

Abstract
BACKGROUND
Lipid-cell tumors are rare, functioning ovarian neoplasms. They have not been reported in women with von Hippel-Lindau syndrome, an autosomal-dominant tumor-suppressor gene mutation that is associated with renal cell carcinoma, and other vascular tumors.
CASES
Two women with von Hippel-Lindau syndrome and kidney tumors were evaluated for secondary amenorrhea, hirsutism, and complex adnexal masses seen on computed tomography. The first patient had known renal cancer and bilateral adnexal masses, one with central necrosis. Because metastatic renal cell cancer could not be excluded on frozen section, bilateral salpingo-oophorectomy was performed. The second patient underwent right salpingo-oophorectomy after human chorionic gonadotropin testing confirmed that the ovarian tumor produced testosterone. Final pathology in both cases revealed testosterone-secreting lipid cell tumors.
CONCLUSION
Lipid cell ovarian tumors should be considered in women with von Hippel-Lindau presenting with adnexal mass, amenorrhea, and hirsuitism.
Lipid cell tumors are rare steroid-producing ovarian neoplasms and are usually unilateral with bilateral involvement found in 6% of cases.1 These tumors generally secrete androgens, and virilization is found in 77% of cases.2 Estrogen, progesterone, and adrenocortical steroids are less common secretory products of lipid cell tumors.3
von Hippel- Lindau syndrome is a rare disorder caused by an autosomal dominant tumor suppressor gene mutation.4 von Hippel- Lindau syndrome is associated with various vascular tumors including retinal angiomas, cerebellar hemangioblastomas, renal cell cancer, pheochromocytomas, and other vascular tumors. We present two cases of young women with von Hippel-Lindau syndrome who presented with virilization, complex ovarian masses, and renal cell carcinoma.
CASE 1
A 28-year-old African-American woman, gravida 0 with von Hippel-Lindau syndrome and a biopsy-proven 2.7 cm renal clear cell carcinoma was referred to the National Institutes of Health (NIH) for participation in a von Hippel-Lindau study.
Her symptoms of masculinization began 10 years ago with clitoromegaly and increasing facial hair growth that required daily shaving. In 2004, biochemical evaluation for her symptoms revealed an elevated serum total testosterone of 189 ng/dL (normal range 20–76 ng/dL), free testosterone, 52 pg/mL (range 1–21 pg/mL), and 17-OH progesterone level, 382 ng/dl (range 30–290 ng/dL). She was diagnosed with congenital adrenal hyperplasia; however, no specific treatment was ever instituted. She was started on birth control pills to treat the hirsutism, but no documentation was available regarding whether this treatment was effective. Her menstrual cycles remained regular after stopping the oral contraceptives, but menses completely ceased 1 year before presentation. Gynecology evaluation before coming to NIH revealed bilateral adnexal masses, and the possibility of ovarian malignancy was raised with the patient.
A metastatic workup performed at NIH included computed tomography (CT) of the chest, abdomen, and pelvis, which revealed multiple cystic pancreatic lesions and two enhancing masses in the body of the pancreas that were suggestive of neuroendocrine tumors. Bilateral adnexal masses, one with central necrosis were also observed. An ultrasound examination of the kidneys showed a lower right kidney mass. Computed tomography scan of the temporal bones and magnetic resonance imaging of the brain were both normal. Magnetic resonance imaging of the cervical, thoracic, and lumbar spine showed no evidence of hemangioblastomas. On transvaginal ultrasonography, a multi-lobed right adnexal mass with areas of necrosis measured 5.6×4.5 cm, but the right ovary could not be discerned from this mass and a large, solid left ovarian mass measured 3×2.6 cm within a left ovary measuring 4×2×2 cm. There was also a moderate amount of free fluid in the pelvis.
The patient was subsequently referred to the Gynecology Consultation Service at NIH for evaluation of secondary amenorrhea, hirsutism, mildly elevated testosterone, and bilateral complex adnexal masses in the context of known kidney cancer. She reported a 4.5-kg (10-lb) weight gain and denied an increase in muscle mass, hoarseness of voice, acne, male-pattern hair loss, temperature intolerance, anxiety, tremors, sweating, palpitations, or headaches.
