

Abstract
Approximately 10% of the patients diagnosed with type 2 diabetes (T2D) have detectable serum levels of glutamic acid decarboxylase 65 autoantibodies (GADA). These patients usually progress to insulin dependency within a few years, and are classified as being latent autoimmune diabetes in adults (LADA). A decrease in the frequency of peripheral blood natural killer (NK) cells has been reported recently in recent-onset T1D and in high-risk individuals prior to the clinical onset. As NK cells in LADA patients have been investigated scarcely, the aim of this study was to use multicolour flow cytometry to define possible deficiencies or abnormalities in the frequency or activation state of NK cells in LADA patients prior to insulin dependency. All patients were GADA-positive and metabolically compensated, but none were insulin-dependent at the time blood samples were taken. LADA patients exhibited a significant decrease in NK cell frequency in peripheral blood compared to healthy individuals (P = 0·0018), as reported previously for recent-onset T1D patients. Interestingly, NKG2D expression was increased significantly (P < 0·0001), whereas killer cell immunoglobulin-like receptor (KIR)3DL1 expression was decreased (P < 0·0001) within the NK cell population. These observations highlight a defect in both frequency and activation status of NK cells in LADA patients and suggest that this immunological alteration may contribute to the development of autoimmune diabetes by affecting peripheral tolerance. Indeed, recent evidence has demonstrated a regulatory function for NK cells in autoimmunity. Moreover, the decrease in NK cell number concords with observations obtained in recent-onset T1D, implying that similar immunological dysfunctions may contribute to the progression of both LADA and T1D.
Keywords: autoimmune, KIR3DL1, LADA, NK cells, NKG2D
Introduction
Type 1 diabetes (T1D) is a multifactorial autoimmune disease affecting up to 1% of the general population that develops most often at an early age. The disease evolves due to T cell-mediated destruction of the pancreatic islet beta-cells which leads to partial or total loss of insulin production, although the mechanism(s) underlying disease development are not understood fully [1].
In the last decade a subgroup of T1D denoted latent autoimmune diabetes in adults (LADA), or as suggested more recently autoimmune diabetes of the adult (ADA), have been described in humans. While patients with LADA are diagnosed initially with type 2 diabetes (T2D) they exhibit clear signs of β cell autoimmunity [2]. Several studies have reported previously that about 10% of the patients diagnosed with type 2 diabetes have circulating autoantibodies against islet cell cytoplasmic antigens [3,4] or glutamic acid decarboxylase (GAD) [2,5–9] in their blood. Thus, even though these patients share the same insulin resistance as individuals with T2D, the presence of autoantibodies characterizes their disease as being autoimmune, as is the case in T1D. These findings indicate that LADA shares features with both T1D and T2D patients. Interestingly, LADA patients display a more severe defect in stimulated β cell capacity compared with T2D patients [10]. Another interesting difference is that LADA is usually diagnosed in adulthood at the age of 35 years or older [9], while T1D is diagnosed most often during childhood and T2D most probably affects individuals older than 40 years.
During the early phase of disease, individuals with LADA retain good metabolic control through diet alone or by taking oral hypoglycaemic drugs, and this state can last for months to years. However, 70% of LADA patients may progress towards insulin dependency within a few years, indicating that in contrast to T1D, LADA involves a slow and progressive loss of β cells. These patients might therefore be instructive in elucidating the initial phase of autoimmune diabetes as well as progression towards insulin deficiency.
Natural killer (NK) cells are an important component of the innate immune system and may be involved in the complex process of autoimmunity due to their ability to both directly kill target cells and to interact with antigen-presenting cells as well as with T cells [11–13]. NK cell function is regulated by a delicate balance between activating and inhibitory signals generated by a diverse array of surface receptors. Human leucocyte antigen (HLA) class I molecules on target cells bind normally to inhibitory receptors expressed by NK cells, and an inhibitory signal that prevents the NK cell from killing a healthy target cell is generated (reviewed in [14,15]). The HLA class I-specific inhibitory receptors expressed on human NK cells include killer cell immunoglobulin-like receptors (KIR)s such as KIR3DL1 (NKB1) and the lectin-like CD94-NKG2A receptors [16,17].
NK cells also express different activating receptors, such as the activating killer cell lectin-like receptor NKG2D. This receptor plays a crucial role in NK cell-mediated immunosurveillance by recognizing different ligands that are expressed on target cells in pathological situations, such as during stress and infection (reviewed in [18]). The NKG2D receptor has thus evolved to function as a sensory receptor that alerts the immune system in the presence of stress, infection or tumour growth. In addition, NK cells also participate in immune regulation via cytokine secretion (reviewed in [19]). Overall, several reports support the idea that a delicate balance between NK cell frequency and function seems necessary to avoid T1D and possibly other autoimmune conditions as well (reviewed in [13]).
Data investigating NK cell numbers and activity in human T1D have been scarce, and published results are conflicting [20–25]. Interestingly, some studies have shown a deficiency in the numbers and/or abnormal function of NK cells in peripheral blood of T1D patients [20,23,26]. Permanent abnormalities in numbers and function were observed in some of these studies [22,25], while in others the stage of the disease was associated with transitory cellular differences [20,26]. A significant decrease in NK cell numbers in recent-onset T1D patients was reported recently supporting an important role for NK cell number and function in the autoimmune process [27].
The aim of this study was to characterize a homogeneous group of patients diagnosed with LADA without insulin requirement that may represent an early stage of the autoimmune process. Our main focus was to investigate the frequency and phenotype of NK cells in peripheral blood of LADA patients and to identify possible deficiencies that might be associated with the onset of diabetes. We report that these pre-insulin-dependent patients had a significant decrease in the frequency of NK cells in peripheral blood compared to healthy individuals.
