

Abstract
Here we report the identification and approximate quantification of cytochrome P450 (CYP) proteins in human liver microsomes as determined by nano-LC-MS/MS with application of the exponentially modified protein abundance index (emPAI) algorithm during database searching. Protocols based on 1D-gel protein separation and 2D-LC peptide separation gave comparable results. In total 18 CYP isoforms were unambiguously identified based on unique peptide matches. Further, we have determined the absolute quantity of two CYP enzymes (2E1 and 1A2) in human liver microsomes using stable-isotope dilution mass spectrometry, where microsomal proteins were separated by 1D-gel electrophoresis, digested with trypsin in the presence of either a CYP2E1- or 1A2-specific stable-isotope labelled tryptic peptide and analysed by LC-MS/MS. Using multiple reaction monitoring (MRM) for the isotope-labelled tryptic peptides and their natural unlabelled analogues quantification could be performed over the range of 0.1 – 1.5 pmol on column. Liver microsomes from four individuals were analysed for CYP2E1 giving values of 88 - 200 pmol/mg microsomal protein. The CYP1A2 content of microsomes from a further three individuals ranged from 165 – 263 pmol/mg microsomal protein. Although, in this proof-of-concept study for CYP quantification, the two CYP-isoforms were quantified from different samples, there are no practical reasons to prevent multiplexing the method to allow the quantification of multiple CYP-isoforms in a single sample.
Keywords: cytochrome P450, microsomes, label-free quantification, stable-isotope dilution mass spectrometry, LC-MS/MS, 2D-LC-MS/MS
Introduction
The liver is the main site of metabolism in human, where a variety of mechanisms are utilised to convert both endogenous and exogenous substances to more readily excreted compounds. However, during metabolism substrates can also be converted into more reactive species which themselves may be toxic. Additionally, many different agents can induce the expression of enzymes involved in metabolism and thereby influence the fate of biomolecules and xenobiotics, including drugs. In this regard a knowledge of “drug – drug” interactions is clearly crucial for the prediction of the outcome of therapy and the possible side-effects of drugs1. Also, in the area of prodrug design knowledge of metabolism is a prerequisite, as prodrugs must be transformed into reactive compounds to exert their effect2. Hence the ability or inability of a certain tissue e.g. tumour tissue, to metabolise a prodrug into the desired form can decide the success of therapy.
In human there are 57 putatively functional cytochrome P450 (CYP) genes (http://drnelson.utmem.edu/CytochromeP450.html), and the major CYP proteins quantitatively expressed in liver are 1A2, 2A6, 2B6, 2C8, 2C9, 2C19, 2D6, 2E1 and 3A4 as determined by immunochemical methods3,4. Of these the most abundant are CYP3A4 (~30%), 2C9 (~20%) and 1A2 (~15%)3. CYP1A2 is of particular interest as it is involved in carcinogen activation3. It is known to activate carcinogens in tobacco smoke into genotoxic compounds. For example arylamines and nitrosamines, which are prominent in tobacco smoke, have been associated with CYP1A2 mediated oxidation to toxic products5. Furthermore, polynuclear aromatic hydrocarbons (PAH) are activated to toxic metabolites via CYP1A2. Additionally, epidemiological studies have shown increased risk of colon cancer in individuals with high CYP1A2 activity but only when coupled with the rapid N-acetyltransferase phenotype and high consumption of “well-done” meat6. CYP1A2 also metabolises various drug substrates, as well as caffeine and melatonin7. CYP2E1 is less abundant than 1A2 making up ~10% of liver CYP enzymes3,4,8 and like CYP 1A2 it is involved in carcinogen activation3,9. CYP2E1 takes part in the metabolism of a large variety of substrates including aromatic compounds, alcohol, halogenated anaesthetics, heterocyclic compounds, some nitrosamines10 and also endogenous compounds such as acetone and fatty acids. In turn expression of CYP2E1 can be induced by many of these substances including alcohol11.
To date, CYP enzymes have been studied using a variety of different bioanalytical methods including immunoblotting, PCR and enzyme activity assays. The studies performed have not provided definitive data on the protein levels of individual CYP-isoforms. Although, the application of RT-PCR to quantify e.g. CYP2E1 mRNA, was successful12, the result does not necessarily translate to the protein level. Western Blot analysis can be performed at the protein level13, but is dependent on the availability of specific antibodies, and may not assure 100% selectivity, additionally only one CYP-isoform can be quantified per analysis. CYP protein levels have also been investigated indirectly by studying their enzymatic activity e.g. CYP2E1 via metabolism of chlorzoxazon14,15. However, there remains the problem that more than one CYP-isoform can catalyse a given reaction. In recent years LC-MS/MS has gained popularity in the qualitative and quantitative determination of proteins16. For CYP analysis LC-MS/MS has been mainly used in a discovery mode, where the goal is one of protein identification17,18. However, quantitative analysis is also possible19-21.
For many years, LC-MS/MS has been used as a standard method for the quantification of small molecules, and currently LC-MS/MS methods are being introduced in proteomics for both absolute and relative protein quantification19-25. Among these, the use of stable-isotope labelled tryptic peptides offers a promising method for absolute quantification of proteins26,27. Simply, proteins are digested with trypsin at the same time as which a stable-isotope labelled tryptic peptide is added to the mixture. The isotope labelled peptide is chosen so as to have an amino acid sequence unique to the protein of interest i.e. a proteotypic peptide, but to contain a heavy-isotope labelled amino acid. Quantification is then performed by comparing the mass spectrometric ion-current of the stable-isotope labelled peptide with its native analogue generated by digestion of the target protein. Assuming tryptic digestion of the target protein is complete; the abundance of the target protein can be inferred from that determined for its tryptic peptide. The use of stable-isotope labelled peptides has the advantage of allowing absolute protein quantification with no additional experimental steps other than addition of the labelled compound and can easily be adapted to an established in-gel digestion method to allow quantification of CYPs. A further advantage is the possibility of multiplexing the experiment i.e. for the quantification of more than one protein.
Although the application of stable-isotope dilution mass spectrometry is recognised to provide the most accurate quantitative data, absolute quantification requires a targeted approach to proteome analysis, in that it involves pre-targeting a preferred analyte. However, high quality data sets are usually generated using an un-targeted approach, in which case it would be extremely valuable to retrospectively obtain quantitative data. It has recently been proposed that application of a protein abundance index (PAI) can provide the necessary solution28. The PAI is defined, for any given protein, as the number of observed peptides (Nobsd) divided by the number of observable peptides (Nobsbl) per protein (Eq 1). For absolute quantification, PAI is converted to emPAI (exponentially modified protein abundance index), which is equal to 10PAI minus 1 (Eq 2), which is proportional to the protein content in a protein mixture29.
In the current study we have investigated the use of emPAI for CYP analysis using two different experimental approaches, one based on 1D-gel separation of proteins and LC-MS/MS analysis of tryptic peptides, and the other on 2D-LC separation of tryptic peptides followed by MS/MS. The data obtained by emPAI was complimented by absolute quantification data obtained by stable-isotope dilution mass spectrometry analysis for two CYPs from human liver microsomal preparations.
Experimental Procedures
The protocol for this study was approved by the Ethics Committee of the Royal Free Hospital and University College School of Medicine. Tissues were accessed from resected discarded masses of liver, which were removed as part of the surgical treatment for hepatic metastases arising from colon cancer30. Only the portions of the liver without pathohistological changes were used in this study. Detailed medical and drug information of the patients is listed in supplementary table S1.