On physical examination, her blood pressure was 144/78 mm Hg, heart rate was 77 beats per minute, and respiratory rate was 19 breaths per minute. She had a body mass index of 37.9 kg/m2. She had a round face, coarse terminal hair growth on the upper lip, chin, and lower cheeks and marked facial hirsutism. Her Ferriman-Gallwey score was 10. Pelvic examination revealed clitoromegaly, and bilateral adnexal masses and mobile pelvic organs were appreciated on bimanual examination.
Repeat endocrine studies revealed elevated androgens (androstenedione of 794 ng/dL and free testosterone of 4.1 pg/mL), a low sex hormone–binding globulin level of 17 mmol/L (range 18–114 mmol/L), and an elevated 17 OH-progesterone level of 767 ng/dl. Dehydroepiandrosterone sulfate, follicle-stimulating hormone, luteinizing hormone, estradiol, thyroid-stimulating hormone, and prolactin were unremarkable. Serum β-hCG, carcinoembryonic antigen, alpha-fetoprotein, and CA 125 tumor markers assessed for ovarian tumors were all within normal ranges. An adrenocorticotropic hormone (ACTH) stimulation test (cosyntropin 250 micrograms given intravenously at 7:30 AM) was performed with measurement of 17 OH-progesterone, cortisol, and androstenedione to confirm or rule out the diagnosis of congenital adrenal hyperplasia and to help in differentiating where the testosterone was coming from (adrenal compared with ovary). The patient demonstrated a lack of stimulation of 17 OH-progesterone on ACTH stimulation testing and a normal cortisol response. The patient’s clinical and biochemical presentation suggested that the cause of the androgen excess was most likely related to ovarian tumors. She was scheduled to undergo an exploratory laparotomy to remove the adnexal masses.
At the time of surgery, the patient had a 2.7-cm right renal mass that was biopsy proven to be consistent with renal clear cell carcinoma. Other than the complex adnexal masses, one of which had central necrosis, there was no other clinical evidence of metastatic renal disease.
At exploratory laparotomy, pelvic washings were obtained. Only the ovarian tumors, but no other intraabdominal masses or evidence of tumor metastasis, were noted. A left ovarian cystectomy was performed to remove a 2.3×1.5×2 cm lobulated, white, yellow mass with dark discoloration that was sent for a frozen section. As the right ovary was totally replaced by a solid, rubbery, yellow mass, a right salpingo-oophorectomy was performed. Intraoperatively, frozen section could not exclude the possibility of metastatic renal cell cancer, although it was possible this was a testosterone-releasing ovarian tumor. Surgical consultation with urologic oncology confirmed that there was no other evidence of metastatic renal cancer in this patient, except for the possibility of ovarian metastases. A left salpingo-oophorectomy was subsequently performed with the intent of removing all potential metastatic renal cancer to avoid leaving any metastatic disease. The final pathology revealed bilateral lipid cell tumors with small lipid tumor rests throughout areas of the normal appearing left ovary that stained positive for inhibin and calretinin and negative for CD10.
Postoperatively, the patient’s total and free testosterone levels dropped, and over a period of 8 weeks, she had lost 4.5 kg (10 lb) and had noticeably less facial hirsutism. Anxious to have her renal cancer treated, the patient underwent cryosurgery for von Hippel-Lindau–related renal cancer at another institution.
CASE 2
A 26-year-old white female with von Hippel-Lindau syndrome, bilateral renal masses, and a history of cervical spine hemangioblastoma was referred to NIH. She was found to have a 5-cm right ovarian mass seen on CT during a workup for von Hippel-Lindau. Multiple renal masses suggestive of renal cell carcinoma were also observed on CT scan. She was referred to the Gynecology Consultative Service for evaluation of the right adnexal mass, hirsutism, and amenorrhea, as well as a breast mass.
The patient reports being born with extra skin in the genital area, which was surgically corrected, but she does not have details of that surgery. She was diagnosed with polycystic ovarian syndrome at age 11. At puberty, she had axillary and pubic hair and normal female breast development and began having menses, but she became amenorrheic at age 14. She took an oral contraceptive for a short period of time but did not have any further menses. She developed significant hirsutism at age 15 and denies current or past use of exogenous androgens.
