

Abstract
The nuclear receptor PPARγ plays important roles in adipogenesis, immune response and in both lipid and carbohydrate metabolism. Although synthetic agonists for PPARγ are widely used as insulin sensitizers, the identity of the natural ligand(s) for PPARγ is still not clear. Suggested natural ligands include 15-deoxy-Δ12,14-PGJ2 and oxidized fatty acids such as 9-HODE and 13-HODE. Crystal structures of PPARγ have revealed the mode of recognition for synthetic compounds. Here we report structures of PPARγ bound to oxidized fatty acids which are likely natural ligands for this receptor. These structures reveal that the receptor can (i) simultaneously bind two fatty acids and (ii) couple covalently with conjugated oxo fatty acids. Thermal stability and gene expression analyses suggest that such covalent ligands are particularly effective activators of PPARγ and thus may serve as potent and biologically relevant ligands.
PPARγ is a nuclear receptor that regulates transcription of target genes in response to binding small lipophilic ligands 1. It has been found to play many diverse roles in biology from serving as a master regulator of adipose differentiation to the regulation of metabolism and inflammation. Indeed PPARγ was one of the first nuclear receptor transcription factors that was recognized to play an important role in regulating human metabolism. This recognition was primarily the result of the unexpected discovery that PPARγ is the direct pharmacological target of thiazolidinedione-based drugs that enhance insulin sensitivity and are in clinical use for the treatment of type II diabetes 2. Despite the identification of synthetic agonists for PPARγ, this receptor is still arguably an orphan nuclear receptor since the identity of its natural, physiological ligand remains controversial. The first ligand that was suggested to be a natural activator for PPARγ was the prostaglandin 15-deoxy-Δ12,14-PGJ2 (PGJ2) 3,4. Whilst there is no doubt that PGJ2 is a potent agonist for PPARγ, the relevance of this ligand has been questioned on the basis that there is an insufficient amount in biological tissues to result in PPARγ activation 5. Unsaturated fatty acids are more abundant than PGJ2 and have also been found to activate PPARγ and hence are candidate natural ligands 6,7. In macrophages, and more recently in other tissues, it has been shown that the oxidized fatty acids 9-HODE and 13-HODE, both present in oxidized LDL particles, are particularly potent agonists 8-10.
Nuclear receptor ligands bind in a cavity within a moderately conserved ligand-binding domain (LBD) towards the carboxy terminus of the nuclear receptor (for reviews see 11,12). A linker joins the LBD to the central DNA-binding domain of the receptor 13. The LBDs of receptors have a conserved structure that consists of three layers of alpha helices. The central layer is incomplete leaving a cavity that can accommodate ligand. The size of this cavity varies according to the disposition of the helices in the outer layers. The ligand binding cavity in PPARγ is particularly large.
Nuclear receptors activate transcription in response to ligand binding through the displacement of co-repressor proteins and the recruitment of co-activator proteins 12. The mechanism appears to involve both a global stabilization of the LBD on ligand binding as well as a specific stabilization of the C-terminal helix 12 14,15. Interestingly, a variety of partial agonists were shown to differentially stabilise distinct regions of the LBD 16. Once bound to ligand, coactivators bind in a helix 12 dependent fashion resulting in a transcriptionally active complex.
Given the importance of PPARγ as a transcription factor regulating key physiological processes and as a drug target for insulin sensitizers, there have been many structures determined in complex with various synthetic agonists 17-20. To date however there have been no structures of PPARγ in complex with proposed natural ligands. Here we present structures of human PPARγ in complex with a variety of proposed and likely natural oxidized fatty acid ligands for PPARγ and reveal the mode of activation by these lipids.
Results
The PPARγ ligand binding domain (aa 204-477) was crystallized in the presence of a variety of fatty acid ligands (Fig. 1a). In all cases the protein crystallized under similar conditions and in the same space group/cell dimensions as the previously reported unliganded homodimeric structure (ref 19, 1prg). In this structure one PPARγ monomer closely resembles the liganded form of the receptor as seen in other structures (e.g. ref 19, 2prg). In the other monomer, the C-terminal helix 12 is displaced and occupies an alternative position. Simulated annealing composite omit maps (CNS 21) were calculated following molecular replacement and preliminary refinement. It was immediately clear that all the structures contained fatty acids in the ligand binding pockets of both monomers. In the monomer in which helix 12 is displaced from the active position, modeling of the ligand conformation was often less confident, or impossible, despite clear evidence for an occupied ligand binding pocket. This suggests that the positioning of the lipid ligand and the stabilization of helix 12 are coupled processes. Here we focus our description on the binding modes of the fatty acids in the monomer with the active conformation of helix 12 since these are generally better ordered and are likely to represent the bona fide mode of ligand binding to the activated receptor.
Figure 1. PPARγ has a versatile fatty acid binding pocket.

(a) Chemical structures of the ligands used in this study. (b) Superposition of 9-HODE, 13-HODE, DHA, 4-HDHA, 5-HEPA, 6-HOTE, 4-oxoDHA and 6-oxoOTE as they bind to PPARγ. Overall, these ligands sample c. 70% (1,004 Å3) of the ligand binding cavity. The architectural cartoon is shown in grey, with helix 12 in yellow.
The central region of the PPARγ ligand binding pocket is surrounded by largely non-polar amino acids. However, at both ends of the cavity there are clusters of polar residues. Many of the synthetic ligands position a carboxylate group, or similar group, adjacent to helix 12 making multiple polar contacts. Figure 1b shows an overlay of the eight fatty acids that we have crystallized in the ligand binding cavity of PPARγ. The combined ligands sample c. 1,004 Å3 of the ligand binding cavity. In the majority of structures, a single fatty acid chain is bound so as to position its carboxylate group adjacent to helix 12, with the aliphatic chain wrapping around helix 3 (Fig.1b). For the oxidized fatty acids, additional polar contacts are made with the hydroxyl or keto groups. However the C18 fatty acids 9-HODE and 13-HODE are exceptions to this general mode of binding.