Materials and methods
Subjects
Blood samples were collected from a total of 20 healthy volunteers (n = 20) and from 46 patients newly diagnosed with LADA (n = 46). These patients were selected based on the following criteria: (i) male or female patients between 30 and 70 years of age; (ii) diagnosis of T2D within the previous 5 years; (iii) presence of glutamic acid decarboxylase 65 autoantibodies (GADA); (iv) requiring diabetes treatment only with diet and oral hypoglycaemic agents; and (v) having no indications of serious diseases or conditions which would exclude them from the trial in the opinion of the investigator. The following parameters were determined after their visit: immunological markers, diabetic status, fasting lipids, haematological and biochemical parameters, physical examinations and reporting of concomitant medication. In addition, values of fasting glucose, fasting and 2-h Sustacal stimulated C-peptide and long-term metabolic control assessed by haemoglobin A1c (HbA1c) was taken into consideration when the diabetes status of each patient was determined. The following data were also recorded for the clinical characterization of these subjects: age, body mass index (BMI), thyroid-stimulating hormone (s-TSH), free triiodothyronine (fT3), free thyroxine (fT4), fB-glucose, fS-insulin and insulin resistance. Our laboratory is number 156 in the Diabetes Antibody Standardization Program (DASP) for GADA and IA-2A measurement, thus the concentration of these autoantibodies was assessed in the serum of each patient. Blood samples were collected into ethylenediamine tetraacetic acid (EDTA) tubes at Malmö University Hospital and processed within 24 h. The study was approved by the Lund University Research Ethics Committee and informed consent was obtained from the participants.
Reagents
For flow cytometric analysis, fluorescence activated cell sorter (FACS) buffer was used containing phosphate-buffered saline (PBS) pH 7·2 (Life Technologies, Paisley, Scotland, UK), supplemented with 2% bovine serum albumin (BSA) (ICN Biomedicals Inc., Aurora, OH, USA) and 2 mM EDTA (Sigma-Aldrich, St Louis, MO, USA). FACS Lysing Solution 2 (BD Biosciences, San Jose, CA, USA) was used to lyse the erythrocytes before analysis. For freezing of peripheral blood mononuclear cells (PBMC), 90% human serum from clotted male whole blood (Sigma-Aldrich) was mixed with 10% dimethylsulphoxide (DMSO) (Sigma-Aldrich). For thawing PBMC, complete RPMI-1640 medium (C-RPMI) was used (Life Technologies) supplemented with 5% v/v pooled human serum from clotted male whole blood (Sigma-Aldrich), 1% sodium pyruvate (Life Technologies), 7·5% sodium bicarbonate (Life Technologies), L-glutamine (ICN Biomedicals Inc.), penicillin–streptomycin–neomycin (PSN) antibiotic mixture (100×; Life Technologies), β-mercaptoethanol (ICN Biomedicals Inc.) and non-essential amino acids (MEM, 100×; Life Technologies).
Whole blood
A small aliquot of blood was analysed using an AC900 AutoCounter (Swelab Instrument AB, Stockholm, Sweden) to determine the absolute numbers of lymphocytes in each sample. In addition, human peripheral blood samples were stained with various monoclonal antibodies to determine the percentage of lymphocyte subsets in each individual using flow cytometry. Briefly, 100 µl of blood was used for each staining and the samples were incubated for 20–30 min at room temperature. Erythrocytes were lysed using BD FACS lysing Solution 2 (BD Bioscience) and the samples were washed with FACS buffer. Cells were resuspended in 300 µl FACS buffer and stored overnight at 4°C until flow cytometric analysis was performed using a FACSCalibur (Becton Dickinson).
PBMC were separated from whole blood using Lymphoprep (Axis-Shield PoC AS, Oslo, Norway) or Vacutainer Cell Preparation Tubes (Becton Dickinson, NJ, USA). The isolated lymphocytes were washed with PBS and frozen in freezing media containing 90% human serum (Sigma-Aldrich) and 10% DMSO (Sigma-Aldrich), which was added dropwise to the cells before they were frozen and stored in liquid nitrogen.
Frozen samples
Staining of PBMC was performed according to the manufacturer's protocol (eBioscience). Briefly, samples were thawed in complete RPMI-1640 medium, washed with staining buffer containing 5% human serum, 0·01% sodium azide in PBS pH 7·2 and stained thereafter with various combinations of anti-human monoclonal antibodies. Following 20 min of incubation at 4°C the cells were washed with staining buffer and analysed using four-colour flow cytometry (FACS Calibur; Becton Dickinson).
Monoclonal antibodies
The following monoclonal anti-human antibodies were used in various combinations for flow cytometric analysis of whole blood: fluorescein isothiocyanate (FITC)-conjugated CD3 and CD25; phycoerythrin (PE)-conjugated CD19 and CD8; peridinin chlorophyll protein (PerCP)-conjugated CD4; CD3/CD16+CD56 Stimultest (FITC/PE) (all from BD Bioscience). The following monoclonal anti-human antibodies were used in various combinations for flow cytometric analysis of PBMC: FITC-conjugated CD56 (ImmunoTools, Friesoythe, Germany); PE-conjugated CD16 (ImmunoTools), PerCP-conjugated CD3 (BD Pharmingen), biotin-conjugated KIR3DL1 (CD158e1) (BioLegend, San Diego, CA, USA), allophycocyanin (APC)-conjugated NKG2D (BD Pharmingen), CD3/CD16+CD56 Stimultest (FITC/PE) (BD Bioscience). The biotin-conjugated antibody was revealed in a second step using streptavidin (SA)-conjugated PerCP (BD Pharmingen).
Antibody measurement
Autoantibodies against GAD65 or IA-2 in plasma were analysed using a radio-binding assay as described previously [28,29].
HLA typing
HLA typing for the HLA-DQB1*0302 T1D risk allele was performed as described previously [30,31]. We used dried blood spot punch-out samples in a 96-well format and conducted polymerase chain reaction (PCR) directly from the filters using biotinylated primers for DQB1, streptavidin-coated 96-well plates and lanthanide-labelled probes for the hybridization, as described previously [30,31]. All reagents were from Wallac Oy, Perkin Elmer Life Science (Turku, Finland).
Genotyping of HLA-Bw locus in LADA patients was performed using allele-specific PCR for HLA-Bw4 and HLA-Bw6. Genomic DNA was extracted from frozen PBMC using MoleStrips™ DNA blood kit (Mole Genetics AS, Lysaker, Norway), according to the manufacturer's recommendation. The HLA-B-specific exon-2 PCR was carried out using the forward primer 5′-GACGACACGCTGTTCGTGA-3′ combined with either the HLA-Bw4-specific primer 5′-GCTCTGGTTGTAGTAGCGGA-3′ or the HLA-Bw6-specific primer 5′-CTCTGGTTGTAGTAGCCGC-3′, as described [32]. PCR products were analysed by electrophoresis on a 2% agarose gel containing 0·5× Tris–borate–EDTA (TBE buffer). The amplified product sizes were 180 base pairs (bp).