The heavy-isotope labelled tryptic peptide GTVVVPTL*DSVLYDNQEFPDPEK (amino acid sequence given by single-letter code, where L* is a leucine containing six 13C and one 15N atom) which has an amino acid sequence unique to CYP2E1 was from Thermo Fisher (Hemel Hempstead, UK). The supplier specified the purity to be 95%. Recombinant CYP2E1 (purity > 85%) was purchased from PanVera (Invitrogen, Paisley, UK). The peptide with the amino acid sequence IGSTPVL*VLSR, unique to CYP1A2, was synthesised by Sigma-Aldrich (Dorset, UK) with purity declared to be 99%. Anti-CYP1A2 (ab22717) and anti-CYP2E1 (ab28146) antibodies were purchase from Abcam (Cambridge, UK). Horseradish peroxidase–linked anti-mouse and anti-rabbit IgG were from GE Healthcare (Amersham, UK). ECL Western blotting substrate was obtained from Pierce (Thermo Scientific, UK). TEAB (triethylammoniumbicarbonate), HEPES (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid), sucrose, sodium chloride and iodoacetamide were obtained from Sigma Chemicals (St. Louis, USA). SDS (sodium dodecyl sulphate) was obtained from Melford Laboratories Ltd. (Ipswich, UK) and formic acid from BDH Laboratory Supplies (Poole, UK). Ammonium bicarbonate and all solvents of HPLC grade were purchased form Fisher Scientific Ltd. (Loughborough, UK).
Preparation of microsomes
Liver microsomes were processed using differential centrifugation essentially as described previously17. Frozen liver tissue was ground in a percussion pestle and mortar and the powdered tissue was homogenised in an ice-cold glass homogeniser with a polytetrafluoroethylene-head pestle. For every 100 mg of tissue 1 mL of homogenisation buffer (0.25 M sucrose, 50 mM HEPES, 100 mM sodium chloride, pH 7.5, containing a Complete protease inhibitor tablet EDTA free, Roche Diagnostics, Welwyn Garden City, UK) was added. After ultracentrifugation the microsomal pellet was suspended in a buffer containing 0.5 M TEAB and 0.1% SDS, pH 8.5, which is also compatible with iTRAQ labelling (unpublished data) and stored at −80°C. A Bradford assay was performed to determine the protein concentration.
SDS-PAGE and Western blot analysis
Microsomal samples were subjected to 1D SDS-polyacrylamide gel electrophoresis (PAGE) using the NUPAGE® electrophoresis system from Invitrogen (Carlsbad, USA). Samples were heated for 10 min at 70°C in NUPAGE® sample buffer and NUPAGE® reducing agent. Alkylation was performed in the dark for 30 minutes at room temperature by the addition of iodoacetamide. About 25 μg of microsomal protein, recombinant protein, or microsomal protein spiked with a known amount recombinant protein was loaded on a NUPAGE® 4-12% Bis-Tris (Bis (2-hydroxyethyl) imino-tris (hydroxymethyl) methane-HCl) pre-cast 1.0 mm 10 well gel. NUPAGE® MOPS (3-(N-morpholino) propane sulfonic acid) SDS running buffer was used. Gels were stained with SimplyBlue™ Safestain (Invitrogen) and destained with water.
For Western blot analysis 25 μg of microsomal protein from each sample was subjected to 10% Tris-glycine SDS-PAGE and subsequently transferred to a nitrocellulose membrane. The membrane was immunoblotted with either a 1:3000 dilution of anti-CYP1A2 antibody or a 1:2000 dilution of anti-CYP2E1 antibody. Western blot analysis was also performed on samples following tissue homogenisation but prior to ultracentrifugation.
In-gel tryptic digestion and peptide extraction
Either the entire lane was cut into 20 - 25 bands, or alternatively bands in the specific CYP mass region (approximately 45 to 62 kDa) were excised as one “large band” at ~50 kDa and two “smaller bands” above and below (see supplemental Figure S1). Each band was washed and destained with 50 mM ammonium bicarbonate in 50% aqueous acetonitrile. Each band was cut into small pieces (1 mm2), dehydrated with acetonitrile and then dried in a Speed Vac for 30 min (Jouan RC10 22, Jouan Nordic, Denmark). Sequencing grade modified trypsin (250 – 375 ng, Promega, Southampton, UK) in 50 mM ammonium bicarbonate was added to each dried band to induce proteolysis. For studies using stable-isotope dilution mass spectrometry, the ammonium bicarbonate buffer additionally contained a known amount of stable-isotope labelled peptide (4 pmol added per band). The samples were left on ice to allow absorption of trypsin (and isotope-labelled peptide) into the dry gel pieces. After 30 minutes the gel pieces were covered with additional 50 mM ammonium bicarbonate and incubated at 37°C overnight. Peptides were extracted by alternating washings with 5% formic acid in 50% aqueous acetonitrile, and 100% acetonitrile. All extracts were combined and dried completely in a speed vac.
For LC-MS/MS analysis samples were reconstituted in either 25 μL of 0.1% formic acid or 50 μL of 100 mM ammonium bicarbonate containing 5% acetonitrile and 0.1% formic (for absolute quantification of CYP2E1), and then centrifuged at 16,000 × g for 5 minutes before 5 μL was injected on-column.
In-solution digestion
Liver microsomal proteins were reduced with 5 mM tris-(2-carboxyethyl)phosphine, alkylated with methyl methanethiosulphate (Applied Biosystems, UK) and subsequently digested with trypsin (Promega, Southampton, UK) (trypsin to substrate 1:10) as previously described 21.
Recombinant CYP2E1 (10 μg) was digested in-solution using 0.2 μg of sequencing grade modified trypsin (Promega, Southampton, UK) in 100 mM aqueous ammonium bicarbonate (trypsin to substrate 1:50). The mixture was incubated overnight at 37°C and stored in aliquots at −20°C.
LC-MS/MS
Analysis using CapLC combined with Q-TOF
To initially identify the CYP profile of human liver microsomes and obtain approximate quantitative data by emPAI, LC-MS/MS analysis was performed using a Micromass CapLC system (Waters, UK) coupled to a Q-TOF Global mass spectrometer (Waters, UK). Peptides derived from in-gel digestion were re-constituted in 25 μL of 0.1% formic acid, and 5 μL aliquots injected. Sample was loaded onto a trap column (5 mm × 0.3 mm, 5 μm, PepMap C18 guard column, Dionex, Camberley, UK) at a flow-rate of 15 μl/min, delivered isocratically with solvent C (0.1% formic acid) by auxiliary pump C. Sample was washed on the trap column for 3 min with solvent C before being switched in-line with the C18 nano-column (150 × 0.075 (i.d) mm, 3 μm C18, Dionex), which was equilibrated with 95% solvent A (0.1% formic acid in 5% acetonitrile), 5% solvent B (0.1% formic acid in 95% acetonitrile) at a flow-rate of approximately 200 nl/min. The proportion of solvent B was increased linearly to 28% over 40 min, then to 80% over 2 min; maintained at 80% solvent B for 15 min (wash phase) then re-equilibrated at 95% solvent A, 5% solvent B for 10 min. The column effluent was continuously directed into the nano-electrospray (nano-ES) source of a Micromass Q-TOF Global mass spectrometer. Data dependent analysis (DDA) was performed using a 1 s MS survey scan (m/z range 420-2000) followed by 1 s MS/MS scans (0.1 s interscan time) on up to three different precursor-ions (intensity threshold 10 counts per second). In the DDA mode, MS/MS spectrum acquisition (in the m/z range 50 - 1800) was allowed for up to a total of 2.2 s on each precursor-ion, or stopped when the signal intensity fell below three counts per second, and a new MS to MS/MS cycle was started. Precursor-ions were excluded from any further MS/MS fragmentation for 45 s (retention time) to minimize repeated identification of the same peptide; singly charged ions were also excluded as precursor-ions for MS/MS.