PPARγ in complex with 9- and 13-HODE
In the structure of PPARγ with 9-HODE (at 2.05Å resolution) two molecules of 9-HODE are clearly observed simultaneously bound in the ligand binding pocket (Fig. 2a-d). The shape of the two lipids is similar, with each fatty acid adopting a C-shaped conformation. The first molecule binds with its head group adjacent to helix 12, linking 4 helical regions of the protein with hydrogen bonds to Ser289 (H3), His323 (H6), His449 (H11) and Tyr473 (H12) (Fig. 2b,d). This is the canonical interaction that is observed in several other PPARγ structures with carboxylate containing ligands. The tail of the 9-HODE fatty acids occupies a region of the cavity that is occupied by particularly potent agonists such as farglitazar. Intriguingly Phe363, which exhibits high temperature factors, and is presumably mobile, in the apo and several liganded structures, is repositioned in this structure so as to interact with both ends of the fatty acid (i.e. carbons 2, 3, 4 and 16, 17, 18). The 9-hydroxyl group of the first 9-HODE ligand makes a hydrogen bond to the carboxylate of the second 9-HODE ligand.
Figure 2. Recognition of 9- and 13-HODE by PPARγ.

(a) Overall view of the two molecules of 9-HODE bound to PPARγ. (b) Refmac weighted map at 0.8 sigma of 9-HODE bound in the canonical position to helix12. (c) Refmac weighted map at 0.8 sigma of the second molecule of 9-HODE bound near the beta-sheet. (d) Details of the amino acid sidechains in contact with the two molecules of 9-HODE. (e) Overall view of 13-HODE bound to PPARγ. (f) Refmac weighted map at 0.8 sigma of 13-HODE bound near the beta- sheet. (g) CNS simulated annealing omit map at 1.0 sigma showing density for a molecule of 13-HODE bound in the canonical position near helix 12. (h) Details of the amino acid sidechains in contact with the ordered 13-HODE ligand.
The second 9-HODE ligand is positioned between helix 3 and the beta-sheet and is distant from helix 12 (Fig. 2a). In addition to hydrogen bonding with the hydroxyl of the first 9-HODE, the carboxylate of the second molecule of 9-HODE forms a salt bridge with Arg288 (Fig. 2c,d). This arginine also makes a salt bridge with Glu295. Interestingly this side chain is repositioned from the apo conformation in which it makes a hydrogen bond to the hydroxyl group of Ser289. The hydroxyl of the second 9-HODE molecule is positioned such that it is approximately equidistant from the carboxylate groups of the glutamic acid residues: Glu291 and Glu295.
In the same way that two 9-HODE ligands bind to PPARγ, there is evidence that two 13-HODE molecules can also bind to the receptor. However, unlike 9-HODE, only one molecule of 13-HODE is well-ordered in the structure at 2.35Å resolution (Fig. 2e-h). In the simulated annealing composite omit map there is density of sufficient volume for a molecule in the canonical position close to helix 12 (Fig. 2g). However, it was not possible to convincingly model any dominant conformation for this molecule. We therefore conclude that it is present in the cavity, but its position is not tightly constrained and it may not stabilize helix 12 as efficiently as the more ordered 9-HODE.
There is clear evidence, however, for a well-ordered 13-HODE molecule bound toward the edge of the cavity between helix 3 and the beta-sheet (Fig. 2e,f,h). The fatty acid adopts a “question mark” shaped conformation with the hydroxyl making a hydrogen bond to its own carboxylic acidic group. The hydroxyl also makes a rather long hydrogen bond with the backbone amide of Ser342. The carboxylic acidic group of 13-HODE makes a salt bridge with Arg288. To make this salt bridge with the ligand, the side chain of Arg288 is positioned quite differently from the complex with 9-HODE such that the salt bridge with Glu295 is broken.
Complexes with DHA, 4-HDHA and 5-HEPA
The simulated annealing composite omit maps clearly show that DHA, 4-HDHA and 5-HEPA all bind in a similar fashion to PPARγ. Their carboxylate groups interact in the canonical fashion in the vicinity of helix 12. Of the three, the 5-HEPA appears to be the best defined after refinement (Fig. 3a). This is likely because the hydroxyl group makes a good hydrogen bond with the hydroxyl of Tyr327. The hydroxyl of 4-HDHA is positioned directly over the aromatic ring of Phe282 and is probably making a favorable interaction with the pi-electron ring. It could also be making a rather long hydrogen bond to Gln286 (Fig. 3a). The aliphatic tail of DHA may have multiple conformations since breaks are observed in the electron density of the refined maps.
Figure 3. Binding of various oxidized fatty acid ligands to PPARγ.

(a) Electron density maps with refined models of 5-HEPA, 4-HDHA, 4-oxoDHA and 6-oxoOTE bound to the PPARγ LBD. Note the 4-oxoDHA and 6-oxoOTE ligands are covalently coupled to Cys285. For orientation the end-on view of helix 3 is indicated by a magenta ellipsoid. The refmac weighted maps are contoured at 0.8 sigma. (b) Maldi-ToF mass spectrometry of ligands bound to PPARγ shows that the oxidized fatty acids are covalently coupled to the LBD. (c) Mechanism of Michael addition for covalent binding of oxidized fatty acid ligands to PPARγ.
Oxo fatty acids couple covalently to PPARγ
The structures of PPARγ bound to the oxo fatty acids 4-oxoDHA and 6-oxoOTE revealed an unambiguous positioning of the ligands. In both cases there was very clear electron density indicating that the ligand is covalently bound at position C8 to the SH group of Cys285 (Fig. 3a). The Cys285 side chain adopts alternative rotamers to accommodate slightly different positioning of the two ligands. The carboxylates of the two ligands are in very similar positions, surrounded by the polar sidechains of Tyr473, His449, His323, Ser289. The serine is positioned almost equidistance from the carboxylate and keto groups of 6-oxoOTE. Other than this the 6-oxoOTE keto group makes no polar contacts. As would be expected, the ketone of 4-oxoDHA is positioned very differently from that of 6-oxoOTE such that it is positioned between, and hydrogen bonds with, the sidechains of Tyr327 and Lys367.