Flow cytometric analysis
Data were analysed using the CellQuest software (Becton Dickinson) and are presented as representative dot plots or diagrams. The median value is indicated in each diagram.
Statistical analysis
Continuous distributions were summarized as medians (range). To assess clinical measurements of LADA patients in relation to their respective NK lymphocyte expression levels, the LADA patients were divided into two groups. Patients with percentage lymphocyte expression levels above the median were defined as having high expression levels and the remaining LADA patients were defined as having low expression levels. Clinical distributions were then compared between patients with high and low lymphocyte expression levels. The same analyses were conducted for clinical measurements of LADA patients in relation to their interferon (IFN)-γ production levels in CD3+ and CD3− populations. Non-parametric Mann–Whitney U-tests were used to test for differences in distribution between two groups.
Graphs of distributions were drawn using statistical program Prism 4 (GraphPad Software, San Diego, CA, USA). Statistical analysis was performed using spss version 14 for windows (http://www.spss.com). Statistical significant difference * at P < 0·05 and ** at P < 0·01.
Results
LADA patients in relation to T1D characteristics
Of the 46 LADA patients studied, 21 of 46 (46%) were positive for the T1D high-risk HLA-DQB1*0302 allele. The clinical characteristics of all LADA patients stratified based on HLA-DQB1*0302 are presented in Table 1. Fasting glucose, fasting and stimulated C-peptide, HbA1c, BMI or GADA levels were not significantly different between LADA patients who had the high-risk T1D HLA allele and patients who did not. However, LADA patients with HLA-DQB1*0302 were noted to be younger (P < 0·01) compared to other patients (Table 1).
Table 1.
Clinical characteristics of latent autoimmune diabetes in adults (LADA) patients with the human leucocyte antigen (HLA)-DQB1*0302 Type 1 diabetes risk allele and patients without the HLA-DQB1*0302 allele.
| Type 1 diabetes risk HLA*DQB1*0302 | ||
|---|---|---|
| Variables | Yes | No |
| n | 21 | 25 |
| Age (years) | 52 (30–65) | 59 (43–69) |
| Males (n) | 19 | 19 |
| GADA (U/ml) | 97 (32–4487) | 56 (38–296 997) |
| IA-2A positive (n) | 5 | 1 |
| IAA | 1 | 1 |
| BMI (kg/m2) | 26 (20–34) | 28 (21–39) |
| HbA1c% | 6·5 (5·1–10·9) | 5·9 (4·6–9·9) |
| P-glucose (mmol/l) pre-Sustacal | 8·5 (5·5–15·8) | 8·0 (5·9–17·4) |
| P c-peptide (nmol/l) pre-Sustacal | 0·6 (0·3–1·6) | 0·7 (0·3–1·8) |
| P c-peptide (nmol/l) post-Sustacal | 1·4 (0·5–3·9) | 1·6 (0·8–5·1) |
BMI, body mass index; GADA, glutamic acid decarboxylase 65 autoantibodies; HbA1c, haemoglobin A1c; IAA, insulin autoantibodies.
Altered NK cell frequency in LADA patients
We first investigated possible differences in the frequency of NK cells in LADA patients in an attempt to determine if NK cells are involved in the establishment of disease. A significant decrease in the percentage and absolute numbers of NK cells, defined as CD3−CD16+CD56+ cells, were observed in LADA patients compared with age-matched healthy control individuals (Fig. 1a,b and Table 2). We did not observe any changes among other major lymphocyte subsets, such as B and T cells (Table 2). No association was determined between the decreased frequency of NK cells and any of the clinical parameters that were documented for each patient. However, these observations suggest that a reduction in circulating NK cells in LADA patients is already apparent prior to insulin deficiency.
Fig. 1.
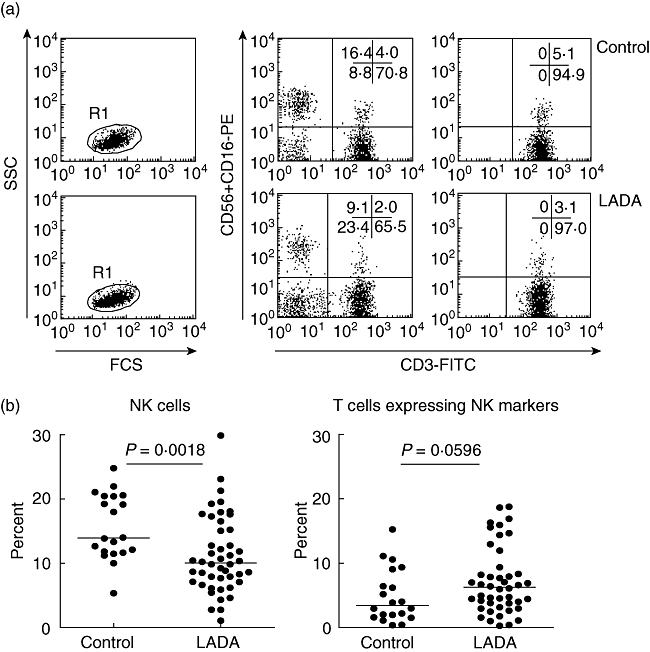
Flow cytometry analysis of natural killer (NK) cells in whole blood. Representative dot plots showing analysis of blood NK cells from the lymphocyte fraction using CD3 and CD16+CD56 as markers (a). The frequency of CD3−CD16+CD56+ NK cells and CD3+ CD16+CD56+ T cells was investigated in blood from latent autoimmune diabetes in adults (LADA) (n = 46) and healthy control individuals (n = 20) (b). Each dot represents a different individual and median values for each group is marked with a horizontal line. Using the Mann–Whitney non-parametric test a significant difference in NK cell frequency was noted between LADA patients and healthy controls (P = 0·0018).
Table 2.