Tryptic peptides derived from in-solution digestion of liver microsomal proteins were also analysed by off-line 2D-LC-MS/MS as described previously21. Briefly, the peptide mixture was first separated by strong cation exchange (SCX) chromatography into 16 fractions. Each fraction was then analysed by LC-MS/MS as above, but with four injections of each fraction and by performing survey scans over different m/z ranges (m/z 420-605, 600-705, 700-805 and 800-1005) for each injection.
Analysis using nano-LC combined with LCQduo
The emPAI method for protein quantification in human liver microsomes was also exploited using data obtained following in-gel digestion and analysis by nano-LC combined with a LCQduo ion-trap mass spectrometer. Nano-LC was carried out using an LC Packings Ultimate Capillary HPLC system with FAMOS™ autosampler (Dionex). Sample (5 μL) was injected via a 20 μL sample loop with 0.1% formic acid as loading buffer delivered by a separate Ultimate Micropump. In order to avoid sample carry-over, every sample injection was followed by three blank injections of 6.4 μL. The sample was loaded onto a pre-column (5 mm × 300 μm PepMap C18 guard column, 3 μm, Dionex). After the sample had been washed for 3.5 min on the pre-column it was transferred to the analytical column (PepMap C18, 75 μm × 150 mm; 3.5 μm, Dionex) which had been equilibrated with 95% solvent A (5% acetonitrile in 0.1% formic acid) and 5% solvent B (80% acetonitrile in 0.1% formic acid) at a flow rate of 200 nL/min.
For the identification of CYP proteins a longer LC gradient programme was used than in experiments devoted to absolute quantification (see below). The longer gradient was 65 min. After 3.5 min of washing of the trap column, the proportion of mobile phase B was increased linearly to 40% over 39 min and raised to 80% in the following 5 min. B was kept at 80% for 7 min after which the column was equilibrated for 10 min with 95% solvent A and 5% B prior the next injection. In the shorter gradient programme (58 min), B was raised to 40% over 35 min and then to 80% in 30 s, and held at 80% for a further 9 min. The column was finally re-equilibrated for 10 min at 95% A and 5% B.
The nano-LC system was connected to the nano-ES source of an LCQduo ion-trap mass spectrometer (Thermo Fisher, Hemel Hempstead, UK) and both mass and MS/MS (i.e. MS2) spectra recorded. For the LC-MS/MS identification method centroid data was collected in the survey scan and data-dependent MS/MS modes. Three microscans were performed, with the maximum ion injection time of 200 ms. Automatic gain control was in operation. Over the 65 min run time four scan events were repeated: first a survey scan was recorded over the m/z range 400-2000, followed by MS/MS spectra of the three most intense ions in the survey scan spectrum. The MS/MS collision energy setting was 35%. The minimum precursor-ion signal required for MS/MS data collection was set to 100,000 cps. An ion was put on an exclusion list for 1.25 min (exclusion mass width 2.00 m/z units) if it appeared as one of the top three most intense ions for more than 3 full mass scans within 30 seconds.
Quantification by MRM using nano-LC combined with LCQduo
The MRM method for the quantification of CYP proteins using the LCQduo was initially validated using recombinant CYP2E1 and the selected stable-isotope labelled tryptic peptide (GTVVVPTL*DSVLYDNQEFPDPEK). Ion-trap instruments have been exploited by others for similar quantification studies26. To optimise the LCQduo ion-trap mass spectrometer for the absolute quantification of CYP2E1, a solution digest of recombinant CYP2E1 and a solution of the stable-isotope labelled peptide (GTVVVPTL*DSVLYDNQEFPDPEK) were directly infused via a syringe pump into the nano-ES source thus circumventing the LC system. MS and MS/MS spectra were acquired and suitable fragment ions for quantification chosen. The precursor-ion selected for CYP2E1 quantification was the [M+2H]2+ ion of the tryptic peptide GTVVVPTLDSVLYDNQEFPDPEK at m/z 1281.8 (theoretical monoisotopic m/z 1281.6), while the [M+2H]2+ for the corresponding stable-isotope labelled peptide was at m/z 1285.3. These peptides co-elute at about 40 min in the LC system (58 min gradient programme, see Figure 2) and their MS/MS spectra shows major fragment ions at m/z 1054.3 for the native and 1057.6 for the stable-isotope labelled peptide, respectively (both y182+, cleavage N-terminal to proline, see fragmentation pattern in Figure 3). The production of these fragment-ions was monitored and provided the basis for the generation of MRM chromatograms (1281.8→1054.3, 1285.3→1057.6) for quantification. During the 58 min run time, 3 scan events were repeated: a full mass scan over m/z 700-1400 was recorded, followed by MS/MS of [M+2H]2+ ions at m/z 1281.8 and 1285.3. The isolation width of the precursor-ion was set to 3.0 and the collision energy was 35%. For the MRM chromatograms the product-ion window was set at 1 m/z unit and peak areas were used in quantitative calculations.
Figure 2.

Reconstructed ion chromatograms for (A) native (1281.8→1054.3), and (B) stable-isotope labelled (1285.3→1057.6) peptides which elute at ~40 min. The native peptide has the amino acid sequence GTVVVPTLDSVLYDNQEFPDPEK and in the stable-isotope labelled peptide leucine-8 contains 13C615N. Data acquired on the LCQduo.
Figure 3.

(A) The MS/MS spectrum of the [M+2H]2+ ion at m/z 1281.8 from the tryptic peptide GTVVVPTLDSVLYDNQEFPDPEK. For MRM experiments the fragmentation channel m/z 1281.8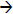 1054.3 i.e. [M+2H]2+ y182+ was monitored. (B) The MS/MS spectrum of the [M+2H]2+ ion at m/z 1285.3 from the stable-isotope labelled peptide GTVVVPTL*DSVLYDNQEFPDPEK, where L* corresponds to leucine labelled with 13C615N. For MRM experiments the fragmentation channel m/z 1285.31057.6 i.e. [M+2H]2+ y182+ was monitored. Data acquired on the LCQduo.
1054.3 i.e. [M+2H]2+ y182+ was monitored. (B) The MS/MS spectrum of the [M+2H]2+ ion at m/z 1285.3 from the stable-isotope labelled peptide GTVVVPTL*DSVLYDNQEFPDPEK, where L* corresponds to leucine labelled with 13C615N. For MRM experiments the fragmentation channel m/z 1285.31057.6 i.e. [M+2H]2+ y182+ was monitored. Data acquired on the LCQduo.
