

Abstract
Motivation: Gene expression patterns can be useful in understanding the structural organization of the brain and the regulatory logic that governs its myriad cell types. A particularly rich source of spatial expression data is the Allen Brain Atlas (ABA), a comprehensive genome-wide in situ hybridization study of the adult mouse brain. Here, we present an open-source program, ALLENMINER, that searches the ABA for genes that are expressed, enriched, patterned or graded in a user-specified region of interest.
Results: Regionally enriched genes identified by ALLENMINER accurately reflect the in situ data (95–99% concordance with manual curation) and compare with regional microarray studies as expected from previous comparisons (61–80% concordance). We demonstrate the utility of ALLENMINER by identifying genes that exhibit patterned expression in the caudoputamen and neocortex. We discuss general characteristics of gene expression in the mouse brain and the potential application of ALLENMINER to design strategies for specific genetic access to brain regions and cell types.
Availability: ALLENMINER is freely available on the Internet at http://research.janelia.org/davis/allenminer.
Contact: davisf@janelia.hhmi.org
Supplementary information: Supplementary data are available at Bioinformatics online.
1 INTRODUCTION
The mouse brain is a complex tissue containing many neuronal and non-neuronal cell types organized in intricate 3D structures. Defining the functional roles of these cell types in the context of their higher order arrangements is an important challenge in neuroscience. Neuronal cell types have been traditionally defined by cell morphology, electrophysiological properties and cell surface markers. Recent studies suggest that genomic transcriptome measurement is also a feasible route to defining functional cell types (Sugino et al., 2006). Besides its utility for classifying cell types, genomics also provides a bridge to genetic strategies to specifically target individual neuronal cell types for optogenetic or pharmacological perturbation (Luo et al., 2008). A variety of gene expression technologies including microarrays (Cahoy et al., 2008; Oldham et al., 2008; Su et al., 2004; Sugino et al., 2006), serial analysis of gene expression (SAGE) (Khattra et al., 2007), bacterial artificial chromosome (BAC) transgenics (Gong et al., 2003) and in situ hybridization (ISH) (Lein et al., 2007) has been used to characterize the expression profiles of brain regions and specific neuronal cell types.
The ISH data collected in the Allen Brain Atlas (ABA) offers the most spatially resolved (∼300 μm) description of gene expression in the adult mouse brain available to date (Lein et al., 2007; Ng et al., 2007). The ABA contains sagittal in situ images for ∼20 000 genes, coronal images for a subset of ∼4000 genes and 3D registration of these images by projection onto a reference atlas (Ng et al., 2007). The registered expression data can be accessed in a number of ways including interactive visualization (BrainExplorer; Lau et al., 2008), a summary description of expression levels in 17 anatomical brain regions, a tool (NeuroBLAST) to identify genes with expression patterns that are spatially similar to that of a query gene, a tool (AGEA) that defines a series of spatial regions based only on gene expression similarity and identifies genes that are enriched in these regions, and manually curated lists of genes that are enriched in 75 ‘fine structures’, such as small brain nuclei and layers of cortex.
The ABA expression data can answer a wide range of neurobiological questions. However, these questions often surpass the currently available query mechanisms. For example, we wished to identify genes that were differentially expressed between the medial and lateral caudoputamen, as behavioral studies have shown that these regions play distinct roles in learning (Corbit and Janak, 2007; Grahn et al., 2008). To enable a range of flexible query mechanisms, we have developed an open-source program, ALLENMINER, that provides an independent high-level interface to 3D ABA data. ALLENMINER offers several unique features including the ability to define custom regions of interest, search for genes that are graded or patterned in regions of interest, and view 3D ABA data on platforms where the BrainExplorer is not available, such as GNU/Linux, by conversion to the Protein Data Bank (PDB) format. The search mechanisms are flexible so that ALLENMINER is generally applicable to expression analyses of the adult mouse brain. For example, enrichment queries can identify regional markers that may be useful in developing transgenic tools to study particular brain regions. Similarly, gradient and patterning queries identify regional expression heterogeneities that may represent molecular correlates of functional and physiological differences within these regions.
We first describe the search strategy implemented in ALLENMINER. We next assess its accuracy by comparing query results to (i) manually curated and (ii) microarray identified lists of regionally enriched genes. We then apply ALLENMINER to identify genes with patterned expression in the caudoputamen and neocortex. Finally, we discuss general characteristics of gene expression in the mouse brain and discuss ALLENMINER's utility for designing genetic strategies to access restricted brain regions and cell types.
