

Abstract
The defective ribosomal product (DRiP) hypothesis of endogenous Ag processing posits that rapidly degraded forms of nascent proteins are a major source of peptide ligands for MHC class I molecules. Although there is broad experimental support for the DRiP hypothesis, careful kinetic analysis of the generation of defined peptide class I complexes has been limited to studies of recombinant vaccinia viruses expressing genes derived from other organisms. In this study, we show that insertion of the SIIN-FEKL peptide into the stalk of influenza A virus neuraminidase (NA) does not detectably modify NA folding, degradation, transport, or sp. act. when expressed in its natural context of influenza A virus infection. Using the 25-D1.16 mAb specific for Kb-SIINFEKL to precisely quantitate cell surface complexes by flow cytometry, we demonstrate that SIINFEKL is generated in complete lockstep with initiation and abrogation of NA biosynthesis in both L-Kb fibroblast cells and DC2.4 dendritic/monocyte cells. SIINFEKL presentation requires active proteasomes and TAP, consistent with its generation from a cytosolic DRiP pool. From the difference in the shutoff kinetics of Kb-SIINFEKL complex expression following protein synthesis versus proteasome inhibition, we estimate that the t1/2 of the biosynthetic source of NA peptide is ~5 min. These observations extend the relevance of the DRiP hypothesis to viral proteins generated in their natural context.
After viral infection, cells rapidly present peptides derived from newly synthesized viral proteins on MHC class I molecules for T cell activation (1). This rapidity of presentation, coupled with the relatively long half-life (t1/2) of most viral proteins, engendered the defective ribosomal product (DRiP) hypothesis of Ag presentation: a subset of newly synthesized proteins are defective in some manner and rapidly degraded intracellularly, yielding peptides for MHC class I Ag presentation to enable rapid immunosurveillance of viral and cellular translation products (2).
More than a decade after the birth of the DRiP hypothesis, there are only a few clues regarding the physical and biochemical properties of DRiPs that give rise to class I peptide ligands (3). Although large amounts of rapidly degraded polypeptides (RDPs) are relatively easy to detect (3–6), their relationship with DRiPs is uncertain (3, 7). The DRiP hypothesis is strongly supported, however, by functional studies that temporally link protein synthesis with peptide generation. Using TAP mobility in the endoplasmic reticulum (ER) membrane as a measure of peptide production, Qian et al. (8) and Reits et al. (9) showed that overall peptide generation from cellular and viral proteins is tightly linked to protein translation. Groettrup and colleagues (10) related peptide generation to levels of mRNA and translation and not amounts of previously synthesized intracellular Ag. Our laboratory has exploited the specificity of 25-D1.16 for Kb-SIINFEKL complexes to demonstrate that SIINFEKL generation is closely tied kinetically to the synthesis of OVA, the natural source of SIINFEKL, and a number of artificial chimeric proteins engineered to express SIINFEKL (3, 11). In all of these studies, we used recombinant vaccinia viruses (VVs) to express SIINFEKL-containing source Ags. It is possible that we grossly overestimated the contribution of DRiPs to Ag processing in these studies due to the use of VV to express non-VV genes. We recently showed that differences in viral translation mechanisms can greatly increase the fraction of DRiPs; expression of influenza A virus (IAV) nuclear protein by an Alphavirus vector resulted in the defective translation of >50% of nuclear protein recovered from cells (12). VV expression is known to modify the Ag processing pathway of some inserted viral gene products compared with their natural infection context (13, 14). Further, the fusion of multiple genes to create chimeric proteins can greatly decrease the fidelity of protein synthesis or protein folding (15).
Ideally, we could study the relationship between viral protein synthesis and class I complex using TCR-like detection reagents specific for natural viral peptides complexed with a class I molecules. Unfortunately, although we were able to generate phage Abs specific for five IAV peptides complexed with Kb or Db, none of these Abs is able to detect complexes synthesized by IAV-infected cells (16). After failing to find suitable reagents from other laboratories, we turned to recombinant A/Puerto Rico/8/34 (PR8) in which the SIINFEKL peptide is either inserted in the stalk of IAV neuraminidase (NA) or used to replace eight amino acids in the stalk (17, 18). These rIAVs are similar to wild type in their ability to replicate in cultured cells or mouse airways, suggesting that SIINFEKL has little effect on NA biogenesis. In this study, we use these rIAVs to rigorously test the DRiP hypothesis.
