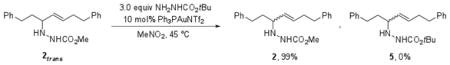 |
(7) |
Mechanistic Discussions
To corroborate the kinetic data observed and to gain insight into the nature of the intermediates in the catalytic cycle, we investigated the mechanism for hydroamination of allenes using density functional theory (DFT) with the B3LYP and M06 functionals. We used the experimental observations as a guide for exploring the geometry and potential energy surface for the reaction. We investigated the catalytic reaction of model (S)-penta-2,3-diene, methyl carbazate and Ph3PAu(I)+. We calculated the free energy of coordination of the allene to Ph3PAu(I)+ to be −9.2 kcal/mol, in favor of the allene-gold complex A.24 Activation of A proceeds through transition state TS1, which lies 4.9 kcal/mol higher in energy than the planar, cationic gold(I) complex 6. Though the methyl carbazate is included in calculations, the nucleophile itself is not associated with TS1 or 6. Malacria and co-workers reported15 that the steric hindrance at the allylic positions determines the energies and stabilities of the possible isomeric forms of the planar, cationic gold(I) complex. In agreement with their analysis, we find that only intermediate 6 is stable. In comparison, intermediates 7 and 8 are not minima and relax to A or A′.
As we moved further along the reaction coordinate, a second transition state (TS2) involving the addition of methyl carbazate to allene-gold specie was found. We find that transition state TS1 leads to adjacent transition state TS2 without an intervening intermediate. On the basis of our experimental observations and calculations, we propose a two-step, no intermediate25 process similar to the one described by Nevado for a Au(I) catalyzed cyclization.26 The geometry of the allene fragment in TS1 and TS2 resemble a bent allene geometry.15 Intermediate B (ΔG = −14.8 kcal/mol) undergoes protodeaureation leading to product (ΔG = −24.1 kcal/mol), in agreement with the irreversibility of the reaction.
The absence or presence of a short-lived intermediate between transition states TS1 and TS2 is affected by the relative concentration and reactivity of the nucleophile, and thus relates directly to the magnitude of chirality transfer. The reaction coordinate bifurcates following TS1, leading to TS2 or intermediate 6. At higher nucleophile concentrations, addition of the nucleophile is fast and the two-step no intermediate pathway is favored, leading to higher levels of chirality transfer. At lower concentrations of nucleophile, TS1 leaks to the planar intermediate 6 with loss of chirality. The operating reaction mechanism is a continuum between a traditional two-step process with loss of chirality and a two-step no-intermediate process with retention of the stereochemistry.
We studied the structural and electronic details of the located transition structures and compared them to Au-allene coordinated complex A. Shown in Figure 7 are key metrical parameters and natural charges derived from natural population analyses of the corresponding M06 wave function. We find that for A, the charge on C3 is least negative (vs. C1), while the charge on C1 is most negative of the allene carbon atoms.
Figure 7.

Structural and natural atomic charge of selected complexes.
On the basis of the kinetic, ligand effect, reversibility, chirality transfer, and computational studies performed, we propose a mechanistic picture for the gold(I)-catalyzed hydroamination of allenes, as shown in Scheme 4. The resting state of the catalyst is consistent with a cationic gold-allene complex that is rapidly exchanging between the two diastereomeric faces of the C2 symmetric allene and is in equilibrium with other cationic gold species, denoted [Au+]. Our computational studies suggest that a bent allene-gold complex is the rate determining transition state (TS1) in the catalytic cycle and our kinetic data provides direct experimental support for these calculations. The overall reaction exhibits zeroth order dependence on the concentration of nucleophile, suggesting that nucleophilic addition must occur after the rate-determining step. This observation is in agreement with calculated free energy of TS2, which is 3.6 kcal/mol lower than that of TS1. In addition, the pseudo first order dependence on the concentration of allene and Ph3PAuNTf2 suggest that the catalytic species is monomeric and that the ratio of allene to catalyst in the transition state is 1:1. Thus, an innersphere mechanism where the nucleophile coordinates to the gold center prior to or during allene activation is unlikely, as such a mechanism would exhibit positive order in nucleophile.
Scheme 4.

Proposed mechanism for hydroamination of 1 with Ph3PAuNTf2.