A similar procedure was applied to optimise the ion-trap for quantification of CYP1A2, where the stable-isotope labelled peptide was IGSTPVL*VLSR. The [M+2H]2+ ions and their y7 fragment-ions (cleavage N-terminal to proline, see supplemental data Figure S2) were selected for MRM analysis monitoring the transitions 571.8→783.5 and 575.3→790.5 for the native and heavy-isotope labelled peptides respectively.
Data analysis
Protein identification and approximate quantification using emPAI
For the identification of proteins in human liver microsomes data was submitted to a Mascot “in-house” search engine (Matrix Science Ltd., London, version 2.2, see supplemental data for parameters used to create peak lists). Database searching was restricted to tryptic peptides of human proteins using the IPI human database (International Protein Index, version 20070707 containing 67922 sequences)31. For analysis of microsomal samples submitted to in-gel digestion the variable modifications carbamidomethyl cysteine, methionine oxidation and N-acetylation (protein) were chosen. For in-solution digestion of microsomal protein, methythio was the chosen cysteine modification. For analysis performed on the Q-TOF mass spectrometer, the precursor and MS/MS tolerance were both set at 0.3 Da (or below), while for analysis performed on the LCQduo the precursor and fragment mass tolerances were set at 1.0 and 0.6 Da respectively. One missed cleavage was allowed. Only peptides giving scores greater than the “identity threshold” were accepted. For proteins identified by only one tryptic peptide, the MS/MS spectrum of the peptide was manually validated. To assess false-positive identifications, an automatic decoy search was performed against a randomised database with a default significance threshold of p < 0.05. For Q-TOF data with precursor and fragment-ion mass tolerance set at 0.3 Da the false discovery rate at the identity threshold was below 2%. For LCQduo data with precursor and fragment-ion tolerance set at 1.0 and 0.6 Da, respectively, the false discovery rate at the identity threshold was below 5%.
Approximate quantification was performed using emPAI values (Eq 1 + 2) which are provided by the Macot 2.2 search engine.
| (1) |
and
| (2) |
where, Nobsd and Nobsbl are the number of observed and observable tryptic peptides per protein respectively. To determine the weight % of a given CYP-isoform from the total CYP content of a sample, Eq (3) is applied:
| (3) |
Where, Mr is the molecular weight of the CYP-isoform and Σ(emPAI × Mr) is the summation of the product of emPAI and Mr for all CYP-isoforms identified.
Quantification using MRM
For absolute quantification, peaks in the MRM chromatograms for the selected transitions were smoothed using a Gaussian algorithm. The ratio of peak areas for the transitions at the appropriate elution times were calculated and used to determine the amount of CYP2E1 or 1A2 in the sample (see Figures 2 and 4).
Figure 4.

(A) The total ion chromatogram of an in-gel digest of human liver microsomes. (B) The reconstructed ion chromatogram for the transition m/z 1281.8→1054.3 corresponding to [M+2H]2+ y182+ for the native tryptic peptide GTVVVPTLDSVLYDNQEFPDPEK unique to CYP2E1. (C) The reconstructed ion chromatogram for the transition m/z 1285.3→1057.6 corresponding to [M+2H]2+ y182+ for the stable-isotope labelled peptide GTVVVPTL*DSVLYDNQEFPDPEK, where L* corresponds to leucine labelled with 13C615N. The ratio of areas under the curves for peaks eluting with retention time ~40 min in (B) and (C) gives a peptide ratio of 0.80 (B/C)which translates to a CYP2E1 concentration of 3.18 pmol/25 μg (127 pmol/mg) microsomal protein. Data acquired on the LCQduo.
Results and Discussion
Identification of CYP enzymes in human liver microsomes by 1D-gel LC-MS/MS and 2D-LC-MS/MS approaches
Our initial question concerned the optimal method for the qualitative analysis of CYP enzymes in human liver microsomes. Evidence in the literature dissuaded us from employing a 2D-gel LC-MS/MS or MALDI-MS approach16, thus these methodologies were not followed. We therefore evaluated two alternative approaches, namely the 1D-gel LC-MS/MS and 2D-LC-MS/MS methods.
Microsome preparations were first validated by Western blotting using an anti-CYP1A2 antibody. CYP1A2 is an enzyme located in the endoplasmic reticulum and hence should be enriched in the microsomal fraction obtained by differential centrifugation32. The majority of CYP1A2 was found in the microsomal fraction, while only a faint band was observed in the mitochondrial fraction. CYP1A2 was not detectable in the cytosolic fraction (Figure 1).
Figure 1.

(A) Western blot analysis of CYP1A2 in subcellular fractions from human liver tissue prepared by differential centrifugation. (B) Western blot analysis of CYP2E1 (left panel) and CYP1A2 (right panel) in liver microsomal fractions prepared from different patients.
CYP enzymes show considerable sequence identity, and their nomenclature is such that they are arranged into families and subfamilies on the basis of percentage amino acid sequence identity. CYPs that share ≥40% identity are assigned to a particular family designated by an Arabic numeral, and those sharing ≥55% identity make up a particular subfamily designated by a letter33. Thus, to identify a given CYP-isoform by database searching using Mascot, at least one peptide must be identified that is of amino acid sequence unique to that isoform. In the results described below CYP-isoform identification was always based on at least one unique tryptic peptide match.
The 1D-gel LC-MS/MS approach to protein identification has the advantage of being very robust and is able to separate CYPs from a complex mixture of proteins. Thus, bands enriched in CYPs can be analysed as discrete entities. In our studies about 25 μg of microsomal protein was subjected to 1D SDS-PAGE followed by tryptic digestion and LC-MS/MS analysis. In experiments performed using the LCQduo CYP1A2, 2A6, 2C8, 2C9, 2E1, 2J2, 3A4 and CYP4A11 and 4F2/3 were identified (see supplemental data, Table S2), while in experiments performed using the Q-TOF CYP4F11 and 8B1 were additionally identified, and it was also possible to differentiate CYP4F2 from 4F3 (supplemental data, Table S3). Table 1 lists the tryptic peptides unique to a given CYP-isoform (proteotypic peptides) which were observed using the 1D-gel LC-MS/MS methodology on these two instruments.
Table 1.
Potential proteotypic peptides suitable for quantification of CYP-isoforms using 1D-gel LC-MS/MS (LCQduo and Q-TOF) and 2D-LC-MS/MS (Q-TOF) formats.