Efficient covalent binding of oxo Fatty acids
To exclude the possibility that the covalent coupling of the oxo ligands seen in the crystal structures was an artifact of crystallization, or exposure to high intensity X-rays, we performed a mass spectroscopic (MALDI-ToF) analysis of the PPARγ LBD after a brief incubation with various ligands. Figure 3b shows that under these conditions (20 minutes at 20°C with a four-fold excess of ligand) about 60% of the receptor binds PGJ2 covalently. For those ligands that contain a conjugated ketone, essentially 100% of the receptor becomes covalently coupled with ligand. Conversely the hydroxy fatty acids show no evidence of covalently coupling to the receptor. These data support the biological relevance of the observations of covalent coupling in the crystal structures for 4-oxoDHA and 6-oxoOTE.
The covalent coupling is the result of a Michael addition (conjugate addition) for which the organic reaction is well established and illustrated in figure 3c. The Michael acceptor contains an electron withdrawing group conjugated to an activated carbon that is then subject to attack by the nucleophile - in this case the SH group of Cys285. Intriguingly, in the case of 4-oxoDHA, there are two conserved sidechains, tyrosine Tyr327 and lysine Lys367, that are positioned such that they can form hydrogen bonds with the keto group of the ligand. It is likely that the tyrosine sidechain catalyses the conjugate addition through enhancing the electron withdrawing effect of the keto group22. The position of these sidechains may play a role in determining the specificity of ligands that efficiently couple to the receptor.
Covalent ligands strongly stabilize the PPARγ LBD
It is well established that ligand binding by nuclear receptors results in the ligand binding domain becoming stabilized to thermal denaturation and furthermore that the degree of stabilization correlates, in many cases with the potency of the ligand. To explore the effect of covalent vs non-covalent ligand binding to PPARγ we used CD at 222 nm to monitor the thermal denaturation of PPARγ (Fig. 4). In the absence of ligand, PPARγ loses it folded structure in a highly cooperative fashion, with a melting temperature of 46°C. When bound to the synthetic agonist rosiglitazone the melting temperature increases to 51°C. In complex with the more potent agonist farglitazar, PPARγ remains folded at much higher temperatures, melting at 60°C.
Figure 4. Thermal denaturation studies of PPARγ LBD.
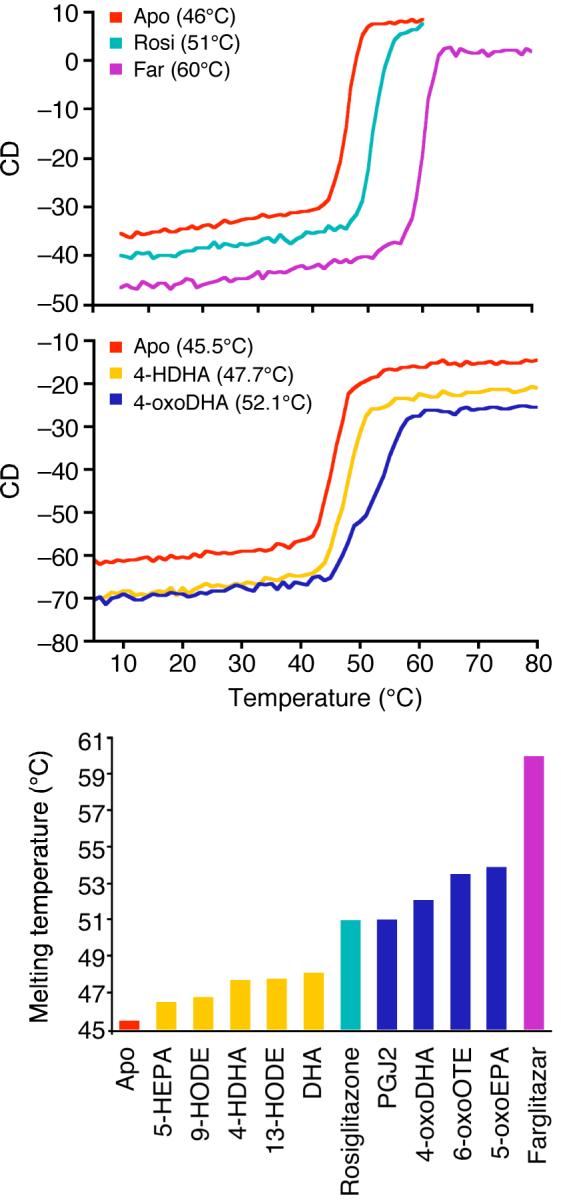
Circular dichroism denaturation curves of the PPARγ LBD show that ligand binding stabilizes the LBD to thermal denaturation. This effect is greater with tight binding ligands such as farglitazar. Note that the covalently bound oxo fatty acids stabilize more strongly than the non-covalently bound fatty acids. Molar ellipticity was monitored at 222 nm. The melting curves were fitted using PRISM.
PPARγ in complex with 4-HDHA is modestly stabilized and melts at 48°C. 4-oxoDHA is a more effective stabilizer of PPARγ such that the receptor has a melting temperature of 52°C (slightly higher than that for rosiglitazone).
The bottom panel in figure 4 summarizes the melting temperatures of all the ligands tested. It is striking that all the complexes with oxo fatty acids denature at a higher temperature than those with hydroxy fatty acids (and also higher than the rosiglitazone bound receptor). Thus these ligands would appear to be excellent stabilizers of the PPARγ structure.
Covalent ligands are potent inducers of PPARγ targets
We used two assays to determine whether ligands that form covalent bonds with PPARγ are more effective activators of transcription than their non-covalent counterparts. In the first assay (left-hand panels in Fig. 5) we measured the activity, in Cos1 cells, of a reporter gene activated by a Gal4 DBD:PPARγ LDB chimera. In the second assay we measured mRNA levels of the PPARγ target gene FABP4/aP2 in dendritic cells, derived from normal peripheral human monocytes23. In both assays, it is clear that the oxo fatty acids that covalently couple to the receptor are up to an order of magnitude more active than the hydroxy fatty acids. This activity represents activation of PPARγ, rather than the RXR partner in the heterodimer, since the various oxidized fatty acids give negligible activity, even at 25μM, with a Gal4DBD:RXRLBD construct (Supplementary Fig. 1). Furthermore RXR selective ligands do not induce FABP4/aP2 expression in dendritic cells (Supplementary Fig. 1). Experiments to determine whether oxo fatty acids might result in a longer-term response than non-covalent ligands were largely inconclusive. However, when ligand was removed after 12 hours, there was some suggestion that covalent ligands might have prolonged activity (Supplementary Fig. 2). Further investigation is required to clarify this issue.