Median and maximum/minimum values of the total number/ml blood and percentage of T cells, B cells, natural killer (NK) cells and T cells expressing NK markers in healthy individuals and latent autoimmune diabetes in adults (LADA) patients.
| Total cell number × 106 |
Percentage |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Cells | Control | n | LADA | n | P-value | Control | n | LADA | n | P-value |
| T cells | 1·383 | 20 | 1·318 | 46 | 0·7430 | 68·82 | 20 | 73·79 | 46 | 0·0533 |
| Max 2·445 | Max 2·391 | Max 87·10 | Max 6·83 | |||||||
| Min 0·7821 | Min 0·5790 | Min 60·16 | Min 48·81 | |||||||
| B cells | 0·1789 | 20 | 0·2148 | 46 | 0·6106 | 11·22 | 20 | 12·65 | 46 | 0·1629 |
| Max 0·4644 | Max 0·6969 | Max 16·17 | Max 4·92 | |||||||
| Min 0·1026 | Min 0·0298 | Min 5·130 | Min 1·570 | |||||||
| NK cells | 0·2647 | 20 | 0·1856 | 46 | 0·0019** | 13·94 | 20 | 9·850 | 46 | 0·0018** |
| Max 0·6443 | Max 0·5082 | Max 24·78 | Max 9·87 | |||||||
| Min 0·1070 | Min 0·0227 | Min 5·350 | Min 1·080 | |||||||
| T cells exp. NK | 0·06095 | 20 | 0·07895 | 46 | 0·0927 | 3·425 | 20 | 6·245 | 46 | 0·0596 |
| Max 0·1286 | Max 0·5286 | Max 15·23 | Max 33·4 | |||||||
| Min 0·0067 | Min 0·0029 | Min 0·370 | Min 0·280 | |||||||
P < 0·01.
Altered expression of the surface receptors NKG2D and KIR3DL1 in LADA patients
NKG2D expression on peripheral lymphocytes was investigated using flow cytometry in LADA patients (n = 37) and healthy individuals (n = 20) for whom frozen PBMC were available. A significant increase in the frequency of NKG2D positive cells was observed in both total lymphocyte and NK cell populations (Fig. 2b). In addition, we found a clear significant increase of the NKG2D receptor expression on total lymphocyte and on NK cell population measured by the mean fluorescence intensity (MFI) (Fig. 2c).
Fig. 2.
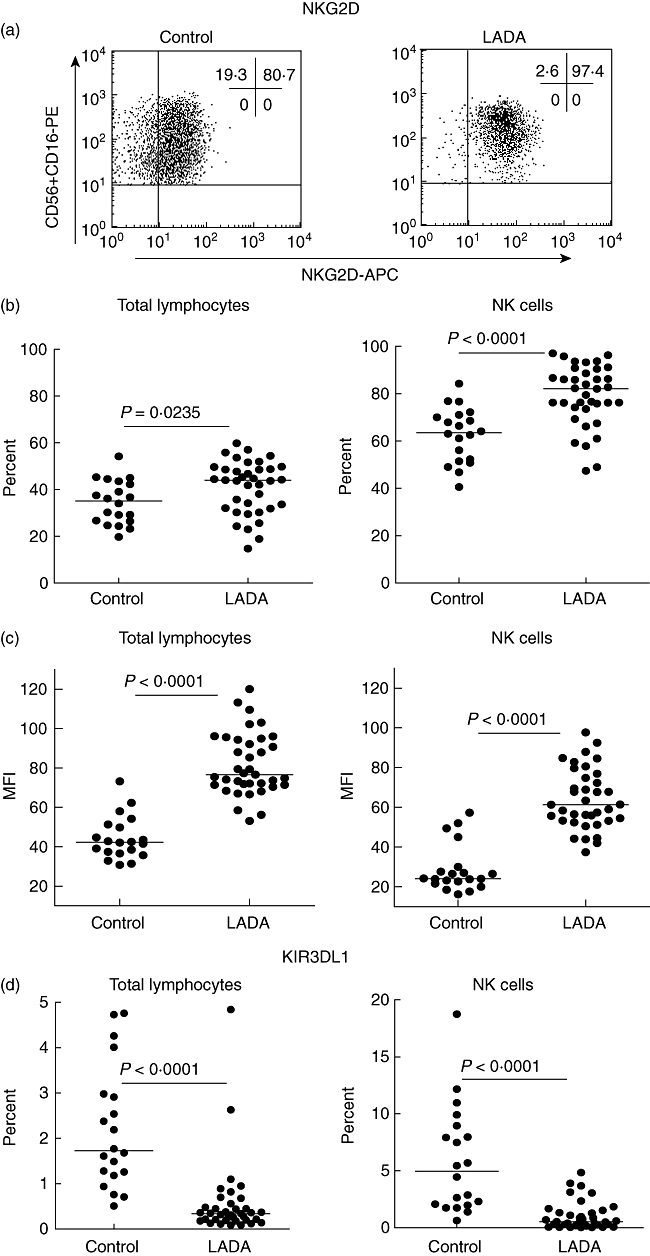
Activating status of lymphocytes and natural killer (NK) cells. (a) Representative dot plots showing NKG2D expression of blood NK cells from the lymphocyte fraction using CD3 and CD16+CD56 as markers in control and latent autoimmune diabetes in adults (LADA) patients. The activating status of blood lymphocytes, especially NK cells, was determined using the expression of the surface markers NKG2D (b,c) and KIR3DL1 (d) from LADA (n = 37) and control (n = 20) individuals from which frozen peripheral blood mononuclear cells (PBMCs) were saved from the first visit. Each dot represents a different individual and the median values for each group is marked with a horizontal line. (b) Using the Mann–Whitney non-parametric test a significant difference in percentage of NKG2D expression was noted between LADA patients and healthy controls both in the total lymphocyte population (b, left panel) (P = 0·0235) and the NK cell population (b, right panel) (P < 0·0001). (c) A significant increase of NKG2D expression at single cell level was observed in LADA patients using the mean fluorescence intensity (MFI) both in the total lymphocyte population (c, left panel) (P < 0·0001) and the NK cell population (c, right panel) (P < 0·0001). (d) A significant difference in percentage of KIR3DL1 expression was also noted between LADA patients and healthy controls in the total lymphocyte population (d, left panel) (P < 0·0001) and the NK cell population (d, right panel) (P < 0·0001).