2 RESULTS
2.1 Identification of regionally enriched genes
The user specifies a region of interest (ROI) by either (i) describing the edges of a cuboid (x, y, z minimum and maximum); or (ii) listing the voxels in the integer coordinate system used by the ABA BrainExplorer (Lau et al., 2008); or by (iii) listing individual or boolean combinations of ABA Reference Atlas brain regions (Fig. 1). An example of this last form of ROI definition is ‘CTX=on,HPF=off,OLF=off’, which selects all voxels in the cortex (CTX) that are not also part of the hippocampal formation (HPF) or olfactory area (OLF). The examples presented in this article use ROIs based on atlas regions, either as individual regions, combinations of regions or sub-regions.
Fig. 1.

ALLENMINER logic. The program begins with a user-specified ROI, optionally partitions it along an axis, and then quantifies expression across the XPR files in the ABA. If multiple ROI have been defined, for example by partitioning, patterning and gradient scores may also be computed.
ALLENMINER processes the ABA 3D expression files (.XPR files), which contain the registration results of the in situ series (Section 4). These files contain the 3D coordinates of voxels that correspond to in situ image pixels with detectable expression. The expression in each voxel is quantified by a series of measures, including the estimated number of expressing cells, cell diameter, grid area spanned by expression and total expression level.
We developed a score to quantify the enrichment of expression in the ROI. ALLENMINER iterates through each 3D expression file and quantifies the expression level, specificity and enrichment in the user-specified ROI (roi_list run mode). The program can compute the expression level in the ROI [expr(roi)] as a sum or per-voxel average of any of the voxel expression measures, or as the number of component voxels with detectable expression. In the analyses presented here, the ROI expression level is defined as the sum of expression levels called for each component voxel. Specificity is computed as the fraction of a gene's total brain expression that occurs in the ROI (Equation 1).
| (1) |
Enrichment is computed as the specificity normalized for the size (number of voxels) of the ROI relative to the whole brain (Equation 2).
| (2) |
In the case of atlas region queries, the total levels [expr(total) and size(total)] are summed over the left-hemisphere ‘Brain’ structure defined by the ABA; otherwise, the total levels are summed over the entire brain. Atlas region queries are accelerated by using a precomputed file containing expression statistics for all genes in all atlas regions (fast_query run mode; 5 min on a single 2.0 GHz Intel Xeon processor). A similar indexing strategy for non-atlas ROI uses a uniform 3D gridding of the atlas to restrict searches to XPR files with expression in the grid sections corresponding to the ROI. For comparison, a non-indexed search completes in 5–10 min when run in parallel on fifty 3.0 GHz Intel Xeon processors (∼4.5 h on a single CPU). As file access dominates the runtime, it is not significantly affected by the specification of multiple ROI.
To demonstrate an ALLENMINER query, we searched for genes enriched in the ventromedial hypothalamus (VMH) compared with the rest of the hypothalamus (HY). As expected, the results suggest that most genes are expressed in the VMH at levels similar to the rest of the HY, although a spectrum is observed that ranges from VMH-depletion (low-enrichment score) to VMH-enrichment (high-enrichment score; Fig. 2). Examples of genes that are enriched in the VMH include Fez family zinc finger 1 (Fezf1; Fig. 2c) and Patched-2 (Ptchd2; Fig. 2d).
Fig. 2.

ALLENMINER identified genes enriched in the VMH. (a) The VMH (purple) is shown in the context of the HY (orange mesh) in the left hemisphere (grey) (figure produced by PyMol, http://pymol.org). (b) Distribution of VMH versus HY enrichment score (Equation 2). In situ images, the 3D registered expression in the HY (inset), and ALLENMINER enrichment scores are shown for two genes that are VMH enriched: (c) Fezf1 and (d) Ptchd2. The positions of the HY (orange outline) and VMH (blue outline) are depicted on the in situ images. The 3D registered expression (inset, yellow circles) in the HY (red) is shown along with the highlighted VMH nuclei (green) (figure produced by BrainExplorer; Lau et al., 2008).
2.2 Comparison to regionally enriched genes manually curated from the ABA
Ideally, we would assess the ALLENMINER enrichment score with a large and completely correct benchmark set of regional expressors (positives) and non-expressors (negatives). However, such a gold standard set does not exist, because every experimental method suffers from unique kinds of false positives and negatives. Here, we take two complementary approaches to benchmark the ALLENMINER enrichment score. First, we compare the results with manually curated lists of genes that are regionally enriched. These lists are extremely accurate, but are limited to ∼100 genes per region due to the human effort required for their creation. For this reason, we additionally benchmark ALLENMINER against the results of genome-wide regional microarray studies. These datasets are more comprehensive, but suffer from experimental errors that are not systematically corrected by human intervention. The results of these two independent approaches to benchmark ALLENMINER, across a total of 18 regions characterized in three studies, give us confidence in the accuracy of ALLENMINER.