Figure 5. Activity of oxidized fatty acids in cell based assays.

The four left hand panels show the activation of a luciferase reporter gene by a Gal4 DBD- PPARγ LBD hybrid protein. The four right hand panels show the activation of transcription of the FABP4/aP2 gene in dendritic cells derived from normal human peripheral monocytes (mRNA levels measured by Q-PCR). A similar pattern of ligand activation is seen in both assays. In all cases the oxo fatty acid exhibits activation at lower concentrations than the hydroxy fatty acids. The rank order of activity is: 6-oxoOTE > 5-oxoEPA > 4-oxoDHA. Error bars indicate standard deviation.
Cys285 is essential for the activity of oxo fatty acids
To confirm that the greater activity of the oxo fatty acids is dependent upon the formation of a covalent bond with Cys285 we tested that ability of 4-oxoDHA and 4-HDHA to activate, in Cos7 cells, PPARγ in which Cys285 had been substituted by a serine or alanine side chain (Fig. 6). Interestingly both the mutants showed reduced activity with both ligands (and also pioglitazone - data not shown). However it was clear that the activity of 4-oxoDHA is less than that of 4-HDHA. This contrasts sharply with the wild-type receptor for which 4-oxoDHA is more potent than 4-HDHA.
Figure 6. Cys285 is essential for the activity of covalent ligands.
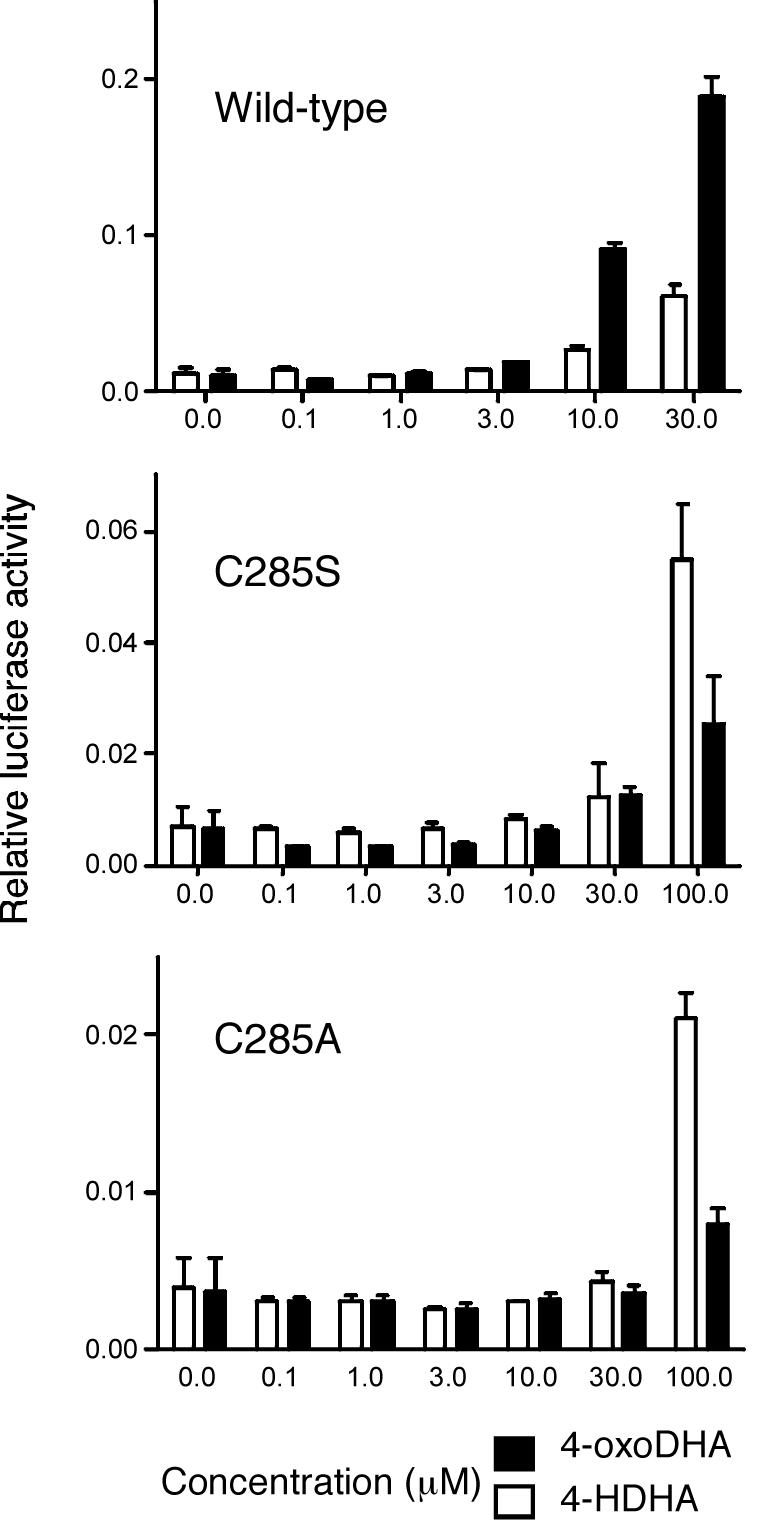
Cell-based transcriptional activation assay using wild type and Cys285 mutants of PPARγ with 4-oxoDHA and 4HDHA. The three panels show the activation of a luciferase reporter gene by a Gal4 DBD- PPARγ LBD hybrid. The 4-oxoDHA is more active than 4-HDHA with the wild type receptor. Both the cysteine mutations impair the activity of the receptor. However it is clear that the activity of 4-oxoDHA is impaired more than that of 4-HDHA supporting the importance of covalent interactions in receptor activation.