Materials and Methods
Cells, Abs, and viruses
Cultures were grown for the mouse cell lines L-Kb and DC2.4 in DMEM and Madin-Darby canine kidney (MDCK) cells in MEM, each containing 7.5% FCS in a 9% CO2 incubator. EL4 and RMA/s cells were cultured in RPMI 1640 medium containing 7.5% FCS and maintained in a 6% CO2 incubator. PR8 and rPR8 strains expressing SIINFEKL in NA (PR8 containing inserted SIINFEKL peptide in NA [insOVA] or PR8 with SIINFEKL replacing 8 amino acids in NA [repOVA]) were grown in 10-d embryonated chicken eggs, and infectious allantoic fluid was titered on MDCK cells. mAbs 25D1-1.16 (anti–Kb-SIINFEKL) and NA2-1C1 (anti-NA1) were labeled with Alexa Fluor 647 or Pacific blue, respectively, using protein labeling kits from Molecular Probes (Eugene, OR) following the manufacturer’s recommended protocols. Rabbit anti-NA serum was generated by multiple immunization with C-terminal NA peptide CSWPDGAELPFSIDK coupled to keyhole limpet hemocyanin (Spring Valley Laboratories, Woodbine, MD). The anti-hemagglutinin (HA) mAbs H17-L2 and Y8-10C2 have been described previously (19). Anti-p97 mouse mAb was from Fitzgerald (Concord, MA). Donkey anti-rabbit secondary Abs labeled with Alexa Fluor 680 were from Molecular Probes, and goat anti-mouse 800CW Abs were from Rockland (Gilbertsville, PA). FITC-anti-rabbit and Texas Red-anti-mouse secondary Abs were from The Jackson Laboratory (Bar Harbor, ME).
Viral infections
L-Kb and DC2.4 cells were resuspended to a concentration of 5 × 106 cells per milliliter in autoclavable minimal essential media, buffered to pH 6.6 whereas EL4 and RMA/s cells were resuspended to the same concentration in media buffered to pH 6.8. Ten to 20 of 50% tissue culture-infective doses IAV were added, and cells were infected for 30 min in a 37°C water bath with occasional agitation. Cells were washed and cultured at 106 cells per milliliter in tissue culture media for the indicated times. In some experiments, brefeldin A (BFA) at 10 μM, cycloheximide (CHX) at 25 μg/ml, or MG132 (10 or 25 μM) was added at the indicated times to the infected cells.
Ag presentation assays and Western blots
After viral infection, cells (~105) were harvested at the indicated times and kept at 4°C until the completion of the experiment. Cells then were labeled with Alexa Fluor 647-coupled 25D-1.16 monoclonal Abs and Pacific blue-coupled anti-NA mAb NA2-1C1. After a 30 min Ab labeling at 4°C, cells were washed in excess PBS and analyzed by flow cytometry using a BD LSR II flow cytometer (BD Biosciences, San Jose, CA). For Western blots, cells were harvested, washed in PBS, and resuspended to a concentration of 107 cells per milliliter in SDS-PAGE sample buffer containing MG132 and a protease inhibitor mixture. Cells were boiled immediately at 95°C for 20 min with occasional, vigorous vortexing until all of the chromatin had been denatured. An equal volume of water containing 50 mM DTT was added to the samples, which were boiled briefly and then resolved by 12% PAGE, blotted onto nitrocellulose, and exposed to rabbit polyclonal serum against NA, followed by anti-rabbit Alexa Fluor 680 secondary Abs. Monoclonal Abs against p97 were used as a loading control. Membranes were then analyzed using an Odyssey infrared imager (LI-COR Biosciences, Lincoln, NE).
Confocal microscopy
MDCK cells (105) were plated on eight-well coverslips (Nunc, Naperville, IL) overnight and then infected with IAV for 30 min. After infection, cells were washed and cultured in complete media. Six hours postinfection (p.i.), cells were fixed in 4% paraformaldehyde and permeabilized with 1% Triton X-100 for 2 min. Cells were washed with PBS and blocked with 5% normal donkey serum. Primary Abs were added to prepared cells for 1 h at room temperature, washed three times, and labeled with appropriate secondary Abs for an additional 30 min. Cells were counterstained with DAPI (5 μg/ml) and visualized on a TCS SP5 (DMI 6000) confocal microscope system (Leica Microsystems, Deerfield, IL).
Statistical analysis
Statistical analysis was performed using GraphPad Prism software (GraphPad, San Diego, CA). To determine the peak time of Kb-SIINFEKL expression from kinetic data, we preformed a regression analysis using the second-order polynomial equation to determine the parameters of the equation. The time to peak height was determined using the following equation:
where x is time and a and b are computed values from the regression analysis.
Results
Kinetics of NA and Kb-SIINFEKL complexes following IAV infection
We first examined the kinetics of cell surface expression of NA and Kb-SIINFEKL complexes after infecting the dendritic cell/macrophage-like cell line DC2.4 with PR8, insOVA, or repOVA (Fig. 1A). To increase the precision and linearity of detection, we simultaneously stained cells with directly conjugated NA2-1C1 (NA-specific) and 25-D1.16 (Kb-SIINFEKL-specific) mAbs. We initially detected NA 3 h p.i. (Fig. 1B), after which NA expression increased linearly with time. Remarkably, Kb-SIINFEKL expression kinetics were nearly identical to those of NA, with complexes first detected 3 h p.i. The specificities of staining for NA2-1C1 and 25-D1.16 were demonstrated, respectively, by their low and kinetically unchanging interactions with uninfected and PR8-infected cells. We extended these findings to fibroblast-like cells by infecting L-Kb cells with the same panel of viruses. We observed nearly identical cell surface expression kinetics for both NA and Kb-SIINFEKL complexes.