| CYP- isoform |
Unique amino acid sequence1 observed from human liver microsomes by LC-MS/MS |
LCQ 1D-gel2 |
Q-TOF 1D-gel2 |
Q-TOF 2D-LC2 |
|---|---|---|---|---|
| CYP1A2 | DITGALFK | x | x | |
| FLWFLQK | x | |||
| MMLFGMGK | x | x | ||
| ASGNLIPQEK | x | |||
| IGSTPVLVLSR | x | x | x | |
| VDLTPIYGLTMK | x | x | ||
| FLTADGTAINKPLSEK | x | x | x | |
| TVQEHYQDFDKNSVR | x | |||
| TVQEHYQDFDK | x | |||
| NTHEFVETASSGNPLDFFPILR | x | |||
| GRPDLYTSTLITDGQSLTFSTDSGPVWAAR | x | |||
| CYP2A6 | GTEVYPMLGSVLR | x | x | x |
| GTGGANIDPTFFLSR | x | x | x | |
| DPSFFSNPQDFNPQHFLNEK | x | x | ||
| CYP2B6 | NLQEINAYIGHSVEK | x | ||
| IAMVDPFFR | x | |||
| CYP2C8 | SFTNFSK | x | ||
| NLNTTAVTK | x | |||
| EALIDNGEEFSGR | x | x | ||
| DQNFLTLMK | x | x | ||
| VQEEIDHVIGR | x | x | x | |
| VQEEAHCLVEELR | x | x | ||
| EHQASLDVNNPR | x | |||
| VKEHQASLDVNNPR | x | |||
| YSDLVPTGVPHAVTTDTK | x | |||
| KLPPGPTPLPIIGNMLQIDVK | x | |||
| CYP2C9 | GIFPLAER | x | ||
| GTTILISLTSVLHDNK | x | |||
| LPPGPTPLPVIGNILQIGIK | x | x | ||
| SHMPYTDAVVHEVQR | x | x | x | |
| GKLPPGPTPLPVIGNILQIGIK | x | |||
| EKHNQPSEFTIESLENTAVDLFGAGTETTSTTLR | x | |||
| CYP2E1 | EALLDYK | x | ||
| YPEIEEK | x | |||
| HEDYNDEK | x | |||
| GDLPAFHAHR | x | x | ||
| GIIFNNGPTWK | x | x | ||
| FKPEHFLNENGK | x | x | ||
| EALLDYKDEFSGR | x | x | ||
| FITLVPSNLPHEATR | x | x | x | |
| VKEHHQSLDPNCPR | x | |||
| DLTDCLLVEMEKEK | x | |||
| GTVVVPTLDSVLYDNQEFPDPEK | x | x | x | |
| EAHFLLEALR | x | x | x | |
| FGPVFTLYVGSQR | x | |||
| YSDYFKPFSTGK | x | |||
| QEMPYMDAVVHEIQR | x | |||
| DRQEMPYMDAVVHEIQR | x | |||
| CYP2J2 | VIGQGQQPSTAAR | x | ||
| FEYQDSWFQQLLK | x | |||
| CYP3A4 | EVTNFLR | x | x | |
| GVVVMIPSYALHR | x | |||
| LSLGGLLQPEKPVVLK | x | x | ||
| LGIPGPTPLPFLGNILSYHK | x | |||
| VWGFYDGQQPVLAITDPDMIK | x | x | ||
| APPTYDTVLQMEYLDMVVNETLR | x | x | ||
| CYP4A11 | VATALTLLR | x | x | x |
| QFAMNELK | x | |||
| CYP4F2 | FDPENIK | |||
| SVINASAAIAPK | x | x | ||
| HVTQDIVLPDGR | x | |||
| CYP4F3 | WQLLASEGSAR | x | ||
| CYP4F11 | TLPTQGIDDFLK | x | x | |
| CYP4F12 | LVHDFTDAVIR | x | ||
| CYP4V2 | EFFQQIIEYTEEYR | x | ||
| SVSEDCEVAGYR | x | |||
| FLEPWLGLGLLTSTGNK | x | |||
| CYP7B1 | SLDILLESMMQNLK | x | ||
| CYP8B1 | NMFEFLK | x | ||
| EEATQVLGEAR | x | |||
| AVREEATQVLGEAR | x | |||
| KFDLLFPR | x | |||
| KLDFGQYAK | x | |||
| WGFGTMQPSHDVR | x | |||
| DKEQDLLQAGELFMEFR | x | |||
| CYP20A1 | TLDPFETMLK | x | ||
| LTPVSAQFQDIEGK | x | |||
| CYP51A1 | NEDLNAEDVYSR | x | ||
| IDDILQTLLDATYK | x | |||
| TFTYLLGSDAAALLFNSK | x | |||
| SPPYIFSPIPFLGHAIAFGK | x |
Unique amino acid sequences were confirmed by performing a Blast Search (http://expasy.org/tools/blast/) against a human protein database. Note, peptides containing chemically reactive residues (C, M, W) and unstable sequences (N-G, N-term Q or N-term N) may not be suitable as proteotypic peptides.
Observation of a unique peptide indicated by x.
Despite the robust nature of the 1D-gel LC-MS/MS method; band cutting, in-gel digestion and peptide extraction are labour intensive, time consuming, are susceptible to spurious contaminations and do not lend themselves readily to automation. We therefore decided to investigate a gel-free approach to the analyse of CYPs. Microsomes from liver samples were prepared and a portion taken for analysis by 1D-gel LC-MS/MS. Further aliquots were subjected to in-solution digestion. An initial experiment was performed on an in-solution digests in which about 0.5 μg of peptides was loaded onto a reversed phase (RP) nano-LC column and separated through a 2 hr gradient and analysed on the Q-TOF (see supplemental data for experimental details). The only CYP identified was 2A6 by the unique peptide sequence of GTGGANIDPTFFLSR. Thereafter, off-line 2D-LC was employed using SCX separation prior to RP chromatography. To limit the number of precursor-ions competing for MS/MS analysis, each SCX fraction was analysed four times by RP-LC-MS/MS using narrow-range survey scans for precursor-ion selection (i.e. 400-605, 600-705, 700-805 and 800-1000 m/z). Using this methodology 18 CYP-isoforms were identified based on 96 peptides, which were spread over 13 SCX fractions. These include all the CYPs identified by the 1D-gel LC-MS/MS approach, and also CYP2B6, 4F12, 4V2, 7B1, 20A1, and 51A1 (Table 1 shows proteotypic peptides for the identified CYP-isoforms, see also supplemental Table S4). Table 2 provides an example of CYPs identified in liver microsomes when analysed by 1D-gel LC-MS/MS on both Q-TOF and LCQ instruments and by 2D-LC-MS/MS on the Q-TOF.
Table 2.
CYP-isoforms identified in a liver microsome sample when analysed by 1D-gel LC-MS/MS on both Q-TOF and LCQ instruments and by 2D-LC-MS/MS on the Q-TOF. emPAI values are given and CYP-isoforms colour coded according to approximate quantity. Dark shading signifies most abundant CYP-isoforms, light shading medium abundance isoforms, and no shading low abundance isoforms.