The effector function of NK cells is controlled by the expression of various surface receptors of the KIR family. Hence, the status of the NK cells was characterized further by studying the expression of the NK-inhibitory receptor KIR3DL1 (NKB1). A significant decrease in KIR3DL1-positive cells was observed in all lymphocytes as well as in the NK cell population when we compared LADA patients with healthy control individuals (Fig. 2d). Because KIR3DL1 recognize the HLA-Bw4 allele [33], we next genotyped this locus in LADA patients. As shown in Fig. 3, the HLA-Bw4/6 genotyping revealed that 22·9% and 57·1% of the typed LADA patients were homozygous for Bw4 and Bw6, respectively, while 20% were heterozygous. According to the major histocompatibility complex (MHC) database (http://www.ncbi.nlm.nih.gov/gv/mhc/), the Bw4 frequency in Europeans is approximately 0·43. The frequency of LADA patients carrying the Bw4 allele (homo- and heterozygous) were approximately 0·43, which correlates with the European Bw4 frequency. There were no significant differences or correlations between the HLA-Bw genotypes and KIR3DL1 expression in either of the cell populations analysed (data not shown).
Fig. 3.
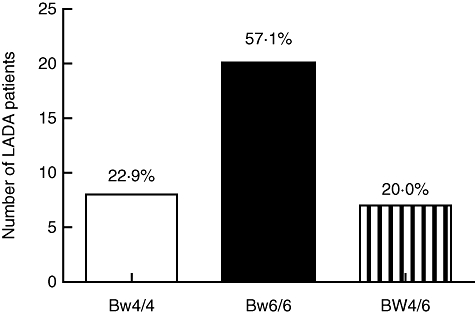
Frequency of human leucocyte antigen (HLA)-Bw alleles in latent autoimmune diabetes in adults (LADA) patients. Genotyping of HLA-Bw locus was performed using allele-specific polymerase chain reaction, as described in Material and methods. The frequency of HLA-Bw4 and HLA-Bw6 were in accordance to the major histocompatibility complex (MHC) database for the European population.
NK cells and NKG2D or KIR3DL1 expression in relation to clinical parameters
NK cell number and KIR3DL1 and NKG2D expression levels were not associated with clinical parameters. However, the percentage of NKG2D expression in all lymphocytes was associated strongly with whether or not the patient had the HLA-DQB1*0302 allele (Table 3). Patients with higher NKG2D expression within the lymphocyte population were more likely to have the DQB1*0302 allele (P < 0·01). HLA was not associated significantly with NK cell number or KIR3DL1 expression.
Table 3.
Natural killer (NK), NKG2D and killer cell immunoglobulin-like receptor (KIR3)DL1 cells in relation to presence of the type 1 diabetes (T1D) risk human leucocyte antigen (HLA)-DQB1*0302.
| LADA patients with T1D risk HLA-DQB1*0302 |
|||
|---|---|---|---|
| Yes |
No |
||
| NK cell subset, NKG2D or KIR3DL1 expression in high levels* | n (% of patients with high levels) | n (% of patients with high levels) | P-value |
| NK cells (% of total) | 10 (52%) | 14 (44%) | 0·57 |
| NKG2D cells (% of total) | 12 (75%) | 7 (32%) | 0·009 |
| NKG2D cells (% of NK cells) | 10 (63%) | 9 (41%) | 0·19 |
| KIR3DL cells (% of total) | 6 (38%) | 13 (59%) | 0·19 |
| KIR3DL cells (% of NK cells) | 6 (38%) | 13 (59%) | 0·19 |
Patients with high levels have percentages above median (see Fig. 2). LADA, latent autoimmune diabetes in adults.
Discussion
NK cells have been implicated in peripheral tolerance and immune regulation [34–36], as well as in T1D pathogenesis [20,23,26]. The purpose of this study was to investigate the frequency and phenotype of NK cells in LADA patients. The major finding of our investigation was a specific alteration of NK cell frequency, suggesting that these cells might be involved in the pathogenesis of autoimmune diabetes in LADA patients.
While a weakness of this study is that it lacks direct functional investigation of NK cells in these patients, the observed discrepancy in NK cells is still in concordance with a number of previously reported studies demonstrating a significant decrease of NK cells in peripheral blood from T1D patients [22,25,27]. Furthermore, differences in NK cell numbers have been reported in patients suffering from other autoimmune syndromes, such as multiple sclerosis (MS) and systemic lupus erythematosus (SLE) (reviewed in [12]), thereby supporting the hypothesis that NK cells play a role in promoting β cell destruction and loss of self-tolerance in autoimmune diseases. As NK cells have been suggested to play an immunoregulatory role in the prevention of autoimmune disease by cross-talking with dendritic cells (DCs) and by down-regulation of T cell responses (reviewed in [19]), the observed NK cell deficiency in both LADA and T1D could contribute to the breakdown of self-tolerance that leads to β cell destruction. The observed decreased number of NK cells in peripheral blood of LADA patients might also indicate that a proportion of NK cells have migrated from the blood to the pancreas or the draining lymph nodes due to the inflammatory response. In turn this would result in a decreased frequency of NK cells in the blood and their accumulation in the infiltrated islets. Unfortunately, the unavailability of pancreatic biopsies makes it impossible to test this hypothesis.
An interesting finding was the increase of the activating receptor NKG2D expression as measured by MFI in CD16+CD56+ NK cells as well as the frequency of NK cells expressing NKG2D in LADA patients. Unexpectedly, Rodacki et al. observed that T1D patients had a decreased expression of NKG2D in NK cells irrespective of disease duration [27], and Ogasawara et al. have demonstrated a reduced NKG2D expression in non-obese diabetic (NOD) mice [37]. Thus, the activating phenotype of NK cells in LADA patients seems to be different compared to T1D patients and NOD mice, and it could be speculated that it might actually be a consequence and not a cause of the disease. However, given the fact that in LADA patients the increased frequency of NK cells expressing the NKG2D receptor is coupled to a significant increase of receptor density at the single cell level (Fig. 2c), we suggest that this finding might represent a pathogenic factor and could therefore account for the slower progression to insulin-dependent diabetes that is observed in these patients compared with individuals who develop T1D.