FIGURE 1.

Presentation of NA peptides occurs at the same rate as NA expression in IAV-infected cells. A, Schematic representation of IAV used. Transmembrane domain amino acids are underlined, and N-linked glycosylation site is bold. SIINFEKL determinant is shown in blue. Sequence replaced by SIINFEKL in rPR8 is shown in red. B, DC2.4 (top) or L-Kb (bottom) cells were infected with 10 50% tissue culture-infective doses of the indicated IAV for 30 min at 37°C at pH 6.6. Postinfection, cells were cultured and subsequently harvested at the indicated time (x-axis) whereupon cells were stained with Abs to NA (left column) or Kb-SIINFEKL (right column). The MFI of the population is reported on the y-axis.
Characterization of NA biosynthesis
Although NA was initially detected at the cell surface 3 h p.i., there could be a considerable lag between the initiation of NA synthesis and its appearance at the cell surface, due to delays in transit through the secretory apparatus (20). To accurately quantitate NA expression, we generated rabbit polyclonal Abs specific for the C-terminal 15 aa. Rabbit antisera proved highly specific for NA in immunoblotting without affinity purification (Fig. 2). Kinetic analysis of NA expression by infected DC2.4 cells revealed a robust amount of NA derived from the input virus, followed by the disappearance of NA due to the release of virus or endosomal proteolysis and the detection of nascent NA beginning 2 h p.i. Treating cells with the protein synthesis inhibitor CHX at the time of infection confirmed that the initial NA signal is due to the input virus and that NA is nearly completely lost by 2 h p.i. Wild type PR8, insOVA, and repOVA yielded generally similar results.
FIGURE 2.
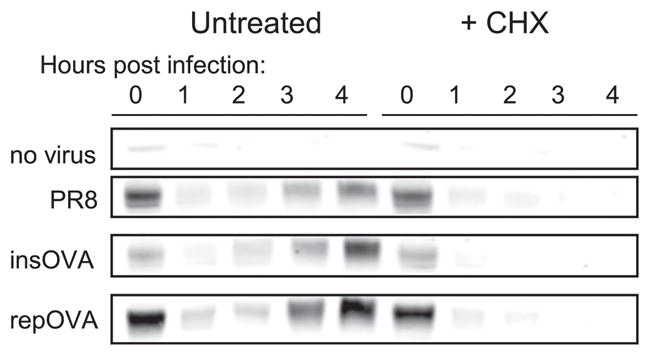
Functional NA protein synthesis begins 2–3 h p.i. DC2.4 cells were infected as in Fig. 1, cells were harvested at the indicated, times and total cell lysates were prepared for Western blot analyses using rabbit polyclonal Abs raised against the C terminus of the NA protein. Cells were either left untreated or incubated with CHX immediately p.i. The prominent band at ~50 kDa corresponding to the anticipated size of the NA monomers is shown.
We further examined NA biogenesis in the three viruses via immunofluorescence of fixed and permeabilized MDCK cells 6 h p.i. Confocal microscopy revealed that rabbit polycolonal Abs detected NA throughout the secretory pathway and cell surface (Fig. 3A), demonstrating that the NA C terminus is available for interaction with Abs as the NA progresses from the ER to the cell surface (note that NA is anchored to the membrane by its N-terminal leader sequence). The polyclonal Ab anti-NA staining pattern was confirmed by costaining cells with either H17-L2 mAb, which, due to its binding to HA only upon trimerization, stains HA in the Golgi complex (GC) and post-GC compartments (Fig. 3B), or H28-E23 mAb, which localizes HA throughout the secretory pathway (note nuclear rim staining characteristic of ER localization) (19). The NA2-1C1 epitope is dependent on NA oligomerization and, similar to PR8 HA trimer-specific mAbs, only detects NA after export from the ER (Fig. 3A, 3B). The staining patterns of cells infected with PR8, insOVA, and repOVA were indistinguishable, consistent with the conclusion that the insertion of SIINFEKL into the stalk of IAV NA does not perturb NA biogenesis.
FIGURE 3.