We first compared ALLENMINER with lists of the 100 genes manually curated from the ABA in situ data to be the most enriched in 12 brain regions: cerebellum, cortex, hippocampus, HY, midbrain, medulla, olfactory bulb, pallidum, pons, retrohippocampal region, striatum and thalamus [Supplementary Table 3 in Lein et al. (2007)]. This comparison assessed how the ALLENMINER enrichment score performs relative to manual curation using the same expression data. Receiver operator characteristic (ROC) analysis was performed to characterize the ability of the score to distinguish between genes curated as enriched (positive set) and the remaining ABA genes (negative set). The optimal score threshold was defined as that which gave rise to the point on the ROC curve closest to the perfect discriminator (true positive rate=1, false positive rate=0). In cases were multiple expression files were available for a gene, the highest enrichment score was used.
The areas under the ROC curves (AUC) demonstrate a 95–99% concordance between ALLENMINER results and the manually curated lists (Table 1; Supplementary Fig. 1). Restricting the ROC analysis to coronal in situ data degraded concordance to 80–96% (data not shown). This reduction in performance is not surprising, since the additional sagittal data overcomes errors in the coronal image series. This comparison suggests that ALLENMINER enrichment queries return accurate results expected from the in situ data.
Table 1.
Comparison of ALLENMINER results to published sets of regionally enriched genes
| Benchmark set | Num | ROC | Optimal |
||
|---|---|---|---|---|---|
| genes | AUC | Score | TPR | FPR | |
| Manually curated ABA listsa | |||||
| Cerebellum | 99 | 0.977 | 3.553 | 0.970 | 0.050 |
| Cortex | 98 | 0.979 | 1.715 | 0.969 | 0.063 |
| Hippocampus | 98 | 0.961 | 3.515 | 0.908 | 0.072 |
| HY | 95 | 0.977 | 1.530 | 0.968 | 0.070 |
| Midbrain | 95 | 0.951 | 1.352 | 0.926 | 0.081 |
| Medulla | 95 | 0.953 | 2.460 | 0.958 | 0.112 |
| OLF | 99 | 0.953 | 2.489 | 0.949 | 0.111 |
| Pallidum | 98 | 0.965 | 1.299 | 0.949 | 0.088 |
| Pons | 97 | 0.951 | 1.580 | 0.969 | 0.103 |
| Retrohippocampal region | 99 | 0.958 | 1.819 | 0.929 | 0.089 |
| Striatum | 97 | 0.991 | 2.514 | 0.959 | 0.019 |
| Thalamus | 99 | 0.982 | 2.553 | 0.960 | 0.045 |
| Published microarray results | |||||
| Amygdalab | 20 | 0.606 | 1.478 | 0.600 | 0.348 |
| Cerebellum | 112 | 0.679 | 1.037 | 0.652 | 0.369 |
| Hippocampus | 53 | 0.699 | 1.441 | 0.660 | 0.368 |
| Olfactory bulb | 68 | 0.735 | 1.371 | 0.647 | 0.275 |
| Periaqueductal gray | 47 | 0.679 | 0.609 | 0.617 | 0.344 |
| VMHc | 37 | 0.792 | 1.412 | 0.703 | 0.185 |
ROC analysis quantified the accuracy of the ALLENMINER enrichment score in discriminating regionally enriched genes, as defined by manual curation or microarray analysis, from the remaining ABA genes. The ‘optimal’ true positive (TPR) and false positive rates (FPR) for the score threshold closest to the perfect classifier (TPR = 1, FPR = 0), along with the AUC, are described for each benchmark set.
aLein et al. (2007).
bZirlinger et al. (2001).
cKurrasch et al. (2007).
2.3 Comparison to regionally enriched genes identified by microarray studies
Next, we compared ALLENMINER with two published microarray studies of regional enrichment in the mouse brain (Kurrasch et al., 2007; Zirlinger et al., 2001). This comparison characterizes not only ALLENMINER accuracy, but also the similarity of regional enrichment detection by the two expression technologies. As the technologies suffer from different kinds of errors, we expected poorer concordance than the previous comparison to manually curated in situ data, although we still expected general agreement.