We have demonstrated that the nucleophillic addition and protodemetalation steps are irreversible under catalytic conditions and occur after the rate limiting transition state. Thus, our reaction can be simplified and modeled using Michaelis-Menten kinetics. The general rate law of the reaction follows the Michaelis-Menten equation (eq 8). The KM for the reaction can be calculated from a Line weaver-Burke plot of our kinetic data and was found to be 3.9(1.5)×103 mM (Supporting Information, Figure S8). Thus under the conditions examined, the concentration of 1 is small relative to 27 KM and the Michaelis-Menten equation can be reduced to eq 9, providing a rate law that is first order in gold and 1.
| (8) |
| (9) |
A Hammett study of the reaction demonstrates that changing the electronics of the Ar3P ligand noticeably affected the rate of reaction and provides quantitative evidence that gold(I) reactivity can be modulated by ligand electronics. In particular, phosphines with electron-withdrawing para substituents increased the rate of the reaction while electron-donating substituents slow the reaction relative to Ph3PAuNTf2. This is consistent with a rate-determining transition state that exhibits a reduction in δ+ character on the phosphine relative to the ground state. Our proposed transition state allows for the delocalization of positive charge from the Ph3PAu+ onto the allenic fragment, and is hence consistent with the ϱ value observed. Furthermore, the inability to regenerate the gold-allene intermediate from the isolated hydroamination products (2) under catalytic conditions suggests that either one or both the addition or the protodemetalation steps are irreversible.
Conclusions
We have reported the first example of intermolecular hydroamination of allenes with a N-N linked nucleophile in the presence of a gold(I) catalyst, Ph3PAuNTf2. This system has allowed us to conduct the first detailed kinetic study of the mechanism of gold(I)-mediated allene activation. Our experiments provide experimental evidence that allene activation is the rate-limiting step in gold(I)-catalyzed addition of hydrazide nucleophiles to allenes. Computational studies show that the reaction examined proceeds via a two-step no-intermediate mechanism and that the second transition state immediately following the rate determining step is not planar, but likely axially chiral. Importantly, the dependence of the degree of chirality transfer on the concentration of the nucleophile suggests that a pathway proceeding through a traditional two-step pathway involving a planar intermediate is also available. This mechanistic study has important implications for the future development of racemic and enantioselective C-X bond forming reactions via addition of a nucleophiles to allenes promoted by gold(I). Using these mechanistic insights, we are currently working toward developing and investigating the mechanism of gold(I)-catalyzed additions of various nucleophiles to allenes.
Supplementary Material
Scheme 2.

Geometries of (S)-penta-2,3-diene and [Ph3PAu]+ as coordination, transition state, and allylic cation complexes.
Scheme 3.

Partial reaction coordinate diagram for Au(I) catalyzed hydroamination of (S)-penta-2,3-diene. Energy values are free energies (ΔG) for the lowest energy isomer at 298 K.
Acknowledgments
We thank the NIHGMS (R01 GM073932) and the Director, Office of Science of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231 for funding and Johnson Mathey for a generous donation of AuCl3. Z. J. Wang thanks the Hertz Foundation for a graduate fellowship. We are also grateful to Dr. Robert Bergman for support during the kinetic studies. Computational facilities were funded by grants from ARO-DURIP and ONR-DURIP.
Footnotes
Supporting Information Available. Experimental procedures, analytic and spectroscopic data for new compounds, spectral and kinetic data, and computational details are including in Supporting Information.
References
- 1.For general reviews, see: Gorin DJ, Sherry BD, Toste FD. Chem Rev. 2008;108:3351. doi: 10.1021/cr068430g.Jiménez-Núñez E, Echavarren AM. Chem Commun. 2007:333. doi: 10.1039/b612008c.Furstner A, Davies PW. Angew Chem, Int Ed. 2007;46:3410. doi: 10.1002/anie.200604335.Hashmi ASK. Chem Rev. 2007;107:3180. doi: 10.1021/cr000436x.Shen HC. Tetrahedron. 2008;64:3885.Li Z, Brouwer C, He C. Chem Rev. 2008;108:3239. doi: 10.1021/cr068434l.