Discussion
A versatile ligand binding pocket
PPARγ has a large Y-shaped ligand-binding pocket with a volume around 1,440Å3. This is markedly larger than the cavities observed in other nuclear receptors, which typically have cavity volumes ranging from 600-1,100Å3. Why PPARγ should need such a large ligand binding pocket is somewhat of a puzzle since no known ligand could come close to filling this cavity. However the set of crystal structures presented here, of oxidized fatty acids binding to PPARγ, reveals that this large cavity confers remarkable versatility in ligand binding such that many different fatty acids can be accommodated in different positions according to the detailed stereochemistry of the particular ligand. Furthermore the cavity can accommodate two copies of certain ligands, satisfying their needs for polar contacts at both ends of the cavity. An overlay of the structures of these fatty acid complexes shows that together, the ligands sample c. 1,004Å3 of the cavity amounting to 70% of the cavity by volume. It can be concluded therefore that the large ligand binding cavity is likely to play a role in PPARγ biology through allowing the receptor to sense not one single ligand but rather a pool of related ligands. This means that the concentration of this pool of ligands is likely to be more relevant than the concentration of each individual ligand, making PPARγ a sensor of oxidized fatty acids.
In addition to highlighting the versatility of the PPARγ ligand binding pocket, these structures also demonstrate the adaptability of the pocket through the rearrangement of side chains on the interior of the ligand binding cavity. Of particular note are the side chains of Phe363 and Arg288. In the structure of 9-HODE bound to PPARγ the aromatic group of Phe363 is rearranged so that it lies between the two ends of the fatty acid, making multiple van der Waals contacts. The role of Arg288 is important since it forms salt bridges with the second fatty acid when two are accommodated in the cavity. Furthermore it is clear that the mobility of Arg288 is key to the ability of the receptor to adapt to the different ligands 9- and 13-HODE in the second site. The biological importance of Arg288 is supported by the finding that this residue is mutated to a histidine in colon cancers 24. Sarraf et al. demonstrated that the mutant receptor has reduced affinity for 9-HODE and 15-HETE, but not for rosiglitazone. Thus the structural studies presented here provide a possible molecular explanation for these critical clinical defects.
It is interesting to note that site between helix 3 and the beta sheet, that accommodates the second 9-HODE and 13-HODE molecules, has been shown to be occupied by synthetic partial agonists for PPARγ 16. This suggests that partial agonists are exploiting part of signaling mechanism used by natural ligands. Since 13-HODE is more ordered in this site than in the canonical position, it is possible that 13-HODE may share some of the properties of synthetic partial agonists and hence suggests a mechanism through which natural ligands could generate graded responses similar to those observed with synthetic agonists.
Given that two molecules of 9-HODE and 13-HODE can bind simultaneously to the receptor this raises the issue as to whether it might be possible for two different fatty acids to bind simultaneously. This would be important since it might allow PPARγ to give a graded response to varying compositions of the cellular pool of fatty acid ligands. The structures show that the 13-HODE adopts a position in the ligand binding cavity that overlaps that of both the 9-HODE molecules. It is clear therefore that if 9- and 13-HODE were to bind simultaneously then there would have to be some rearrangement of their binding position. However the versatility of fatty acid binding that is apparent in these structures suggests that it is very likely that the ligand binding cavity will simultaneously accommodate distinct ligands if the cellular pool of ligands contains the appropriate mix of fatty acids.
Is covalent binding of ligands to PPARγ relevant in vivo?
The observation that oxo fatty acids can couple covalently to a cysteine residue conserved not only in PPARγ, but also in PPARα and δ (these receptors will very likely also couple covalently to oxo fatty acids), raises the question as to whether this mode of ligand binding is physiologically relevant. This ultimately depends on two issues: (i) do covalent ligands effectively activate PPARγ and (ii) are covalent ligands sufficiently abundant (and regulated) so as to serve a signalling molecules. The first issue we have addressed by showing that, like synthetic agonists, covalent ligands are effective stabilizers of the LBD and that they are able to activate both reporter genes and natural PPARγ target genes in relevant cells. The second issue is not so clear. Like oxo fatty acids, PGJ2 is also a good activator for PPARγ and has also been found to couple covalently to the receptor 25,26. However, the biological relevance of PGJ2 as natural agonist has recently been questioned on the basis that the natural abundance of PGJ2 in cells is insufficient to activate PPARγ 5.
Nitro-fatty acids are also candidates for natural covalent ligands for PPARγ 27. The nitrate group is strongly electron withdrawing and therefore conjugated nitro fatty acids would be expected to react readily with Cys285. Indeed relatively high concentrations (c. 500nM) of nitro fatty acids have been reported in plasma 27. However nitro fatty acids would be expected to be very short-lived inside the cell since they are readily coupled, in the absence of an enzyme, with the high endogenous concentrations of glutathione 28. This may mean that nitro fatty are not likely ligands, but on the other hand it might mean that the levels of nitro fatty acids can be tightly regulated through controlling their release.
Oxo fatty acids are not readily modified by glutathione except in combination with the enzyme glutathione-S-transferase. They might therefore be expected to have a rather longer lifetime than nitro fatty acids and perhaps serve as better indicators of metabolic status. Probably the most abundant oxo fatty acids in vivo are 5-oxoEPA and 5-oxoETE 29,30. These are metabolites of the dietary fatty acids eicosapentaenoic acid and arachidonic acid. 5-oxoEPA and 5-oxoETE have been shown to be readily synthesized by neutrophils and peripheral blood mononuclear cells 29-31 and might reach concentrations of 10-150μM, which would certainly be sufficient to activate PPARγ.