The decreased expression of the inhibitory receptor KIR3DL1 that is expressed by a proportion of NK cells was also an intriguing finding in lymphocytes from LADA patients. The interaction of KIR3DL1 with specific HLA-B antigens (e.g. HLA-Bw4 allele) inhibits cytotoxicity and prevents target cell lysis and death [33,38,39]. The absence of KIR3DL1 expression but also KIR ligands may thus lower the threshold for cell activation. Activating receptors can initiate a response which is not balanced by inhibitory receptor signals from KIR3DL1, resulting in increased inflammatory responses and susceptibility to autoimmune diseases. Hence, the observed increase in the activation receptor NKG2D expression and decrease in the inhibitory receptor KIR3DL1 expression may indicate an alteration of the balance between inhibitory and activating NK receptors, which in turn could be involved in the loss of tolerance leading to β cell destruction.
Our results also indicated that some LADA patients have an increased number of T cells expressing NK markers (CD16 and/or CD56) compared to healthy individuals. We have no explanation for this, but it is tempting to speculate that this may represent an active state of the disease when effector autoreactive T cells are appearing with specific cytotoxicity towards pancreatic beta-cells. Further investigations of T cells expressing NK markers in peripheral blood from LADA patients are warranted to evaluate the importance of these lymphocytes in disease development.
In mice, a deficiency of CD4+CD25+ regulatory T cells correlates with an increase of NK cell numbers in secondary lymphoid organs [40,41]. The presence of CD4+CD25+ regulatory T cells thus seems necessary and sufficient to repress both NK cell recruitment, as well as their ability to kill NK cells using direct cytotoxic activity via perforin and granzyme B (reviewed in [42]). This is an interesting observation that indicates a close interplay between NK cells and CD4+CD25+ regulatory T cells in modulating the outcome of immune responses. It has been reported recently that the major effector molecule used by CD4+CD25+ regulatory T cells to suppress cell activity is membrane-bound transforming growth factor (TGF)-β and that regulation of the NK cell activating receptor NKG2D is a TGF-β-dependent process [43–45]. These results suggest that the observed increase in NKG2D expression in NK cells from LADA patients might be due to an alteration in the ability of CD4+CD25+ regulatory T cells to inhibit NK cell activation and effector functions [43–48].
Interestingly, NK cells have also been implicated in immune regulation, and the concept of regulatory NK cells has emerged (reviewed in [49]). NK cells can, in fact, act as regulatory cells and kill various other cell types such as DCs, T cells, B cells and endothelial cells through cytotoxicity (reviewed in [49]). Recently, Brillard et al. demonstrated that NK cells activated by interleukin (IL)-2 could inhibit CD28-mediated conversion of CD4+CD25− T cells into CD4+CD25+ forkhead box P3 (FoxP3+) regulatory T cells using an IFN-γ-dependent pathway [50]. This prevented the development of CD4+CD25+ regulatory T cells and promoted immune responses. Furthermore, NKG2D expressed on NK cells can mediate the killing of activated CD4+CD25+ regulatory T cells expressing the NKG2D ligand ULBP1 [51]. This observation indicates a central role for NK/CD4+CD25+ regulatory T cell interaction in control of immune responses. Consequently, a derangement of immunological interactions that can occur between NK and CD4+CD25+ regulatory T cells could contribute to the progression of β cell autoimmunity. To test this suggestive hypothesis, the analysis of the phenotype and frequency of CD4+CD25+ regulatory T cells in LADA patients is ongoing and will be reported elsewhere. Nevertheless, our observation of a decrease in NK cells in LADA is in accordance with previous data obtained in recent-onset T1D patients [27], suggesting that these cells may contribute to autoimmunity in both LADA and T1D.
In conclusion, the present study highlights an important deviation in the cellular arm of the immune system in patients diagnosed with LADA who have not progressed into insulin-dependent diabetes, and suggests that NK cell alterations may contribute to the development of autoimmune diabetes by affecting peripheral tolerance.
Acknowledgments
We thank Jeanette Arvastsson for valuable technical assistance. This work has been supported by grants from the Swedish Research Council, the Juvenile Diabetes Research Foundation, the Marianne and Marcus Wallenberg Foundation, the Knut and Alice Wallenberg Foundation and UMAS funds.
Disclosure
None.
References
- 1.McDevitt HO, Unanue ER. Autoimmune diabetes mellitus – much progress, but many challenges. Adv Immunol. 2008;100:1–12. doi: 10.1016/S0065-2776(08)00801-8. [DOI] [PubMed] [Google Scholar]
- 2.Tuomi T, Groop LC, Zimmet PZ, Rowley MJ, Knowles W, Mackay IR. Antibodies to glutamic acid decarboxylase reveal latent autoimmune diabetes mellitus in adults with a non-insulin-dependent onset of disease. Diabetes. 1993;42:359–62. doi: 10.2337/diab.42.2.359. [DOI] [PubMed] [Google Scholar]
- 3.Irvine WJ, McCallum CJ, Gray RS, Duncan LJ. Clinical and pathogenic significance of pancreatic-islet-cell antibodies in diabetics treated with oral hypoglycaemic agents. Lancet. 1977;1:1025–7. doi: 10.1016/s0140-6736(77)91258-2. [DOI] [PubMed] [Google Scholar]