- 2.(a) Gorin DJ, Toste FD. Nature. 2007;446:395. doi: 10.1038/nature05592. [DOI] [PubMed] [Google Scholar]; (b) Shapiro ND, Toste FD. Synlett. 2010:675. doi: 10.1055/s-0029-1219369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.For examples, see: Zhang Z, Liu Cong, Kinder RE, Han X, Qian H, Widenhoefer RA. J Am Chem Soc. 2006;128:9066. doi: 10.1021/ja062045r.Nishina N, Yamamoto Y. Tetrahedron. 2009;65:1799.Duncan AN, Widenhoefer RA. Synlett. 2010:419. doi: 10.1055/s-0029-1218555.Weber D, Tarselli MA, Gagné MR. Angew Chem Int Ed. 2009;48:5733. doi: 10.1002/anie.200902049.
- 4.For selected examples, see: Hyland CT, Hegedus LS. J Org Chem. 2006;71:8658. doi: 10.1021/jo061340r.Zhang Z, Bender CF, Widenhoefer RA. J Am Chem Soc. 2007;129:14148. doi: 10.1021/ja0760731.Liu C, Widenhoefer RA. Org Lett. 2007;9:1935. doi: 10.1021/ol070483c.Piera J, Krumlinde P, Strübing D, Bäckvall JE. Org Lett. 2007;9:2235. doi: 10.1021/ol070793v.Hamilton GL, Kang EJ, Mba M, Toste FD. Science. 2007;317:496. doi: 10.1126/science.1145229.For an example of a gold(I) trimer precatalyst, see: Sherry BD, Maus L, Laforteza BN, Toste FD. J Am Chem Soc. 2006;128:8132. doi: 10.1021/ja061344d.
- 5.Lalonde RL, Wang JZ, Mba M, Lackner AD, Toste FD. Angew Chem Int Ed. 2009;49:598. doi: 10.1002/anie.200905000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nishina N, Yamamoto Y. Angew Chem Int Ed. 2006;45:3144. doi: 10.1002/anie.200600331. [DOI] [PubMed] [Google Scholar]
- 7.For proposed outersphere mechanisms, see: Widenhoefer RA, Han Q. Eur J Org Chem. 2006:4555.and Reference 3a. For proposed innersphere mechanisms, see: Zeng X, Soleilhavoup M, Bertrand G. Org Lett. 2009;11:3166. doi: 10.1021/ol901418c.and reference 3b. For evidence of gold-catalyzed outersphere hydroamination of alkenes, see: LaLonde RL, Brenzovich WE, Jr, Benitez D, Tkatchouk E, Kelley K, Goddard WA, III, Toste FD. Chem Sci. 2010;1:226. doi: 10.1039/C0SC00255K.
- 8.(a) Schmidbaur H. Gold Bulletin. 2000;33:3. [Google Scholar]; (b) Scherbaum F, Grohmann A, Huber Brigitte, Kruger C, Schmidbaur H. Angew Chem Int Ed. 1988;27:1544. [Google Scholar]; (c) Cheong PH, Morganelli P, Luzung MR, Houk KN, Toste FD. J Am Chem Soc. 2008;130:4517. doi: 10.1021/ja711058f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.(a) Mezailles N, Ricard L, Gagosz F. Org Lett. 2005;7:4133. doi: 10.1021/ol0515917. [DOI] [PubMed] [Google Scholar]; (b) Toullec PY, Blarre T, Michelet V. Org Lett. 2009;11:2888. doi: 10.1021/ol900864n. [DOI] [PubMed] [Google Scholar]
- 10.(a) Grisé CM, Barriault L. Org Lett. 2006;8:5905. doi: 10.1021/ol062582g. [DOI] [PubMed] [Google Scholar]; (b) Grisé CM, Rodrigue EM, Barriault L. Tetrahedron. 2008;64:797. [Google Scholar]; (c) Seidel G, Mynott R, Fürstner A. Angew Chem Int Ed. 2009;48:2510. doi: 10.1002/anie.200806059. [DOI] [PubMed] [Google Scholar]
- 11.Mizushima E, Hayashi T, Tanaka M. Org Lett. 2003;5:3349. doi: 10.1021/ol0353159.Also see reference 3b and 7b.
- 12.Mauleon P, Krinsky JL, Toste FD. J Am Chem Soc. 2009;131:4513. doi: 10.1021/ja900456m.Benitez D, Tkatchouk E, Gonzales AZ, Goddard WA, III, Toste FD. Org Lett. 2009;11:4798. doi: 10.1021/ol9018002.Deutsch C, Gockel B, Hoffman-Roder A, Krause N. Synlett. 2007;11:1790.For an example with gold(III), see: Morita N, Krause N. Org Lett. 2004;6:4121. doi: 10.1021/ol0481838.