The concept of covalent ligands for PPARγ raises a number of interesting issues since it means that we should no longer think in terms of equilibrium kinetics to describe ligand activation of PPARγ. It also means that an activated receptor presumably remains active until it is degraded by the proteosome. However, off-rates are probably very low for at least some conventional ligands binding to other nuclear receptors, since they bind with very high affinity. It may therefore be that the covalent ligands for PPARγ allow this receptor to mimic the behavior of more conventional receptors whilst retaining the ability to bind multiple substrates. Further studies using covalent ligands will need to address whether they result in differences in the behavior of the receptor, such as increased rate of turnover or specific co-regulator requirements.
In conclusion the structures presented here, of oxidized fatty acids bound to PPARγ, along with the associated functional assays, suggest mechanisms through which the activity of this important nuclear receptor might be regulated in vivo. In particular two concepts are highlighted by these studies. First, PPARγ can bind multiple fatty acids (in certain cases simultaneously) and thus sense the concentration of a pool of lipids. Second, PPARγ can covalently couple to certain oxidized fatty acids, which suggests a rather different signaling paradigm since PPARγ could capture such activators at fairly low concentration and the covalent complex may be able to activate transcription for a longer duration.
Methods
Protein expression and purification
The human PPARγ LBD (aa 204-477) was expressed using a modified pET30a vector with an N-terminal 6xHis tag cleavable by TEV protease. Details of the expression and purification are given in the Supplementary Methods.
Crystallisation, structure determination, ligand assignment and modeling
9-(S)-HODE and 13-(S)-HODE were purchased from Cayman Chemical. DHA was a kind gift from Maruha Corporation (Tsukuba, Japan). 4-HDHA and 4-oxoDHA were prepared as previously described 32,33. 5-HEPA / 5-oxoEPA and 6-HOTE / 6-oxoOTE were prepared from EPA and γ-linoleic acid (respectively) following strategies analogous to those described previously 32,33.
All crystals were obtained through co-crystallization with the relevant ligand. Detailed crystallization conditions are given in the Supplementary Methods. Diffraction data were collected at the synchrotron ESRF ID 14.3 (wavelength - 0.931Å). Data were indexed, integrated and scaled using MOSFLM 34 and the CCP4 35 suite of programs. The structures were solved using molecular replacement, rebuilt and refined using COOT 36, REFMAC 37 and CNS 21. At multiple stages simulated annealing omit maps and simulated annealing composite omit maps (calculated in CNS v1.2) were used to guide the modeling of ligand. Where clear contiguous density did not return in the weighted maps at 0.8 sigma, then the ligand was not modeled. The ligand molecules were not actually included in the protein model until the final stages of refinement. This approach gave confidence in the modeling of the fatty acid and allowed the best possible unbiased assignment of the ligands. All ligands were modeled with occupancy 1.0. Data and refinement statistics are shown in Table 1.
Mass spectrometry
20 μL of 250 μM PPARγ (20 mM Tris pH 7.4, 1 mM TCEP, 0.5 mM EDTA) were incubated with four-fold excess ligands for 20 min at room temperature. Samples were loaded under aqueous conditions onto a MoBiCol spin column (VH Bio Ltd., Gateshead, UK) containing 0.67 mg of POROS 50 R2 media (Applied Biosystems, Warrington, UK). The samples were eluted using 50μl of 50% v/v acetonitrile with 0.1% v/v trifluoroacetic acid.
The eluant was mixed in a 1:1 ratio with a 10 mg ml-1 solution of sinapinic acid (Sigma, Dorset, UK) and 0.5 μl was spotted onto a stainless steel MALDI target plate. Mass spectra were obtained using a Voyager DE-STR MALDI-ToF mass spectrometer (Applied Biosystems) in positive ion linear mode over the mass range 4,000-40,000 Da. Calibration was performed using the singly-charged, doubly-charged and dimer masses of horse heart myoglobin (Sigma).
CD monitored thermal denaturation
25 μM of PPARγ was incubated with equimolar ligand. Circular dichroism was monitored at 222 nm, with data points collected every minute per degree celcius as the sample temperature was increase from 5-80°C (1°C per minute) and with a spectropolarimeter (Jasco J-715) with a temperature controller (Jasco PTC-348WI). An approximate melting temperature was obtained by fitting the data to a single-site sigmoidal dose response curve (GraphPad Prism).
Transient transfections and reporter gene assays
For experiments performed in Cos1 cells (shown in Fig. 5): The MH-100-TK-luc reporter gene (containing Gal binding sites) along with Gal-DBD fused hsPPARγ -LBD and pCMX beta gal were transfected into COS1 cells using jetPEI reagent (Qbiogene) according to manufacturer’s instructions and essentially as described by 8. Transfected cells were treated with the indicated ligands or vehicle 6 hours following transfection. Cells were lysed and assayed for reporter expression 24 h after transfection. Luciferase activity was measured using “The Luciferase Assay System” (Promega), β-galactosidase activity was determined as described in 8. Measurements were carried out using a Wallac Victor 2 multilabel counter. Luciferase activity of each sample was normalized to the β-galactosidase activity.
For experiments performed in Cos7 cells (shown in Fig. 6): COS-7 cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 5% v/v fetal bovine serum (FBS). Cells were seeded on 24-well plates (2 × 104 cells in 0.5 mL medium per well). After 24 h, DNA/Trans IT-LT1 reagent (Mirus, Madison, USA) mixture [0.18 μg of a reporter plasmid containing four copies of MH100 GAL4 binding site (MH100×4-TK-Luc), 0.05 μg of wild-type or mutant GAL4-hPPARγ chimera expression plasmid (pSG5-GAL-hPPARγ), and 0.02 μg of the internal control plasmid containing sea pansy luciferase expression constructs (pRL-CMV), and 0.75 μL of Trans IT-LT1 reagent per well] was prepared according to the manufacturer’s procedures, and added to each well. After 8 h-incubation, the cells were treated with either the ligand or vehicle and cultured for 16 h. Cells in each well were harvested with a cell lysis buffer, and the luciferase activity was measured with a luciferase assay kit (Toyo Ink, Inc., Japan). Transactivation measured by the luciferase activity was normalized with the internal control. All experiments were done in triplicate.