- 4.Groop LC, Bottazzo GF, Doniach D. Islet cell antibodies identify latent type I diabetes in patients aged 35–75 years at diagnosis. Diabetes. 1986;35:237–41. doi: 10.2337/diab.35.2.237. [DOI] [PubMed] [Google Scholar]
- 5.Hagopian WA, Karlsen AE, Gottsater A, et al. Quantitative assay using recombinant human islet glutamic acid decarboxylase (GAD65) shows that 64K autoantibody positivity at onset predicts diabetes type. J Clin Invest. 1993;91:368–74. doi: 10.1172/JCI116195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zimmet PZ, Tuomi T, Mackay IR, et al. Latent autoimmune diabetes mellitus in adults (LADA): the role of antibodies to glutamic acid decarboxylase in diagnosis and prediction of insulin dependency. Diabet Med. 1994;11:299–303. doi: 10.1111/j.1464-5491.1994.tb00275.x. [DOI] [PubMed] [Google Scholar]
- 7.Niskanen LK, Tuomi T, Karjalainen J, Groop LC, Uusitupa MI. GAD antibodies in NIDDM. Ten-year follow-up from the diagnosis. Diabetes Care. 1995;18:1557–65. doi: 10.2337/diacare.18.12.1557. [DOI] [PubMed] [Google Scholar]
- 8.Turner R, Stratton I, Horton V, et al. UKPDS 25: autoantibodies to islet-cell cytoplasm and glutamic acid decarboxylase for prediction of insulin requirement in type 2 diabetes. UK Prospective Diabetes Study Group. Lancet. 1997;350:1288–93. doi: 10.1016/s0140-6736(97)03062-6. [DOI] [PubMed] [Google Scholar]
- 9.Tuomi T, Carlsson A, Li H, et al. Clinical and genetic characteristics of type 2 diabetes with and without GAD antibodies. Diabetes. 1999;48:150–7. doi: 10.2337/diabetes.48.1.150. [DOI] [PubMed] [Google Scholar]
- 10.Carlsson A, Sundkvist G, Groop L, Tuomi T. Insulin and glucagon secretion in patients with slowly progressing autoimmune diabetes (LADA) J Clin Endocrinol Metab. 2000;85:76–80. doi: 10.1210/jcem.85.1.6228. [DOI] [PubMed] [Google Scholar]
- 11.Colucci F, Caligiuri MA, Di Santo JP. What does it take to make a natural killer? Nat Rev Immunol. 2003;3:413–25. doi: 10.1038/nri1088. [DOI] [PubMed] [Google Scholar]
- 12.Baxter AG, Smyth MJ. The role of NK cells in autoimmune disease. Autoimmunity. 2002;35:1–14. doi: 10.1080/08916930290005864. [DOI] [PubMed] [Google Scholar]
- 13.Johansson S, Berg L, Hall H, Hoglund P. NK cells: elusive players in autoimmunity. Trends Immunol. 2005;26:613–18. doi: 10.1016/j.it.2005.08.008. [DOI] [PubMed] [Google Scholar]
- 14.Lanier LL. Activating and inhibitory NK cell receptors. Adv Exp Med Biol. 1998;452:13–18. doi: 10.1007/978-1-4615-5355-7_2. [DOI] [PubMed] [Google Scholar]
- 15.Long EO. Regulation of immune responses through inhibitory receptors. Annu Rev Immunol. 1999;17:875–904. doi: 10.1146/annurev.immunol.17.1.875. [DOI] [PubMed] [Google Scholar]
- 16.Yokoyama WM, Plougastel BF. Immune functions encoded by the natural killer gene complex. Nat Rev Immunol. 2003;3:304–16. doi: 10.1038/nri1055. [DOI] [PubMed] [Google Scholar]
- 17.Parham P. MHC class I molecules and KIRs in human history, health and survival. Nat Rev Immunol. 2005;5:201–14. doi: 10.1038/nri1570. [DOI] [PubMed] [Google Scholar]
- 18.Lopez-Larrea C, Suarez-Alvarez B, Lopez-Soto A, Lopez-Vazquez A, Gonzalez S. The NKG2D receptor: sensing stressed cells. Trends Mol Med. 2008;14:179–89. doi: 10.1016/j.molmed.2008.02.004. [DOI] [PubMed] [Google Scholar]
- 19.Zhang C, Zhang J, Tian ZG. The regulatory effect of natural killer cells: do ‘NK-reg cells’ exist? Cell Mol Immunol. 2006;3:241–54. [PubMed] [Google Scholar]
- 20.Negishi K, Waldeck N, Chandy G, et al. Natural killer cell and islet killer cell activities in type 1 (insulin-dependent) diabetes. Diabetologia. 1986;29:352–7. doi: 10.1007/BF00903343. [DOI] [PubMed] [Google Scholar]
- 21.Nair MP, Lewis EW, Schwartz SA. Immunoregulatory dysfunctions in type I diabetes: natural and antibody-dependent cellular cytotoxic activities. J Clin Immunol. 1986;6:363–72. doi: 10.1007/BF00915375. [DOI] [PubMed] [Google Scholar]
- 22.Hussain MJ, Alviggi L, Millward BA, Leslie RD, Pyke DA, Vergani D. Evidence that the reduced number of natural killer cells in type 1 (insulin-dependent) diabetes may be genetically determined. Diabetologia. 1987;30:907–11. doi: 10.1007/BF00295872. [DOI] [PubMed] [Google Scholar]
- 23.Herold KC, Huen A, Gould L, Traisman H, Rubenstein AH. Alterations in lymphocyte subpopulations in type 1 (insulin-dependent) diabetes mellitus: exploration of possible mechanisms and relationships to autoimmune phenomena. Diabetologia. 1984;27(Suppl.):102–5. doi: 10.1007/BF00275660. [DOI] [PubMed] [Google Scholar]
- 24.Scheinin T, Maenpaa J, Kontiainen S. Immune responses to insulin and lymphocyte subclasses at diagnosis of insulin-dependent diabetes and one year later. Immunobiology. 1990;180:431–40. doi: 10.1016/s0171-2985(11)80304-9. [DOI] [PubMed] [Google Scholar]
- 25.Wilson RG, Anderson J, Shenton BK, White MD, Taylor RM, Proud G. Natural killer cells in insulin dependent diabetes mellitus. BMJ (Clin Res Ed) 1986;293:244. doi: 10.1136/bmj.293.6541.244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lorini R, Moretta A, Valtorta A, et al. Cytotoxic activity in children with insulin-dependent diabetes mellitus. Diabetes Res Clin Pract. 1994;23:37–42. doi: 10.1016/0168-8227(94)90125-2. [DOI] [PubMed] [Google Scholar]
- 27.Rodacki M, Svoren B, Butty V, et al. Altered natural killer cells in type 1 diabetic patients. Diabetes. 2007;56:177–85. doi: 10.2337/db06-0493. [DOI] [PubMed] [Google Scholar]
- 28.Schranz DB, Bekris L, Landin-Olsson M, et al. A simple and rapid microSepharose assay for GAD65 and ICA512 autoantibodies in diabetes. Diabetes Incidence Study in Sweden (DISS) J Immunol Methods. 1998;213:87–97. doi: 10.1016/s0022-1759(98)00025-8. [DOI] [PubMed] [Google Scholar]