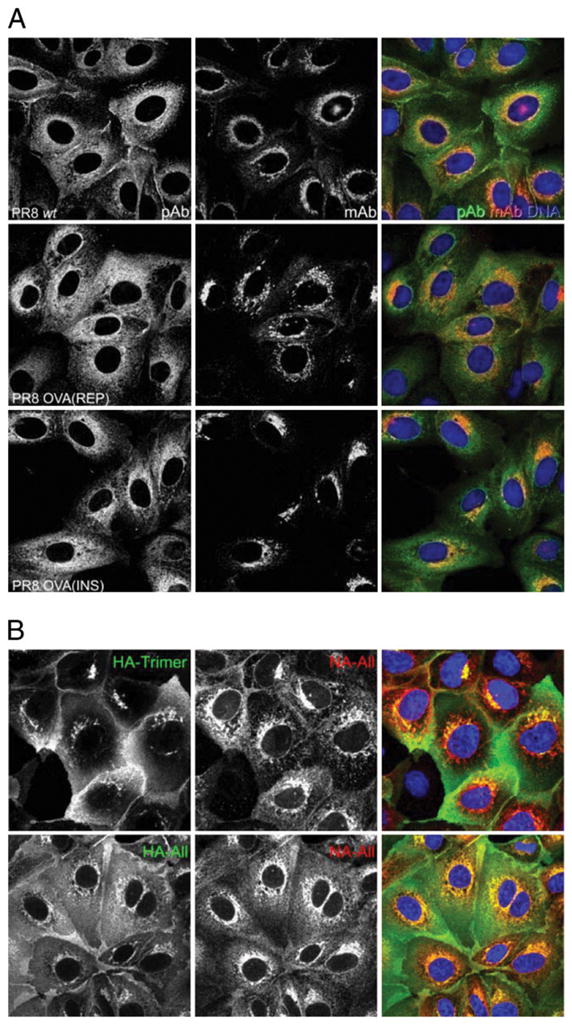
Intracellular NA trafficking is unaffected by the SIINFEKL epitope. A, MDCK cells were infected with the indicated PR8 viruses for 30 min at 37°C and then cultured for 6 h. Cells were fixed and permeabilized with 0.5% Triton X-100, then stained with rabbit polyclonal NA Abs (green) or a mouse mAb (red) that recognizes the mature, dimerized form of NA. Nuclear staining is shown in blue. B, The GC and post-GC compartments of infected cells were identified by staining with mouse mAb H17-L2 against the IAV HA trimer (green, top). The entire secretory pathway was identified by staining cells with the HA-specific mAb Y8-10C2 (HA-All, green, bottom). Corresponding NA staining with rabbit polyclonal Abs (NA-All) is shown in red.
To further investigate this point, we examined the trafficking kinetics of NA from the ER to the cell surface by treating cells with either BFA or CHX 3.5 h p.i. and monitoring cell surface expression by flow cytometry with NA2-1C1 (Fig. 4A). BFA treatment immediately abrogated the time-dependent increase in NA expression, demonstrating that each virus expresses a negligible post-GC pool of NA that can traffic to the cell surface following BFA-induced retrograde transport of GC elements and abrogation of ER export (21). After CHX treatment, there was a 1 h lag before NA expression reached a plateau. This lag reflects the time required for all of the NA to exit the ER and reach the plasma membrane and is consistent with the 1 h lag between detection of NA biosynthesis and NA cell surface expression. Importantly, each of the three viruses demonstrated highly similar kinetics. Because misfolded protein typically exhibits retarded trafficking in the ER, these data again support the neutral effect of SIINFEKL insertion on NA biogenesis.
FIGURE 4.

NA stability of enzymatic activity is unaffected by the SIINFEKL epitope. A, DC2.4 cells were infected with the indicated virus (black line) or mock-infected (open circle and dotted line) and cultured. Either BFA (blue line) or CHX (orange line) was added to the infected cells 3.5 h p.i., and samples were collected for FACS analysis at the indicated times. The y-axis is the MFI of cells stained with an anti-NA Ab. B, DC2.4 cells were infected with the indicated IAV and cultured for 4 h, at which time CHX and BFA were added, and cells were collected at the indicated times for Western blot analyses for NA and p97 (loading control). NA levels were normalized to p97 and are depicted in the graphs. C, Infected cells were used as the source of enzyme for fluorometric analyses of NA activity. The observed Vmax (left) and the Vmax normalized to the cell surface expression of NA (right) of the reaction are reported on the y-axis. D, Western blot analyses of NA protein from total cell lysates of IAV-infected cells with (+) or without (−) MG132. The prominent band for monomeric NA is at ~50 kDa, and residual NA dimers are also detected (~95kDa).
What is the effect of SIINFEKL insertion on the metabolic stability of NA? Nascent NA enters the secretory pathway and is released from the cell in budding virions, complicating traditional pulse-chase experiments. To circumvent this problem, 3 h p.i. we treated infected cells with CHX (to block further NA synthesis) and BFA (to prevent export of NA from the early secretory pathway) and harvested cells every 30 min for 1.5 h for immunoblot-based NA quantitation. SIINFEKL insertion had no discernible destabilizing effect on NA (Fig. 4B).
We next examined the effect of SIINFEKL insertion on NA enzymatic activity. We measured NA activity using a fluorogenic substrate at 5 h p.i. with the three IAVs. The specific activities for the three viruses (calculated by dividing Vmax values by the corresponding mean fluorescence intensity (MFI) of NA expression determined by FACS analysis) are essentially identical (Fig. 4C), demonstrating that SIINFEKL insertion does not affect the NA enzymatic activity.
Finally, we used the ability of rabbit anti-NA polyclonal Abs to detect denatured NA via immunoblotting to determine whether NA insertion modifies the generation of NA RDPs (Fig. 4D). To detect RDPs degraded by proteasomes, we treated cells for 3 h with the proteasome inhibitor MG132 starting at 2 h p.i. when NA biosynthesis begins. The only effect of MG132 was to reduce the amount of NA detected (confirmed by flow cytometry as described below). Our failure to detect proteasome-dependent RDPs for any of the three viruses provides further evidence that SIINFEKL insertion does not increase the DRiP rate for NA.