| CYP-isoform | Total peptides |
Unique peptides |
emPAI |
Weight% of total identified CYPs |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| LCQ 1D-gel |
Q-TOF 1D-gel |
Q-TOF 2D-LC |
LCQ 1D-gel |
Q-TOF 1D-gel |
Q-TOF 2D-LC |
LCQ 1D-gel |
Q-TOF 1D-gel |
Q-TOF 2D-LC |
LCQ 1D-gel |
Q-TOF 1D-gel |
Q-TOF 2D-LC |
|
| CYP1A2 | 4 | 6 | 8 | 3 | 5 | 8 | 0.28 | 0.53 | 0.68 | 7.2 | 15.6 | 7.2 |
| CYP2A6 | 9 | 11 | 10 | 3 | 2 | 3 | 0.78 | 1.4 | 1.15 | 19.4 | 41.6 | 12.4 |
| CYP2B6 | -- | -- | 1 | -- | -- | 1 | -- | -- | 0.06 | -- | -- | 0.6 |
| CYP2C8 | 6 | 2 | 7 | 4 | 1 | 5 | 0.38 | 0.16 | 0.51 | 9.4 | 5.2 | 5.2 |
| CYP2C9 | 6 | 4 | 9 | 3 | 1 | 5 | 0.57 | 0.35 | 1.15 | 14 | 10.4 | 12.4 |
| CYP2E1 | 8 | 2 | 9 | 6 | 1 | 9 | 0.66 | 0.16 | 1.02 | 16.5 | 5.2 | 11.4 |
| CYP2J2 | 1 | -- | 1 | 1 | -- | 1 | 0.06 | -- | 0.06 | 1.5 | -- | 0.6 |
| CYP3A4 | 11 | 4 | 11 | 3 | 1 | 1 | 1.13 | 0.33 | 1.49 | 28.6 | 10.4 | 16.5 |
| CYP4A11 | 2 | 3 | 8 | 1 | 1 | 1 | 0.13 | 0.23 | 0.69 | 3.4 | 6.5 | 7.2 |
| CYP4F2 | -- | -- | 8 | -- | -- | 3 | -- | -- | 0.86 | -- | -- | 9.3 |
| CYP4F11 | -- | 2 | 3 | -- | 1 | 1 | -- | 0.15 | 0.25 | -- | 5.2 | 3.1 |
| CYP4F12 | -- | -- | 3 | -- | -- | 1 | -- | -- | 0.25 | -- | -- | 3.1 |
| CYP4V2 | -- | -- | 2 | -- | -- | 2 | -- | -- | 0.12 | -- | -- | 1.0 |
| CYP7B1 | -- | -- | 1 | -- | -- | 1 | -- | -- | 0.12 | -- | -- | 1.0 |
| CYP8B1 | -- | -- | 6 | -- | -- | 6 | -- | -- | 0.42 | -- | -- | 4.1 |
| CYP20A1 | -- | -- | 1 | -- | -- | 1 | -- | -- | 0.07 | -- | -- | 0.6 |
| CYP51A1 | -- | -- | 4 | -- | -- | 4 | -- | -- | 0.34 | -- | -- | 4.1 |
Approximate quantification of CYPs using emPAI
The term emPAI is defined in Eq (2) above29. It has been incorporated into the Mascot search engine to provide an estimate of protein abundance in complex mixtures at the same time as protein identification. Table 2 provides an example of emPAI values determined using the 1D-gel LC-MS/MS (LCQduo and Q-TOF) and 2D-LC-MS/MS approaches, and also the weight percentage of each CYP-isoform out of the total CYP content of microsomal protein as calculated using Eq (3) above. Based on these values (see also supplemental data, Tables S2-S4), the identified CYP-isoforms can be classified into three categories, i.e. of high, medium and low abundance. CYPs 1A2, 2A6, 2C9, 2E1 and 3A4 were found to be the most abundant CYPs in human liver microsomes, each accounting for ~10% or greater of the total CYP protein in liver. CYPs 2C8, 4A11 and 4F2, were less abundant accounting for ~5 to 10% of total CYPs in liver microsomes. The low abundance CYPs include 2B6, 2J2, 4F11, 4F12, 4V2, 7B1, 8B1, 20A1 and 51A1, which are present at less than 5% of the total CYPs. This data is in agreement with previous studies performed by immunoblotting, where the most abundant CYPs (>10%) were identified as 3A4, 2C9, 1A2 and 2E1, and those of medium abundance (<10%) were 2C8, 2A6 and 2B63,5. Not surprisingly our data also agrees with that obtained in an earlier study performed by 1D-gel LC-MS/MS where peptide counting was employed as a method of approximate quantification17. Admittedly, the variation in protein level for a given CYP isoform determined using emPAI on the three instrument set-ups is rather large (Table 2). The experimentally determined emPAI value is clearly affected by the analytical method used. However, abundant housekeeping proteins, such as actin and myosin were shown to have consistent emPAI values when analysed using either the 1D-gel LC-MS/MS or 2D-LC-MS/MS approach (agreement >70%, data not shown). Thus, as with many mass spectrometric approaches, the accuracy of the emPAI method appears biased towards the more abundant proteins.
Selection of stable-isotope labelled tryptic peptide for protein quantification by stable-isotope dilution mass spectrometry
Although emPAI gives a quantitative estimate and allows classification of CYP proteins into categories according to approximate quantity, which agrees with that obtained by immunochemistry, the data shows considerable variation when e.g. the same sample is analysed by differing methods or on different instruments (Table 2). This was noted in the original publication describing emPAI, which also indicated that the accuracy of the method may lead to average errors of a factor of 2 to 4 29. To obtain more accurate quantification data stable-isotope labelling is recommended. Here we have performed absolute protein quantification using stable-isotope dilution mass spectrometry at the peptide level, which relies on quantification of a peptide derived from tryptic digestion of the target protein and extrapolation of peptide abundance to protein abundance. The quantified tryptic peptide should be of amino acid sequence unique to the target protein (proteotypic peptide). Quantification is performed by the addition of a stable-isotope labelled analogue of the defined peptide to a tryptic digest of the target protein. Thus, the peptide for quantification should be unique to the CYP of interest and reproducibly observed in proteomic analysis (see Table 1). In this work we initially defined the CYP isoform-specific proteotypic peptides which are observed in LC-MS/MS experiments performed on tryptic digests of human liver microsomes (Table 1), and then performed a proof-of-concept study by investigating the levels of the CYP-isoforms 2E1 and 1A2 in human liver microsomes. CYP proteins are membrane bound proteins and thus provide more of an analytical challenge than those found in the cytosol. Additionally, the considerable overlap of amino acid sequence between CYP-isoforms can make selection of an appropriate unique and readily observed tryptic peptide difficult. Here we describe the initial validation of the absolute quantification method for CYP proteins using CYP2E1 as a test compound and then its exploitation for the quantification of CYP1A2. The peptide chosen for CYP2E1 quantification was of amino acid sequence GTVVVPTLDSVLYDNQEFPDPEK, and in the stable-isotope labelled analogue leucine-8 contained six 13C and one 15N atom. It is noteworthy that this peptide having a mass larger than the average observed tryptic peptide and eluting late in the chromatogram stands out from the “background” of tryptic peptides in a complex mixture, thus reducing the possibility of contamination of the MRM by co-eluting peptides (or other components) of similar m/z. Both unlabelled and labelled peptides give abundant [M+2H]2+ ions at m/z 1281.8 and 1285.3, respectively. The MRM transitions m/z 1281.81054.3 and m/z 1285.31057.6 were monitored for the native and stable-isotope labelled peptide, respectively. These transitions offer the twin advantages of sensitivity (i.e. abundant fragment ions, see Figure 3) and selectivity (completely different ions involved in both transitions) thereby avoiding cross-talk between reaction channels. Figure 4 shows the chromatograms obtained in a quantification experiment and the reconstructed ion chromatograms (RICs) for the two selected MRM transitions.
Limits of quantification
Recombinant CYP2E1 was found to be accurately quantifiable by stable-isotope dilution mass spectrometry over a range of 0.1 – 1.5 pmol on-column (see also supplemental data, Figure S3). The limit of quantification (LOQ) as defined by a signal to noise ratio of 3 to 1 and minimum peak intensity of 10,000 cps was determined to be 0.1 pmol on-column. As can be seen from the above the nano-LC system linked to the LCQduo gave a rather limited dynamic range. At the upper end of the range this is a consequence of overloading the ion-trap which was observed even with automatic gain control in operation, and at the lower end of the range by the comparative insensitivity of this rather “mature” instrument. The LOQ of 0.1 pmol on-column translated to 1 pmol on gel as only 10% of the sample was actually injected. If sensitivity was at a premium, 100% injection of sample would lead to an on-gel detection LOQ of 0.1 pmol.