On physical examination, the patient’s blood pressure was 140/85 mm Hg, heart rate was 99 beats per minute, and respiratory rate was 20 breaths per minute. She had a body mass index of 40.4 kg/m2. She had male pattern arm, leg, and chest hair, male escutcheon, and facial hair with a Ferriman-Gallwey score of 18. The breast mass was mobile and cystic. Pelvic examination revealed normal female genitalia with no evidence of surgical scars. The clitoris was normal size, and adnexa were free and mobile without pain.
Mammography revealed no evidence of malignancy. Transvaginal ultrasonography revealed an enlarged right ovary with normal ovarian tissue with a few small follicles. Most of the ovary was replaced by a 4–5-cm echo dense mass. The left ovary had multiple follicles and was of normal size.
Endocrine studies revealed a serum total testosterone of 139 ng/dL and free testosterone of 4.2 ng/dL, and androstenedione level was 621 ng/dL (normal range 40–150 ng/dL). Seventeen-OH progesterone level was 393 ng/dL. An ACTH stimulation test was performed and excluded congenital adrenal hyperplasia. An human chorionic gonadotropin (hCG) stimulation test was performed to distinguish metastatic disease from an ovarian androgen-secreting tumor, as metastatic disease should not respond to hCG stimulation. Ovarian vein sampling 30 minutes after 250 units of hCG was administered intravenously confirmed that elevated levels of testosterone were coming from the right ovary (total testosterone 710 right, 214 left, 214 peripheral ng/dL; free testosterone 26.8 right, 7 left, 7.1 peripheral ng/dL). Baseline values for total testosterone were 636 right, 329 left, 170 peripheral ng/dL; free testosterone was 21.6 right, 10.4 left, 5 peripheral ng/dL.
The patient underwent laparoscopy, which revealed a significantly enlarged right ovary consistent with prior imaging studies. The left ovary, fallopian tubes, and uterus appeared normal. A right salpingo-oophorectomy was performed. The cut surface revealed a yellow nodule measuring 4×3.5 cm with a homogenous surface (Fig. 1). Immunohistochemical staining for inhibin and calretinin were diffusely and strongly positive. The final pathology revealed a lipid cell tumor of the ovary.
Fig. 1.
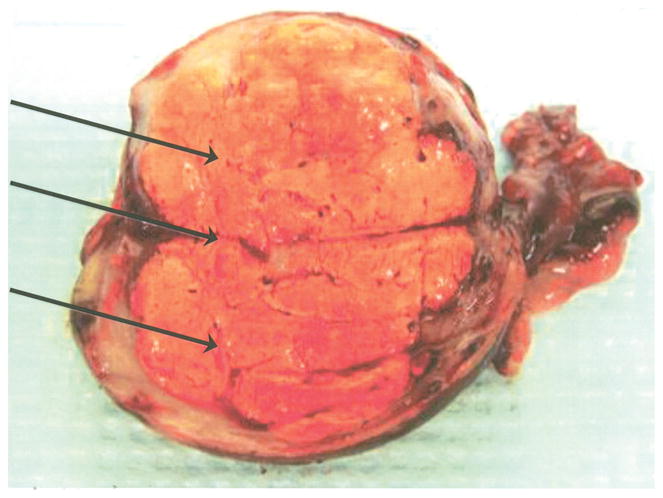
Bivalved ovary from patient with von Hippel-Lindau syndrome depicting a solid yellow tumor (arrows).
Wagner. Lipid Cell Tumors in von Hippel-Lindau. Obstet Gynecol 2010.
One year postoperatively, the patient returned to NIH for follow-up and for kidney cancer surgery. Two renal masses were excised, and the final pathology indicated clear cell renal carcinoma. The left ovary continues to appear normal on imaging, and testosterone remains in the normal range.