Taken together, these data strongly support the conclusion that SIINFEKL insertion has minimal effects on the generation of NA DRiPs and therefore that repOVA and insOVA expressed in the context of IAV infection are accurate proxies for the behavior of natural IAV gene products.
Kinetic analysis of Kb-SIINFEKL generation upon abrogation of protein synthesis or proteasome-mediated degradation
We next characterized Kb-SIINFEKL generation from NA. Kb-SIINFEKL expression is TAP-dependent, based on background levels of staining with 25-D1.16 following rPR8 infection of RMA/S cells, which lack functional TAP due to the introduction of a stop codon in the TAP2 gene [Fig. 5; note that the parental RMA cells are identical to EL4 cells (22)]. Kb-SIINFEKL presentation is also proteasome-dependent (Fig. 6A), because adding the proteasome inhibitor MG132 to DC2.4 cells 2 h p.i. completely abrogates 25-D1.16 staining, in contrast to its moderate effect on NA expression, which is reduced 2.5-fold as measured by either flow cytometry or immunoblotting (Figs. 4D, 6A). Similarly, treating cells with CHX 1.5 h p.i. with rPR8 completely inhibited Kb-SIINFEKL expression, demonstrating that peptides are generated exclusively from biosynthesized NA and not NA on input virions (Fig. 6B).
FIGURE 5.
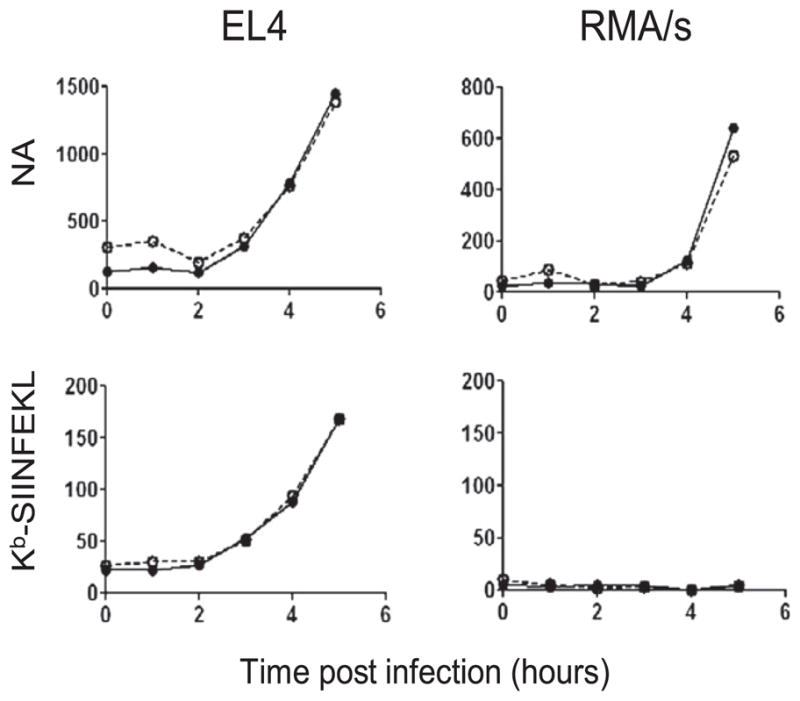
Functional TAP is necessary for the presentation of NA peptides. EL4 (left) or TAP-deficient RMA/s (right) were infected with the indicated PR8 containing insOVA (filled circle) or repOVA (open circle) and harvested at the indicated times for analysis of NA expression (top) and Kb-SIINFEKL (bottom). The background Ab staining of uninfected cells was subtracted from infected cells to generate the MFIs reported (y-axis).
FIGURE 6.

NA presentation requires active translation and proteasome activity. A, DC2.4 cells were infected with the indicated viruses and cultured for 2 h before the addition of 10 μM MG132 or DMSO and then cultured for an additional 3 h. Cells were analyzed by FACS for either cell surface NA (top) or Kb-SIINFEKL (bottom). B, DC2.4 cells were infected with insOVA (left) or repOVA (right) and cultured for 1.5 h before the addition of CHX. Culture of infected cells continued, and Kb-SIINFEKL expression was determined by FACS.
Finally, we made a detailed study of Kb-SIINFEKL kinetics by harvesting L-Kb cells at 10 min intervals to finely measure differences in Kb-SIINFEKL complex expression following the addition of BFA, CHX, or MG132 at 3.5 h p.i. (Fig. 7A). After the addition of the inhibitor, Kb-SIINFEKL surface expression increased to a plateau and then declined as the turnover in surface complexes outnumbered the number of complexes delivered by the secretory pathway. We used regression analysis based on the second-order polynomial equation to determine the time required to achieve maximal Kb-SIINFEKL expression following inhibitor addition.
FIGURE 7.