- 29.Grubin CE, Daniels T, Toivola B, et al. A novel radioligand binding assay to determine diagnostic accuracy of isoform-specific glutamic acid decarboxylase antibodies in childhood IDDM. Diabetologia. 1994;37:344–50. doi: 10.1007/BF00408469. [DOI] [PubMed] [Google Scholar]
- 30.Ilonen J, Reijonen H, Herva E, et al. Rapid HLA-DQB1 genotyping for four alleles in the assessment of risk for IDDM in the Finnish population. The Childhood Diabetes in Finland (DiMe) Study Group. Diabetes Care. 1996;19:795–800. doi: 10.2337/diacare.19.8.795. [DOI] [PubMed] [Google Scholar]
- 31.Sjoroos M, Iitia A, Ilonen J, Reijonen H, Lovgren T. Triple-label hybridization assay for type-1 diabetes-related HLA alleles. Biotechniques. 1995;18:870–7. [PubMed] [Google Scholar]
- 32.Steffens-Nakken HM, Zwart G, van den Bergh FA. Validation of allele-specific polymerase chain reaction for DNA typing of HLA-B27. Clin Chem. 1995;41:687–92. [PubMed] [Google Scholar]
- 33.Gumperz JE, Litwin V, Phillips JH, Lanier LL, Parham P. The Bw4 public epitope of HLA-B molecules confers reactivity with natural killer cell clones that express NKB1, a putative HLA receptor. J Exp Med. 1995;181:1133–44. doi: 10.1084/jem.181.3.1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Baecher-Allan C, Brown JA, Freeman GJ, Hafler DA. CD4+CD25high regulatory cells in human peripheral blood. J Immunol. 2001;167:1245–53. doi: 10.4049/jimmunol.167.3.1245. [DOI] [PubMed] [Google Scholar]
- 35.Horwitz DA, Gray JD, Ohtsuka K, Hirokawa M, Takahashi T. The immunoregulatory effects of NK cells: the role of TGF-beta and implications for autoimmunity. Immunol Today. 1997;18:538–42. doi: 10.1016/s0167-5699(97)01149-3. [DOI] [PubMed] [Google Scholar]
- 36.Kos FJ. Regulation of adaptive immunity by natural killer cells. Immunol Res. 1998;17:303–12. doi: 10.1007/BF02786453. [DOI] [PubMed] [Google Scholar]
- 37.Ogasawara K, Hamerman JA, Hsin H, et al. Impairment of NK cell function by NKG2D modulation in NOD mice. Immunity. 2003;18:41–51. doi: 10.1016/s1074-7613(02)00505-8. [DOI] [PubMed] [Google Scholar]
- 38.Litwin V, Gumperz J, Parham P, Phillips JH, Lanier LL. NKB1: a natural killer cell receptor involved in the recognition of polymorphic HLA-B molecules. J Exp Med. 1994;180:537–43. doi: 10.1084/jem.180.2.537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gumperz JE, Parham P. The enigma of the natural killer cell. Nature. 1995;378:245–8. doi: 10.1038/378245a0. [DOI] [PubMed] [Google Scholar]
- 40.Terme M, Chaput N, Combadiere B, et al. Regulatory T cells control dendritic cell/NK cell cross-talk in lymph nodes at the steady state by inhibiting CD4+ self-reactive T cells. J Immunol. 2008;180:4679–86. doi: 10.4049/jimmunol.180.7.4679. [DOI] [PubMed] [Google Scholar]
- 41.Giroux M, Yurchenko E, St-Pierre J, et al. T regulatory cells control numbers of NK cells and CD8alpha+ immature dendritic cells in the lymph node paracortex. J Immunol. 2007;179:4492–502. doi: 10.4049/jimmunol.179.7.4492. [DOI] [PubMed] [Google Scholar]
- 42.Zimmer J, Andres E, Hentges F. NK cells and Treg cells: a fascinating dance cheek to cheek. Eur J Immunol. 2008;38:2942–5. doi: 10.1002/eji.200838813. [DOI] [PubMed] [Google Scholar]
- 43.Ghiringhelli F, Menard C, Terme M, et al. CD4+CD25+ regulatory T cells inhibit natural killer cell functions in a transforming growth factor-beta-dependent manner. J Exp Med. 2005;202:1075–85. doi: 10.1084/jem.20051511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Smyth MJ, Teng MW, Swann J, et al. CD4+CD25+ T regulatory cells suppress NK cell-mediated immunotherapy of cancer. J Immunol. 2006;176:1582–7. doi: 10.4049/jimmunol.176.3.1582. [DOI] [PubMed] [Google Scholar]
- 45.Barao I, Hanash AM, Hallett W, et al. Suppression of natural killer cell-mediated bone marrow cell rejection by CD4+CD25+ regulatory T cells. Proc Natl Acad Sci USA. 2006;103:5460–5. doi: 10.1073/pnas.0509249103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Trzonkowski P, Szmit E, Mysliwska J, et al. CD4+CD25+ T regulatory cells inhibit cytotoxic activity of T CD8+ and NK lymphocytes in the direct cell-to-cell interaction. Clin Immunol. 2004;112:258–67. doi: 10.1016/j.clim.2004.04.003. [DOI] [PubMed] [Google Scholar]
- 47.Romagnani C, Della Chiesa M, Kohler S, et al. Activation of human NK cells by plasmacytoid dendritic cells and its modulation by CD4+ T helper cells and CD4+ CD25hi T regulatory cells. Eur J Immunol. 2005;35:2452–8. doi: 10.1002/eji.200526069. [DOI] [PubMed] [Google Scholar]
- 48.Nishikawa H, Kato T, Tawara I, et al. Accelerated chemically induced tumor development mediated by CD4+CD25+ regulatory T cells in wild-type hosts. Proc Natl Acad Sci USA. 2005;102:9253–7. doi: 10.1073/pnas.0503852102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vivier E, Tomasello E, Baratin M, et al. Functions of natural killer cells. Nat Immunol. 2008;9:503–10. doi: 10.1038/ni1582. [DOI] [PubMed] [Google Scholar]
- 50.Brillard E, Pallandre JR, Chalmers D, et al. Natural killer cells prevent CD28-mediated Foxp3 transcription in CD4+CD25- T lymphocytes. Exp Hematol. 2007;35:416–25. doi: 10.1016/j.exphem.2006.12.004. [DOI] [PubMed] [Google Scholar]
- 51.Roy S, Barnes PF, Garg A, et al. NK cells lyse T regulatory cells that expand in response to an intracellular pathogen. J Immunol. 2008;180:1729–36. doi: 10.4049/jimmunol.180.3.1729. [DOI] [PubMed] [Google Scholar]