Quantification of CYP2E1 in liver microsomes
Table 3 shows the absolute content of 2E1 in each of four liver samples analysed (5 - 8 analysis for each sample) calculated in pmol/mg of microsomal protein. The coefficients of variation (CV) values were in the range of 2 to 30%. This degree of variance results from the relatively high standard deviations which are a consequence of using a “secondary” stable-isotope labelled standard, i.e. a labelled tryptic peptide added at an intermediate step in the experiment, as opposed to using a “primary” isotope-labelled standard, e.g. isotope labelled CYP2E1 protein added to liver homogenate during the initial protein isolation step.
Table 3.
Quantification of CYP2E1 and 1A2 in human liver microsomes.
Calculated CYP2E1 and 1A2 amounts are presented as means (±standard deviation). Coefficients of variation (CV) is stated as a percentage.
| Patient | 1 | 2 | 3 | 4 | 6 | 7 | 8 |
|---|---|---|---|---|---|---|---|
| Gender | Male | Female | Female | Female | Female | Male | Male |
| Age | 41 | 73 | 60 | 79 | 64 | 78 | 45 |
| Amount of microsomal protein loaded on gel (μg) |
25 | 26.5 | 24 | 24 | 24.5 | 28 | 24.5 |
| CYP2E1 (pmol/mg of microsomal protein) | 129.76 ±2.81 | 144.85±41.63 | 200.51±51.99 | 88.12±22.65 | nd | nd | nd |
| CYP1A2 (pmol/mg of microsomal protein) |
nd1 | nd | nd | nd | 165.43±66.75 | 240.76±25.53 | 262.6±76.18 |
| CV (%) | 2 | 29 | 26 | 26 | 40 | 11 | 29 |
not determined
The quantitative method described here permitted the absolute determination of the protein quantity of CYP2E1 in human liver microsomes. The 2E1 content in the studied individuals ranged from 88 to 200 pmol/mg microsomal protein. The degree of variance was confirmed by Western blot analysis (see Figure 1). Guengerich et al13 (performed an immunochemical study on 36 normal human liver samples and detected the amount of CYP2E1 varied by two orders of magnitude with a mean level of 119 pmol/mg microsomal protein. It should be noted that quantitative differences between individuals are mostly not “encoded”, but rather CYP expression is influenced by diet, drugs and circadian regulation. In later studies Shimada et al4 and Rodrigues8 determined the CYP2E1 content of human liver microsomes to be 22 pmol/mg proteins (~7% of total CYPs) and 49 pmol/mg protein (~9 % of total CYPs) respectively. Shimada et al4 additionally determined the total CYP concentration in human liver microsomes to be ~200 - 1000 pmol/mg protein. Taken together, and considering that CYP2E1 represents about 10% of total microsomal CYP protein3,4,8 the data presented in our study is in agreement with published values.
Quantification of CYP1A2 in liver microsomes
Having obtained satisfactory data for the quantification of CYP2E1 in human liver microsomes, attention was shifted to a further CYP-isoform i.e. CYP 1A2. A peptide with the sequence IGSTPVLVLSR was selected as an optimum candidate for CYP1A2 quantification on account of its mass spectrometry compatible length (11 amino acids) and the presence of a proline residue in the sequence expected to generate an intense y7 ion (supplemental data Figure S2). This was found to be a proteotypic peptide using both mass spectrometers and the 1D-gel-LC-MS/MS and 2D-LC-MS/MS approaches (Table 1). The amount of CYP1A2 was determined in samples from three patients (9 analysis for each sample). Despite the very limited linear dynamic range of the MRM method for CYP1A2 quantification of 0.3 – 1 pmol on-column, which translates to 3 pmol – 10 pmol on-gel (with 10% of sample loaded on-column) or 120 – 400 pmol/mg of microsomal protein, the quantities of CYP1A2 in human microsomes fell within this range. The values determined were between 165 and 263 pmol/mg microsomal proteins (Table 3). The inter-patient variation was also validated by western blot (Figure 1) and agrees with the mass spectrometry measurements. Previous studies by immunochemistry suggest the level of 1A2 were in the region of 30 – 50 pmol/mg microsomal protein4,8,13.
Further consideration when using stable-isotope labelled peptides to infer protein quantification
Choosing the optimal (tryptic) peptide as an internal standard is the crucial step when using peptide-based stable-isotope dilution mass spectrometry for protein quantification. The chosen (tryptic) peptide must not only have an amino acid sequence unique to the protein of interest but also be of a size (molecular weight) and hydrophobicity suitable for analysis by LC-MS/MS. Such peptides have been christened “proteotypic peptides”34. Additionally, labile amino acid residues and sequences must be avoided. Not surprisingly, not all (tryptic) peptides with a unique amino acid sequences meet these requirements. Furthermore, ionisation efficiency varies between peptides depending on their amino acid sequence, and when analysed as part of a complex mixture a theoretically satisfactory internal standard peptide may not be experimentally observed. It is thus advisable to check for their appearance in previously acquired experimental data (e.g. Table 1). This way the observation of candidate peptides can be confirmed.
In this proof of principle study performed using the 1D-gel-LC-MS/MS approach we limited absolute quantitative analysis to two of the more abundant CYP proteins found to give unique peptides. In principle it would be possible to synthesise other unique peptides for the minor CYPs and use these for absolute quantification. Now that the 1D-gel-LC-MS/MS method has been successfully validated such studies are being planned.
Conclusions
In this study CYP-isoforms from human liver tissue were qualitatively and quantitatively analysed by LC-MS/MS. Eighteen CYP-isoforms were identified based on unique peptide matches and their approximate relative abundance determined by application of emPAI. The absolute amounts of CYP2E1 and 1A2 were also measured by stable-isotope dilution mass spectrometry using unique tryptic peptides, which has advantage of high intrinsic specificity. Although in this proof-of-concept study only one CYP-isoform was analysed in a single experiment, there are no technical reasons to prevent multiplexing of the method to allow the quantification of multiple CYP-isoforms. In fact on modern triple quadrupole instruments many tens of peptides can be analysed by MRM in a given run. It is very important, however, to note that the accuracy of this method relies on the complete trypsin digestion of the CYP protein of interest, and the application of the method over a concentration range showing linear response. Overloading of the mass spectrometry detector, in this case the ion-trap, must be avoided. In summary, we have demonstrated that CYP-isoforms can be analysed by both 1D-gel and 2D-LC approaches. In future studies detergents assisted in-solution digestion will be evaluated to allow the absolute quantification of multiple CYP-isoforms in one experiment in a gel-free format.
Supplementary Material
Acknowledgements
This work was supported by the UK Biotechnology and Biological Sciences Research Council (BBSRC grant no. BB/C515771/1 and BB/C511356/1). We thank Dr Catherine S Lane for provision of microsomes.