Rapid shutoff of Ag presentation after the addition of MHC class I Ag presentation inhibitors. A, L-Kb cells were infected with insOVA or repOVA and cultured for 3.5 h before the addition of 10 μM BFA, 25 μg/ml CHX, or 25 μM MG132. After the addition of the inhibitors (arrow), samples were taken every 10 min for FACS analysis of Kb -SIINFEKL expression. The time to peak Kb-SIIN-FEKL expression after inhibitor addition was determined and averaged for four experiments. B, Table with the SE indicated.
The results of four experiments with both insOVA and repOVA viruses are averaged in Fig. 7B. As expected, BFA shut off Ag presentation first, followed by MG132 and lastly CHX. From the difference in the times needed to achieve maximal expression following CHX and MG132 addition, we can estimate that ~12 min is required to completely degrade the DRiPs that give rise to antigenic peptides. This implies a t1/2 of <4 min for bona fide antigenic DRiPs (three half-lives are needed to achieve ~90% reduction in substrate, which is probably our detection limit). Because peptides are degraded with a t1/2 on the order of 10 s (23), the difference of 9 min between maximal Kb-SIINFEKL expression following MG132 and BFA implies that Kb-SIINFEKL complexes are transported from the ER to the BFA-resistant portion of the secretory pathway extremely rapidly (t1/2 of ~2 min, if the process follows first-order kinetics).
Taken together, these data demonstrate that in both professional and nonprofessional APCs rapidly degraded DRiPs provide virtually all of the antigenic peptides generated from a minimally manipulated viral protein expressed in the context of its natural infection.
Discussion
NA is a plasma membrane protein with no known mechanism of returning to the cytosol once exported from the early secretory pathway, and it is perhaps expected that antigenic peptides derive from DRiPs. Still, because ~50% of cellular translation products are imported into the ER (24), this pool represents a major source of antigenic peptides, and indeed, ER-targeted proteins are well represented among defined target Ags for antiviral CD8+ T cells. The role of DRiPs in generating peptides from ER-targeted proteins has been questioned in the case of tyrosinase (25), so it is important to firmly establish the route of peptide generation from this abundant pool of Ags.
We note that beyond demonstrating the proteasome and TAP dependence indicative of cytosolic processing our data do not address the mechanism of peptide generation from NA DRiPs, particularly with regard to the contribution of DRiPs that fail to be exported into the ER versus proteins exported from the ER via the ER-associated degradation pathway. Because there is evidence for the participation of both pathways in the processing of ER targeting proteins (26–29), it is entirely possible that both pathways contribute to NA Ag processing. Future studies will be required to address this important issue. Our findings do not rule out the generation of a minor level of peptides from functional NA that is somehow exported to the cytosol.
Our findings extend a number of previous studies demonstrating the close kinetic link between viral Ag synthesis and peptide generation. Because speed is of the essence in destroying virus-infected cells before they can release their progeny, rapid presentation clearly has evolutionary value to the host in handing virus infections. Ultimately, however, the rapidity of recognition may be a useful by-product of a more important driving force in the evolution of the Ag processing machinery: immunosurveillance of tumor cells. To enable detection of aberrant gene products independent of their levels of expression, evolution may have homed in on translation. The immunoribosome hypothesis proposes that cells possess a mechanism for selecting mRNAs on a Noah’s Ark type basis (i.e., quality rather than quantity) to translate proteins in a dedicated manner as a source of antigenic peptides (30, 31). Although speed is unlikely to be important in recognizing tumor cells, rapid recognition of newly synthesized genes will result from nearly every translation-based mechanism of peptide generation. Although DRiPs are clearly a major source of antigenic peptides, it is important to recognize that peptides are also generated from natural protein turnover.
Having clearly established the central role of DRiPs in Ag processing, it is of central importance to develop strategies to identify the cellular gene products involved in the DRiP pathway. It would not be surprising if there were considerable overlap with the cross-presentation pathway, which in rapidly generating peptides from minute quantities of internalized Ags in the face of overwhelming amounts of cellular proteins bears striking similarities to the DRiP pathway.
Acknowledgments
We thank Glennys Reynoso for providing outstanding technical assistance.
This work was supported by the Division of Intramural Research of the National Institute of Allergy and Infectious Diseases of the National Institutes of Health.
Abbreviations in this paper
- BFA
brefeldin A
- CHX
cycloheximide
- DRiP
defective ribosomal product
- ER
endoplasmic reticulum
- GC
Golgi complex
- HA
hemagglutinin
- IAV
influenza A virus
- insOVA
PR8 containing inserted SIINFEKL peptide in NA
- MDCK
Madin-Darby canine kidney
- MFI
mean fluorescence intensity
- NA
neuraminidase
- p.i
postinfection
- PR8
A/Puerto Rico/8/34
- RDP
rapidly degraded polypeptide
- repOVA
PR8 with SIINFEKL replacing 8 amino acids in NA
- VV
vaccinia virus
Footnotes
Disclosures
The authors have no financial conflicts of interest.