- 13.(a) Hashmi ASK, Ramamurthi TD, Rominger F. J Organomet Chem. 2009;694:592. [Google Scholar]; (b) Shi Y, Ramgren SD, Blum SA. Organometallics. 2009;28:1275. [Google Scholar]; (c) Mohr F, Falvello LR, Laguna M. Eur J Inorg Chem. 2006:833. [Google Scholar]; (d) Porter KA, Schier A, Schmidbaur H. Organometallics. 2003;22:4922. [Google Scholar]; (e) Herrero-Gómez E, Nieto-Oberhuber C, López J, Benet-Buchholz J, Echavarren AM. Angew Chem Int Ed. 2006;45:5455. doi: 10.1002/anie.200601688. [DOI] [PubMed] [Google Scholar]
-
14.The composition of the cationic gold species observed under catalytic conditions could not be determined as both Ph3PAu+ and 3 show phosphorous NMR signals around 30 ppm. However, in the absence of nucleophile, the Keq for the equilibrium between 1, Ph3PAuNTf2, and 4 was found to be 1.32 at 50 °C using 31P NMR techniques with PPh3 internal standard.
 This value is within an order of magnitude to the Keq reported for structurally similar gold(I) π-alkene complexes. For example, see Brown TJ, Dickens MG, Widenhoefer RA. J Am Chem Soc. 2009;131:6350. doi: 10.1021/ja9015827.
This value is within an order of magnitude to the Keq reported for structurally similar gold(I) π-alkene complexes. For example, see Brown TJ, Dickens MG, Widenhoefer RA. J Am Chem Soc. 2009;131:6350. doi: 10.1021/ja9015827.
- 15.Gandon V, Lemiére G, Hours A, Fensterbank L, Malacria M. Angew Chem Int Ed. 2008;47:7534. doi: 10.1002/anie.200802332. [DOI] [PubMed] [Google Scholar]
- 16.The pseudo first order rate constants (kobs) measured reflects the rate of formation of both trans and cis hydroamination products.
- 17.For examples involving showing nucleophilic addition as the first irreversible step in the catalytic cycle, see ref. 3b and Kinder RE, Zhang Z, Widenhoefer RA. Org Lett. 2008;10:3157. doi: 10.1021/ol8010858.For studies involving hydroamination of dienes and olefins, see Kovacs G, Ujaque G, Lledos A. J Am Chem Soc. 2008;130:853. doi: 10.1021/ja073578i.and ref 7c.
- 18.Also see Supporting Information, Figure S4.
- 19.Mauleon P, Zeldin RM, Gonzalez AZ, Toste FD. J Am Chem Soc. 2009;131:6348. doi: 10.1021/ja901649s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.(a) Sherry BD, Toste FD. J Am Chem Soc. 2004;126:15978. doi: 10.1021/ja044602k. [DOI] [PubMed] [Google Scholar]; (b) Lee JH, Toste FD. Angew Chem Int Ed. 2007;46:912. doi: 10.1002/anie.200604006. [DOI] [PubMed] [Google Scholar]
- 21.(a) Zhang Z, Widenhoefer RA. Org Lett. 2008;10:2079. doi: 10.1021/ol800646h. [DOI] [PubMed] [Google Scholar]; (b) Patil NT, Lutete LM, Nishina N, Yamamoto Y. Tetrahedron Lett. 2006;47:4749. [Google Scholar]
- 22.Myers AG, Zheng B. J Am Chem Soc. 1996;118:4492. [Google Scholar]
- 23.Hadfield MS, Lee A. Org Lett. 2010;12:484. doi: 10.1021/ol902675k. [DOI] [PubMed] [Google Scholar]
- 24.We calculate isomer A′ to be ~1 kcal/mol higher in free energy than A.
- 25.Singleton DA, Hang C, Szymanski MJ, Meyer MP, Leach AG, Kuwata KT, Chen JS, Greer A, Foote CS, Houk KN. J Am Chem Soc. 2003;125:1319. doi: 10.1021/ja027225p. [DOI] [PubMed] [Google Scholar]
- 26.Garayalde D, Gomez-Bengoa E, Huang XG, Goeke A, Nevado C. J Am Chem Soc. 2010;132:4720. doi: 10.1021/ja909013j. [DOI] [PubMed] [Google Scholar]
- 27.The concentration of substrate examined in our kinetic experiments ranged from 90.1 mM to 150 mM.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.