COMMENT
von Hippel-Lindau syndrome is an autosomal dominant inherited syndrome that is characterized by a variety of benign and malignant vascular tumors in multiple organs. Clinical disease usually develops during the reproductive years of life. The most common lesions associated with von Hippel-Lindau are retinal angiomas and cerebellar hemangioblastomas, and although rare, the development of renal cell carcinoma or pheochromocytoma is of great concern.4 Papillary cystadenomas of the broad ligament and adnexa have also been reported in women with von Hippel-Lindau and are usually asymptomatic. They are thought to be of mesonephric origin and can be misinterpreted as malignant neoplasms.5,6 Bilateral metastatic renal carcinoma to the ovary is rare7 and has not been reported in patients with von Hippel-Lindau, but it can be mistaken for primary ovarian tumors. We report the first two cases of testosterone-secreting lipid cell tumors of the ovary in von Hippel-Lindau syndrome. We performed a literature search on PubMed (November 1976 to February 2009) to ascertain that these are the first two reported cases of lipid cell tumors of the ovary in patients with von Hippel-Lindau syndrome. The search encompassed all types of articles published at any date. The search was limited to include articles published in English and with human female patients. The following query was used: (“lipids”[MeSH Terms] OR “lipids”[All Fields] OR “lipid”[All Fields]) AND (“cell”[All Fields] OR “cells”[MeSH Terms] OR “cells”[All Fields]) AND (“tumor”[All Fields] OR “neoplasms”[MeSH Terms] OR “neoplasms”[All Fields] OR “tumor”[All Fields])) AND (VHL[All Fields] OR “von Hippel-Lindau”[All Fields]) AND (“humans”[MeSH Terms] AND “female”[MeSH Terms] AND English[lang]).
Testosterone-secreting lipid cell ovarian tumors have not been previously reported in women with von Hippel-Lindau. These are rare functioning ovarian neoplasms and are commonly seen in reproductive-aged women.8 Lipid cell tumors usually have a unilateral presentation, and bilateral involvement has been found in only 6% of cases.1
Clinical manifestations are a result of their endocrine activity. Virilization occurs in 77% of cases, while evidence of estrogenic excess and Cushing’s syndrome occurs in 23% and 10% of cases, respectively.2 They are usually benign, slow-growing tumors, and malignancy occurs in about 20% of cases.3
Lipid cell tumors are solid and well-circumscribed and on gross inspection have a yellow to orange-tan appearance with areas of hemorrhage, necrosis, and cystic degeneration. Their cut surface is often soft and lobulated. Histologic examination often reveals cells resembling Leydig cells, luteinized ovarian stromal cells, or adrenal cortical cells.8 Although their origin remains a mystery, they are thought to arise from specialized ovarian stromal cells within the medullary part of the ovary.8 Varying proportions of the two major steroid-producing cell types of these tumors can result in different clinical manifestations.
Management usually involves performing a unilateral salpingo-oophorectomy for reproductive-aged women with early disease, and a total abdominal hysterectomy with bilateral salpingo-oophorectomy is indicated for advanced, extra-ovarian disease and for women beyond reproductive age. Both of our patients had disease confined to the ovaries. Because our first patient had central necrosis on one ovary and small lipid tumor rests throughout both ovaries on final pathology, she would most likely have developed recurrent disease and would have needed additional surgery if she had not undergone a bilateral salpingo-oophorectomy. The second patient had one affected ovary. Unilateral ovarian involvement was confirmed by ovarian vein sampling. At surgery, no disease was seen outside of the ovaries. Based on ultrasound findings, the absence of a significant rise in androgens following hCG and intraoperative findings, the likelihood of a small tumor in the contralateral ovary was minimal. Surgical removal of this tumor results in rapid biochemical resolution, and serial serum testosterone levels are useful to detect recurrence.
In our first patient, the history of biopsy-proven renal cancer in a patient with von Hippel- Lindau syndrome, the central necrosis of the ovarian mass, and the intraoperative frozen section reading that could not exclude renal cell cancer metastatic to the ovaries illustrates the challenges in making the preoperative or intraoperative diagnosis of lipid cell tumor, especially in a patient with another known malignancy and genetic disorder. In this case, considering metastatic renal disease in these bilateral ovarian masses was warranted. Furthermore, lipid cell tumor with a large percentage of adrenal cortical cells may be mistakenly diagnosed as an adrenal or renal metastasis.