References
- 1.Esquivel F, Yewdell J, Bennink J. RMA/S cells present endogenously synthesized cytosolic proteins to class I-restricted cytotoxic T lymphocytes. J Exp Med. 1992;175:163–168. doi: 10.1084/jem.175.1.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yewdell JW, Antón LC, Bennink JR. Defective ribosomal products (DRiPs): a major source of antigenic peptides for MHC class I molecules? J Immunol. 1996;157:1823–1826. [PubMed] [Google Scholar]
- 3.Qian SB, Princiotta MF, Bennink JR, Yewdell JW. Characterization of rapidly degraded polypeptides in mammalian cells reveals a novel layer of nascent protein quality control. J Biol Chem. 2006;281:392–400. doi: 10.1074/jbc.M509126200. [DOI] [PubMed] [Google Scholar]
- 4.Wheatley DN, Giddings MR, Inglis MS. Kinetics of degradation of “short-” and “long-lived” proteins in cultured mammalian cells. Cell Biol Int Rep. 1980;4:1081–1090. doi: 10.1016/0309-1651(80)90045-4. [DOI] [PubMed] [Google Scholar]
- 5.Schubert U, Antón LC, Gibbs J, Norbury CC, Yewdell JW, Bennink JR. Rapid degradation of a large fraction of newly synthesized proteins by proteasomes. Nature. 2000;404:770–774. doi: 10.1038/35008096. [DOI] [PubMed] [Google Scholar]
- 6.Vabulas RM, Hartl FU. Protein synthesis upon acute nutrient restriction relies on proteasome function. Science. 2005;310:1960–1963. doi: 10.1126/science.1121925. [DOI] [PubMed] [Google Scholar]
- 7.Yewdell JW. Plumbing the sources of endogenous MHC class I peptide ligands. Curr Opin Immunol. 2007;19:79–86. doi: 10.1016/j.coi.2006.11.010. [DOI] [PubMed] [Google Scholar]
- 8.Qian SB, Reits E, Neefjes J, Deslich JM, Bennink JR, Yewdell JW. Tight linkage between translation and MHC class I peptide ligand generation implies specialized antigen processing for defective ribosomal products. J Immunol. 2006;177:227–233. doi: 10.4049/jimmunol.177.1.227. [DOI] [PubMed] [Google Scholar]
- 9.Reits EA, Vos JC, Grommé M, Neefjes J. The major substrates for TAP in vivo are derived from newly synthesized proteins. Nature. 2000;404:774–778. doi: 10.1038/35008103. [DOI] [PubMed] [Google Scholar]
- 10.Khan S, de Giuli R, Schmidtke G, Bruns M, Buchmeier M, van den Broek M, Groettrup M. Cutting edge: neosynthesis is required for the presentation of a T cell epitope from a long-lived viral protein. J Immunol. 2001;167:4801–4804. doi: 10.4049/jimmunol.167.9.4801. [DOI] [PubMed] [Google Scholar]
- 11.Princiotta MF, Finzi D, Qian SB, Gibbs J, Schuchmann S, Buttgereit F, Bennink JR, Yewdell JW. Quantitating protein synthesis, degradation, and endogenous antigen processing. Immunity. 2003;18:343–354. doi: 10.1016/s1074-7613(03)00051-7. [DOI] [PubMed] [Google Scholar]
- 12.Berglund P, Finzi D, Bennink JR, Yewdell JW. Viral alteration of cellular translational machinery increases defective ribosomal products. J Virol. 2007;81:7220–7229. doi: 10.1128/JVI.00137-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Neumeister C, Nanan R, Cornu TI, Lüder CGK, ter Meulen V, Naim H, Niewiesk S. Measles virus and canine distemper virus target proteins into a TAP-independent MHC class I-restricted antigen-processing pathway. J Gen Virol. 2001;82:441–447. doi: 10.1099/0022-1317-82-2-441. [DOI] [PubMed] [Google Scholar]
- 14.Johnstone C, Guil S, Rico MA, García-Barreno B, López D, Melero JA, Del Val M. Relevance of viral context and diversity of antigen-processing routes for respiratory syncytial virus cytotoxic T-lymphocyte epitopes. J Gen Virol. 2008;89:2194–2203. doi: 10.1099/vir.0.2008/002485-0. [DOI] [PubMed] [Google Scholar]
- 15.Yen HC, Xu Q, Chou DM, Zhao Z, Elledge SJ. Global protein stability profiling in mammalian cells. Science. 2008;322:918–923. doi: 10.1126/science.1160489. [DOI] [PubMed] [Google Scholar]
- 16.Lev A, Takeda K, Zanker D, Maynard JC, Dimberu P, Waffarn E, Gibbs J, Netzer N, Princiotta MF, Neckers L, et al. The exception that reinforces the rule: crosspriming by cytosolic peptides that escape degradation. Immunity. 2008;28:787–798. doi: 10.1016/j.immuni.2008.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jenkins MR, Webby R, Doherty PC, Turner SJ. Addition of a prominent epitope affects influenza A virus-specific CD8+ T cell immunodominance hierarchies when antigen is limiting. J Immunol. 2006;177:2917–2925. doi: 10.4049/jimmunol.177.5.2917. [DOI] [PubMed] [Google Scholar]