Monocyte isolation and dendritic cell generation
Human peripheral monocytes were obtained from platelet free Buffy coats isolated from healthy donors by Ficoll-Hypaque (Pharmacia) gradient centrifugation and immunomagnetic cell separation using anti-CD14-conjugated microbeads (VarioMACS; Miltenyi Biotec). Monocytes were resuspended into 24-well culture plates or tissue flaks at a density of 106 cells per ml and cultured in RPMI 1640 (Sigma-Aldrich) supplemented with 10% v/v Lipoprotein deficient FBS (Invitrogen) containing 800 U/ml GM-CSF (PeproTech) and 500 U per ml IL-4 (PeproTech) and Penicillin and Streptomycin (Sigma-Aldrich). Cells were treated by the indicated ligands or vehicle and were harvested after 12 hours of treatment.
Real Time quantitative PCR
Total RNA was isolated using TRIZOL reagent (Invitrogen). Reverse transcription was performed at 25°C for 10 min, 42°C for 2 h, and 72°C for 5 minutes using Superscript II reverse transcriptase (Invitrogen) and random primers (3 μg μl-1; Invitrogen). Quantitative PCR was performed using real-time PCR (ABI PRISM 7900; Applied Biosystems): 40 cycles at 95°C for 12 s and at 60°C for 30 s using Taqman assays for the indicated genes. All PCR reactions were carried out in triplicate. Information on the individual assays is available upon request.
Supplementary Material
Acknowledgements
Protein mass spectrometry was carried out by Dr Andrew R. Bottrill and Mrs Shairbanu Ibrahim of the PNACL Proteomics Facility at the University of Leicester. The Schwabe laboratory is supported by the Wellcome Trust. The Nagy laboratory is supported by the Howard Hughes Medical Institute; the Wellcome Trust and the Hungarian Scientific Research Fund. We acknowledge the ESRF for provision of synchrotron radiation facilities and thank the staff on beamline ID14.3.
Footnotes
Accession codes. Protein Data Bank: Coordinates and structure factors for the structures presented in this manuscript have been deposited with accession codes 2vsr (PPARγ:9-HODE); 2vst (PPARγ:13-HODE); 2vv0 (PPARγ:DHA); 2vv1 (PPARγ:4-HDHA); 2vv3 (PPARγ:4-oxoDHA); 2vv2 (PPARγ:5-HEPA); 2vv4 (PPARγ:6-oxoOTE).
References
- 1.Tontonoz P, Spiegelman BM. Fat and beyond: The diverse biology of PPARgamma. Annu Rev Biochem. 2008;77 doi: 10.1146/annurev.biochem.77.061307.091829. doi:10.1146/annurev.biochem.77.061307.091829. [DOI] [PubMed] [Google Scholar]
- 2.Lehmann JM, et al. An antidiabetic thiazolidinedione is a high affinity ligand for peroxisome proliferator-activated receptor gamma (PPAR gamma) J Biol Chem. 1995;270:12953–6. doi: 10.1074/jbc.270.22.12953. [DOI] [PubMed] [Google Scholar]
- 3.Kliewer SA, et al. A prostaglandin J2 metabolite binds peroxisome proliferator-activated receptor gamma and promotes adipocyte differentiation. Cell. 1995;83:813–9. doi: 10.1016/0092-8674(95)90194-9. [DOI] [PubMed] [Google Scholar]
- 4.Forman BM, et al. 15-Deoxy-delta 12, 14-prostaglandin J2 is a ligand for the adipocyte determination factor PPAR gamma. Cell. 1995;83:803–12. doi: 10.1016/0092-8674(95)90193-0. [DOI] [PubMed] [Google Scholar]
- 5.Powell WS. 15-Deoxy-delta12,14-PGJ2: endogenous PPARgamma ligand or minor eicosanoid degradation product? J Clin Invest. 2003;112:828–30. doi: 10.1172/JCI19796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kliewer SA, et al. Fatty acids and eicosanoids regulate gene expression through direct interactions with peroxisome proliferator-activated receptors α and γ. Proc Nat Acad Sci. 1997;94:3418–4323. doi: 10.1073/pnas.94.9.4318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Krey G, Braissant O, FL’Horset, Kalkhoven E. Fatty Acids, Eicosanoids, and Hypolipidemic Agents Identified as Ligands of Peroxisome Proliferator-Activated Receptors by Coactivator-Dependent Receptor Ligand Assay. Mol Endocrinol. 1997;11:779–791. doi: 10.1210/mend.11.6.0007. [DOI] [PubMed] [Google Scholar]
- 8.Nagy L, Tontonoz P, Alvarez JG, Chen H, Evans RM. Oxidized LDL regulates macrophage gene expression through ligand activation of PPARgamma. Cell. 1998;93:229–40. doi: 10.1016/s0092-8674(00)81574-3. [DOI] [PubMed] [Google Scholar]
- 9.Marx N, Bourcier T, Sukhova GK, Libby P, Plutzky J. PPARgamma activation in human endothelial cells increases plasminogen activator inhibitor type-1 expression: PPARgamma as a potential mediator in vascular disease. Arterioscler Thromb Vasc Biol. 1999;19:546–51. doi: 10.1161/01.atv.19.3.546. [DOI] [PubMed] [Google Scholar]
- 10.Schild RL, et al. The activity of PPAR gamma in primary human trophoblasts is enhanced by oxidized lipids. J Clin Endocrinol Metab. 2002;87:1105–10. doi: 10.1210/jcem.87.3.8284. [DOI] [PubMed] [Google Scholar]
- 11.Renaud JP, Moras D. Structural studies on nuclear receptors. Cell Mol Life Sci. 2000;57:1748–69. doi: 10.1007/PL00000656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nagy L, Schwabe JW. Mechanism of the nuclear receptor molecular switch. Trends Biochem Sci. 2004;29:317–24. doi: 10.1016/j.tibs.2004.04.006. [DOI] [PubMed] [Google Scholar]
- 13.Khorasanizadeh S, Rastinejad F. Nuclear-receptor interactions on DNA-response elements. Trends Biochem Sci. 2001;26:384–90. doi: 10.1016/s0968-0004(01)01800-x. [DOI] [PubMed] [Google Scholar]