Reference List
- 1.Guengerich FP. AAPS. J. 2006;8:E101–E111. doi: 10.1208/aapsj080112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Patterson LH, Murray GI. Curr. Pharm. Des. 2002;8:1335–1347. doi: 10.2174/1381612023394502. [DOI] [PubMed] [Google Scholar]
- 3.Guengerich FP. Mol. Interv. 2003;3:194–204. doi: 10.1124/mi.3.4.194. [DOI] [PubMed] [Google Scholar]
- 4.Shimada T, Yamazaki H, Mimura M, Inui Y, Guengerich FP. J. Pharmacol. Exp. Ther. 1994;270:414–423. [PubMed] [Google Scholar]
- 5.Guengerich FP, Shimada T, Yun CH, Yamazaki H, Raney KD, Thier R, Coles B, Harris TM. Environ. Health Perspect. 1994;102(Suppl 9):49–53. doi: 10.1289/ehp.94102s949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lang NP, Butler MA, Massengill J, Lawson M, Stotts RC, Hauer-Jensen M, Kadlubar FF. Cancer Epidemiol. Biomarkers Prev. 1994;3:675–682. [PubMed] [Google Scholar]
- 7.Sansen S, Yano JK, Reynald RL, Schoch GA, Griffin KJ, Stout CD, Johnson EF. J. Biol. Chem. 2007;282:14348–14355. doi: 10.1074/jbc.M611692200. [DOI] [PubMed] [Google Scholar]
- 8.Rodrigues AD. Biochem. Pharmacol. 1999;57:465–480. doi: 10.1016/s0006-2952(98)00268-8. [DOI] [PubMed] [Google Scholar]
- 9.Guengerich FP, Kim DH, Iwasaki M. Chem. Res. Toxicol. 1991;4:168–179. doi: 10.1021/tx00020a008. [DOI] [PubMed] [Google Scholar]
- 10.Tanaka E, Terada M, Misawa S. J. Clin. Pharm. Ther. 2000;25:165–175. doi: 10.1046/j.1365-2710.2000.00282.x. [DOI] [PubMed] [Google Scholar]
- 11.Cederbaum AI. Mt. Sinai J. Med. 2006;73:657–672. [PubMed] [Google Scholar]
- 12.Haufroid V, Toubeau F, Clippe A, Buysschaert M, Gala JL, Lison D. Clin. Chem. 2001;47:1126–1129. [PubMed] [Google Scholar]
- 13.Guengerich FP, Turvy CG. J. Pharmacol. Exp. Ther. 1991;256:1189–1194. [PubMed] [Google Scholar]
- 14.Tanaka E, Kurata N, Yasuhara H. J. Clin. Pharm. Ther. 2003;28:157–165. doi: 10.1046/j.1365-2710.2003.00486.x. [DOI] [PubMed] [Google Scholar]
- 15.Wang Z, Hall SD, Maya JF, Li L, Asghar A, Gorski JC. Br. J. Clin. Pharmacol. 2003;55:77–85. doi: 10.1046/j.1365-2125.2003.01731.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang Y, Al-Gazzar A, Seibert C, Sharif A, Lane C, Griffiths WJ. Biochem. Soc. Trans. 2006;34:1246–1251. doi: 10.1042/BST0341246. [DOI] [PubMed] [Google Scholar]
- 17.Lane CS, Nisar S, Griffiths WJ, Fuller BJ, Davidson BR, Hewes J, Welham KJ, Patterson LH. Eur. J. Cancer. 2004;40:2127–2134. doi: 10.1016/j.ejca.2004.04.029. [DOI] [PubMed] [Google Scholar]
- 18.Nisar S, Lane CS, Wilderspin AF, Welham KJ, Griffiths WJ, Patterson LH. Drug Metab Dispos. 2004;32:382–386. doi: 10.1124/dmd.32.4.382. [DOI] [PubMed] [Google Scholar]
- 19.Jenkins RE, Kitteringham NR, Hunter CL, Webb S, Hunt TJ, Elsby R, Watson RB, Williams D, Pennington SR, Park BK. Proteomics. 2006;6:1934–1947. doi: 10.1002/pmic.200500432. [DOI] [PubMed] [Google Scholar]
- 20.Lane CS, Wang Y, Betts R, Griffiths WJ, Patterson LH. Mol. Cell Proteomics. 2007;6:953–962. doi: 10.1074/mcp.M600296-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang Y, Muneton S, Sjovall J, Jovanovic JN, Griffiths WJ. J. Proteome. Res. 2008;7:1606–1614. doi: 10.1021/pr7006076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gevaert K, Van DP, Ghesquiere B, Impens F, Martens L, Helsens K, Vandekerckhove J. Proteomics. 2007;7:2698–2718. doi: 10.1002/pmic.200700114. [DOI] [PubMed] [Google Scholar]
- 23.Gygi SP, Aebersold R. Curr. Opin. Chem. Biol. 2000;4:489–494. doi: 10.1016/s1367-5931(00)00121-6. [DOI] [PubMed] [Google Scholar]
- 24.Nesvizhskii AI, Vitek O, Aebersold R. Nat. Methods. 2007;4:787–797. doi: 10.1038/nmeth1088. [DOI] [PubMed] [Google Scholar]
- 25.Righetti PG, Campostrini N, Pascali J, Hamdan M, Astner H. Eur. J. Mass Spectrom. (Chichester, Eng) 2004;10:335–348. doi: 10.1255/ejms.600. [DOI] [PubMed] [Google Scholar]
- 26.Barnidge DR, Dratz EA, Martin T, Bonilla LE, Moran LB, Lindall A. Anal. Chem. 2003;75:445–451. doi: 10.1021/ac026154+. [DOI] [PubMed] [Google Scholar]
- 27.Gerber SA, Rush J, Stemman O, Kirschner MW, Gygi SP. Proc. Natl. Acad. Sci. U. S. A. 2003;100:6940–6945. doi: 10.1073/pnas.0832254100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rappsilber J, Ryder U, Lamond AI, Mann M. Genome Res. 2002;12:1231–1245. doi: 10.1101/gr.473902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ishihama Y, Oda Y, Tabata T, Sato T, Nagasu T, Rappsilber J, Mann M. Mol. Cell Proteomics. 2005;4:1265–1272. doi: 10.1074/mcp.M500061-MCP200. [DOI] [PubMed] [Google Scholar]
- 30.Hewes JC, Riddy D, Morris RW, Woodrooffe AJ, Davidson BR, Fuller B. J. Hepatol. 2006;45:263–270. doi: 10.1016/j.jhep.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 31.Kersey PJ, Duarte J, Williams A, Karavidopoulou Y, Birney E, Apweiler R. Proteomics. 2004;4:1985–1988. doi: 10.1002/pmic.200300721. [DOI] [PubMed] [Google Scholar]
- 32.Watkins PB, Wrighton SA, Maurel P, Schuetz EG, Mendez-Picon G, Parker GA, Guzelian PS. Proc. Natl. Acad. Sci. U. S. A. 1985;82:6310–6314. doi: 10.1073/pnas.82.18.6310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nebert DW, Gonzalez FJ. Annu. Rev. Biochem. 1987;56:945–993. doi: 10.1146/annurev.bi.56.070187.004501. [DOI] [PubMed] [Google Scholar]
- 34.Mallick P, Schirle M, Chen SS, Flory MR, Lee H, Martin D, Ranish J, Raught B, Schmitt R, Werner T, Kuster B, Aebersold R. Nat. Biotechnol. 2007;25:125–131. doi: 10.1038/nbt1275. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.