Because lipid cell tumors appear to be “great impersonators,” a definitive diagnosis can only be made with the aid of special immunohistochemical stains. Lipid tumors show strong and diffuse calretinin expression and also express inhibin, although less strongly. They are consistently positive for Melan-A and negative for melanoma antigen recognized by T-cells9 Primary clear cell carcinoma of the ovary is cytokeratin 7 positive10 and stains for mesothelin with high specificity but low sensitivity.11 Primary renal cell carcinoma is cytokeratin 7 negative and stains positive for CD10 and renal cell carcinoma marker.10
We report two cases of testosterone-releasing lipid tumors of the ovary in women with von Hippel-Lindau syndrome. Diagnosis may be suspected based on mildly elevated testosterone, amenorrhea, and hirsutism.
Acknowledgments
Supported in part by the Urologic Oncology Branch, NCI, Surgical Pathology Branch, NCI, Clinical Endocrinology Branch, NIDDK, Program in Reproductive and Adult Endocrinology, Eunice Kennedy Shriver NICHD, and the Clinical Center, NIH.
Footnotes
Financial Disclosure
The authors did not report any potential conflicts of interest.
References
- 1.Dengg K, Fink F, Heitger A, Tabarelli M, Kreczy A, Glatzl J, et al. Precocious puberty due to a lipid cell tumour of the ovary. Eur J Pediatr. 1993;152:12–4. doi: 10.1007/BF02072508. [DOI] [PubMed] [Google Scholar]
- 2.Taylor HB, Norris HJ. Lipid cell tumors of the ovary. Cancer. 1967;20:1953–62. doi: 10.1002/1097-0142(196711)20:11<1953::aid-cncr2820201123>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 3.Montag TW, Murphy RE, Belinson JL. Virilizing malignant lipid cell tumor producing erythropoietin. Gynecol Oncol. 1984;19:98–103. doi: 10.1016/0090-8258(84)90164-1. [DOI] [PubMed] [Google Scholar]
- 4.Losner RR, Glenn GM, Walther M, Chew EY, Libutti SK, Linehan WM, et al. von Hippel-Lindau disease. Lancet. 2003;361:2059–67. doi: 10.1016/S0140-6736(03)13643-4. [DOI] [PubMed] [Google Scholar]
- 5.Gaffey MJ, Mills SE, Boyd JC. Aggressive papillary tumor of middle ear/temporal bone and adnexal papillary cystadenoma. Manifestations of von Hippel-Lindau disease. Am J Surg Pathol. 1994;18:1254–60. doi: 10.1097/00000478-199412000-00009. [DOI] [PubMed] [Google Scholar]
- 6.Gersell DJ, King TC. Papillary cystadenoma of the mesosal-pinx in von Hippel-Lindau disease. Am J Surg Pathol. 1988;12:145–9. doi: 10.1097/00000478-198802000-00008. [DOI] [PubMed] [Google Scholar]
- 7.Vara A, Madrigal B, Veiga M, Diaz A, Garcia J, Calvo J. Bilateral ovarian metastases from renal clear cell carcinoma. Acta Oncol. 1998;37:379–80. doi: 10.1080/028418698430610. [DOI] [PubMed] [Google Scholar]
- 8.Case records of the Massachusetts General Hospital. Weekly clinicopathological exercises. Case 22–1982. A nine-year-old girl with virilization and a calcified pelvic mass. N Engl J Med. 1982;306:1348–55. doi: 10.1056/NEJM198206033062208. [DOI] [PubMed] [Google Scholar]
- 9.Deavers MT, Malpica A, Ordonez NG, Silva EG. Ovarian steroid cell tumors: an immunohistochemical study including a comparison of calretinin with inhibin. Int J Gynecol Pathol. 2003;22:162–7. doi: 10.1097/00004347-200304000-00008. [DOI] [PubMed] [Google Scholar]
- 10.Cameron RI, Ashe P, O’Rourke DM, Foster H, McCluggage WG. A panel of immunohistochemical stains assists in the distinction between ovarian and renal clear cell carcinoma. Int J Gynecol Pathol. 2003;22:272–6. doi: 10.1097/01.PGP.0000071044.12278.43. [DOI] [PubMed] [Google Scholar]
- 11.Leroy X, Farine MO, Buob D, Wacrenier A, Copin MC. Diagnostic value of cytokeratin 7, CD10 and mesothelin in distinguishing ovarian clear cell carcinoma from metastasis of renal clear cell carcinoma. Histopathology. 2007;51:874–6. doi: 10.1111/j.1365-2559.2007.02874.x. [DOI] [PubMed] [Google Scholar]