- 14.Johnson BA, et al. Ligand-induced stabilization of PPARgamma monitored by NMR spectroscopy: implications for nuclear receptor activation. J Mol Biol. 2000;298:187–94. doi: 10.1006/jmbi.2000.3636. [DOI] [PubMed] [Google Scholar]
- 15.Kallenberger BC, Love JD, Chatterjee VK, Schwabe JW. A dynamic mechanism of nuclear receptor activation and its perturbation in a human disease. Nat Struct Biol. 2003;10:136–40. doi: 10.1038/nsb892. [DOI] [PubMed] [Google Scholar]
- 16.Bruning JB, et al. Partial agonists activate PPARgamma using a helix 12 independent mechanism. Structure. 2007;15:1258–71. doi: 10.1016/j.str.2007.07.014. [DOI] [PubMed] [Google Scholar]
- 17.Cronet P, et al. Structure of the PPARalpha and -gamma ligand binding domain in complex with AZ 242; ligand selectivity and agonist activation in the PPAR family. Structure. 2001;9:699–706. doi: 10.1016/s0969-2126(01)00634-7. [DOI] [PubMed] [Google Scholar]
- 18.Gampe RT, Jr., et al. Asymmetry in the PPARgamma/RXRalpha crystal structure reveals the molecular basis of heterodimerization among nuclear receptors. Mol Cell. 2000;5:545–55. doi: 10.1016/s1097-2765(00)80448-7. [DOI] [PubMed] [Google Scholar]
- 19.Nolte RT, et al. Ligand binding and co-activator assembly of the peroxisome proliferator-activated receptor-gamma. Nature. 1998;395:137–43. doi: 10.1038/25931. [DOI] [PubMed] [Google Scholar]
- 20.Pochetti G, et al. Insights into the mechanism of partial agonism: crystal structures of the peroxisome proliferator-activated receptor gamma ligand-binding domain in the complex with two enantiomeric ligands. J Biol Chem. 2007;282:17314–24. doi: 10.1074/jbc.M702316200. [DOI] [PubMed] [Google Scholar]
- 21.Brunger AT. Version 1.2 of the Crystallography and NMR system. Nat Protoc. 2007;2:2728–33. doi: 10.1038/nprot.2007.406. [DOI] [PubMed] [Google Scholar]
- 22.Oakley AJ, et al. The three-dimensional structure of the human Pi class glutathione transferase P1-1 in complex with the inhibitor ethacrynic acid and its glutathione conjugate. Biochemistry. 1997;36:576–85. doi: 10.1021/bi962316i. [DOI] [PubMed] [Google Scholar]
- 23.Szatmari I, et al. Activation of PPARgamma specifies a dendritic cell subtype capable of enhanced induction of iNKT cell expansion. Immunity. 2004;21:95–106. doi: 10.1016/j.immuni.2004.06.003. [DOI] [PubMed] [Google Scholar]
- 24.Sarraf P, et al. Loss-of-function mutations in PPAR gamma associated with human colon cancer. Mol Cell. 1999;3:799–804. doi: 10.1016/s1097-2765(01)80012-5. [DOI] [PubMed] [Google Scholar]
- 25.Shiraki T, et al. Alpha,beta-unsaturated ketone is a core moiety of natural ligands for covalent binding to peroxisome proliferator-activated receptor gamma. J Biol Chem. 2005;280:14145–53. doi: 10.1074/jbc.M500901200. [DOI] [PubMed] [Google Scholar]
- 26.Soares AF, et al. Covalent binding of 15-deoxy-delta12,14-prostaglandin J2 to PPARgamma. Biochem Biophys Res Commun. 2005;337:521–5. doi: 10.1016/j.bbrc.2005.09.085. [DOI] [PubMed] [Google Scholar]
- 27.Schopfer FJ, et al. Fatty acid transduction of nitric oxide signaling. Nitrolinoleic acid is a hydrophobically stabilized nitric oxide donor. J Biol Chem. 2005;280:19289–97. doi: 10.1074/jbc.M414689200. [DOI] [PubMed] [Google Scholar]
- 28.Baker LM, et al. Nitro-fatty acid reaction with glutathione and cysteine. Kinetic analysis of thiol alkylation by a Michael addition reaction. J Biol Chem. 2007;282:31085–93. doi: 10.1074/jbc.M704085200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Erlemann KR, Rokach J, Powell WS. Oxidative stress stimulates the synthesis of the eosinophil chemoattractant 5-oxo-6,8,11,14-eicosatetraenoic acid by inflammatory cells. J Biol Chem. 2004;279:40376–84. doi: 10.1074/jbc.M401294200. [DOI] [PubMed] [Google Scholar]
- 30.Powell WS, Gravel S, Gravelle F. Formation of a 5-oxo metabolite of 5,8,11,14,17-eicosapentaenoic acid and its effects on human neutrophils and eosinophils. J Lipid Res. 1995;36:2590–8. [PubMed] [Google Scholar]
- 31.Ting-Beall HP, Needham D, Hochmuth RM. Volume and osmotic properties of human neutrophils. Blood. 1993;81:2774–80. [PubMed] [Google Scholar]
- 32.Itoh T, Murota I, Yoshikai K, Yamada S, Yamamoto K. Synthesis of docosahexaenoic acid derivatives designed as novel PPARgamma agonists and antidiabetic agents. Bioorg Med Chem. 2006;14:98–108. doi: 10.1016/j.bmc.2005.07.074. [DOI] [PubMed] [Google Scholar]
- 33.Yamamoto K, et al. Identification of putative metabolites of docosahexaenoic acid as potent PPARgamma agonists and antidiabetic agents. Bioorg Med Chem Lett. 2005;15:517–22. doi: 10.1016/j.bmcl.2004.11.053. [DOI] [PubMed] [Google Scholar]
- 34.Leslie AG. The integration of macromolecular diffraction data. Acta Crystallogr D Biol Crystallogr. 2006;62:48–57. doi: 10.1107/S0907444905039107. [DOI] [PubMed] [Google Scholar]
- 35.The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr. 1994;50:760–3. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 36.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–32. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 37.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–55. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.