- 18.Mintern JD, Bedoui S, Davey GM, Moffat JM, Doherty PC, Turner SJ. Transience of MHC class I-restricted antigen presentation after influenza A virus infection. Proc Natl Acad Sci USA. 2009;106:6724–6729. doi: 10.1073/pnas.0901128106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yewdell JW, Yellen A, Bächi T. Monoclonal antibodies localize events in the folding, assembly, and intracellular transport of the influenza virus hemagglutinin glycoprotein. Cell. 1988;52:843–852. doi: 10.1016/0092-8674(88)90426-6. [DOI] [PubMed] [Google Scholar]
- 20.Hausmann J, Kretzschmar E, Garten W, Klenk HD. Biosynthesis, intracellular transport and enzymatic activity of an avian influenza A virus neuraminidase: role of unpaired cysteines and individual oligosaccharides. J Gen Virol. 1997;78:3233–3245. doi: 10.1099/0022-1317-78-12-3233. [DOI] [PubMed] [Google Scholar]
- 21.Doms RW, Russ G, Yewdell JW. Brefeldin A redistributes resident and itinerant Golgi proteins to the endoplasmic reticulum. J Cell Biol. 1989;109:61–72. doi: 10.1083/jcb.109.1.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.van Hall T, van Bergen J, van Veelen PA, Kraakman M, Heukamp LC, Koning F, Melief CJ, Ossendorp F, Offringa R. Identification of a novel tumor-specific CTL epitope presented by RMA, EL-4, and MBL-2 lymphomas reveals their common origin. J Immunol. 2000;165:869–877. doi: 10.4049/jimmunol.165.2.869. [DOI] [PubMed] [Google Scholar]
- 23.Reits E, Griekspoor A, Neijssen J, Groothuis T, Jalink K, van Veelen P, Janssen H, Calafat J, Drijfhout JW, Neefjes J. Peptide diffusion, protection, and degradation in nuclear and cytoplasmic compartments before antigen presentation by MHC class I. Immunity. 2003;18:97–108. doi: 10.1016/s1074-7613(02)00511-3. [DOI] [PubMed] [Google Scholar]
- 24.Shaffer AL, Shapiro-Shelef M, Iwakoshi NN, Lee AH, Qian SB, Zhao H, Yu X, Yang L, Tan BK, Rosenwald A, et al. XBP1, downstream of Blimp-1, expands the secretory apparatus and other organelles, and increases protein synthesis in plasma cell differentiation. Immunity. 2004;21:81–93. doi: 10.1016/j.immuni.2004.06.010. [DOI] [PubMed] [Google Scholar]
- 25.Ostankovitch M, Robila V, Engelhard VH. Regulated folding of tyrosinase in the endoplasmic reticulum demonstrates that misfolded full-length proteins are efficient substrates for class I processing and presentation. J Immunol. 2005;174:2544–2551. doi: 10.4049/jimmunol.174.5.2544. [DOI] [PubMed] [Google Scholar]
- 26.Ostankovitch M, Altrich-Vanlith M, Robila V, Engelhard VH. N-glycosylation enhances presentation of a MHC class I-restricted epitope from tyrosinase. J Immunol. 2009;182:4830–4835. doi: 10.4049/jimmunol.0802902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Skipper JCA, Hendrickson RC, Gulden PH, Brichard V, Van Pel A, Chen Y, Shabanowitz J, Wolfel T, Slingluff CL, Jr, Boon T, et al. An HLA-A2-restricted tyrosinase antigen on melanoma cells results from post-translational modification and suggests a novel pathway for processing of membrane proteins. J Exp Med. 1996;183:527–534. doi: 10.1084/jem.183.2.527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bacik I, Snyder HL, Antón LC, Russ G, Chen W, Bennink JR, Urge L, Otvos L, Dudkowska B, Eisenlohr L, Yewdell JW. Introduction of a glycosylation site into a secreted protein provides evidence for an alternative antigen processing pathway: transport of precursors of major histocompatibility complex class I-restricted peptides from the endoplasmic reticulum to the cytosol. J Exp Med. 1997;186:479–487. doi: 10.1084/jem.186.4.479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schlosser E, Otero C, Wuensch C, Kessler B, Edelmann M, Brunisholz R, Drexler I, Legler DF, Groettrup M. A novel cytosolic class I antigen-processing pathway for endoplasmic-reticulum-targeted proteins. EMBO Rep. 2007;8:945–951. doi: 10.1038/sj.embor.7401065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yewdell J. To DRiP or not to DRiP: generating peptide ligands for MHC class I molecules from biosynthesized proteins. Mol Immunol. 2002;39:139–146. doi: 10.1016/s0161-5890(02)00097-4. [DOI] [PubMed] [Google Scholar]
- 31.Yewdell JW, Nicchitta CV. The DRiP hypothesis decennial: support, controversy, refinement and extension. Trends Immunol. 2006;27:368–373. doi: 10.1016/j.it.2006.06.008. [DOI] [PubMed] [Google Scholar]