The microarray identified genes were mapped to the ABA through UCSC genome tables (Section 4). The first microarray dataset identified 299 ABA genes that were enriched at least 3.5-fold in the amygdala (AMY), cerebellum (CB), hippocampus (HIP), olfactory bulb (MOB) or periaqueductal gray (PAG) of 3-week-old female mice (Zirlinger et al., 2001). The second study identified 37 ABA genes that were at least 2.2-fold enriched in the fetal VMH compared with the rest of the HY (Kurrasch et al., 2007). ALLENMINER enrichment queries were performed using the corresponding atlas regions (AMY, CB, HIP, MOB, PAG; VMH versus HY). The fetal VMH microarray study was chosen because the results were exceptionally well-curated by hand and in several cases were verified by ISH. Although, in general, gene expression can change across developmental stages, this particular comparison of fetal microarray and adult ABA data on the VMH is valid because neurons have already migrated and formed a mature and functional VMH cluster by the time the animal is born (Kurrasch et al., 2007; McClellan et al., 2006). The high concordance we observe in our comparison further confirms the highly similar gene expression profiles in fetal and adult VMH.
ROC analysis demonstrated a 61–80% concordance between ALLENMINER results and the microarray data (Table 1). This agrees well with a published genome-wide comparison of ABA and microarray expression data in the GNF (Su et al., 2004) and Terragenomics (Zapala et al., 2005) compendia of mouse tissue expression data that found an absence/presence call agreement of 58–72% (Lee et al., 2008). Surprisingly, the adult ALLENMINER results exhibited better concordance with the fetal VMH study than with the adult brain regions analyzed in the Zirlinger study (Zirlinger et al., 2001).
2.4 False negatives
Visual inspection of in situ images for the 12 microarray identified genes that exhibited an ALLENMINER VMH-versus-HY enrichment score of <1.5, the optimal threshold as identified by ROC analysis (Table 1), suggested four reasons for these apparent false negatives (Supplementary Table 1 and Supplementary Fig. 2). First, several genes exhibited VMH expression below the threshold required by the ABA pipeline to call many expressing pixels, and therefore had no registered VMH voxels (Kcnj5, Mst1r, Nmbr, Ntn2l, Sema3a; Supplementary Fig. 2a). Second, tissue damage, bubbles and other in situ artifacts produced several false negatives (Mst1r, Nmbr; Supplementary Fig. 2b).
Third, the VMH was often very sparsely sampled by in situ sections or even skipped entirely (Supplementary Fig. 2c). In two image series (A2bp1, Satb2), a gene appeared to express strongly in the VMH, but the sagittal slice was 3D registered onto the edge of the atlas VMH definition rather than the VMH itself. However, it is not clear from the images if there was indeed VMH expression, or rather expression in an adjacent structure, such as the tubercular nuclei that lies ventral to the VMH.
Finally, it is possible that several of the apparent false negatives are actually true negatives caused by variation of gene expression across development. For instance, Vgll2, the most enriched gene in the fetal VMH (8.2-fold), exhibited no detectable adult expression (Supplementary Fig. 2d). This agrees with a published in situ study that found a dramatic reduction in expression from the significant enrichment observed at birth (developmental stage P0) to subtle expression at stage P7, and complete abolition at stage P21 (Kurrasch et al., 2007). Although, it is difficult to clearly distinguish true from false negatives, expression was also not evident on the in situ images for B3gnt3, B530002L08 and Card14. Similarly, Titf1 expression appears to be strong throughout the HY and not particularly enriched in the VMH.
2.5 False positives
Visual inspection of in situ images for genes identified by ALLENMINER, but not by microarray, to be VMH-enriched (18.5% estimated false positive rate; Table 1) suggested several potential causes of false positives (Supplementary Fig. 3). The most prevalent cause appears to be in situ image artifacts that range from small particulate aggregates registered as single voxels (e.g. Fbxl7; Supplementary Fig. 3a) to fluid bubble artifacts registered as a curve of expressing voxels (e.g. Gna15; Supplementary Fig. 2b). Particularly in the absence of expression in the surrounding tissue, even a small number of artifactual voxels registered in the ROI produces a high-enrichment score. Imposing a minimum threshold on the number of expressing voxels or the expression level in the ROI may minimize the effects of these artifacts. However, this strategy is problematic for small nuclei, since a small number of artifactual voxels is difficult to distinguish from true low-level expression, especially given the sparse sampling in small ROI. For example, Cholecystokinin B receptor (Cckbr; 11 VMH voxels, enrichment score = 12.53), Malic enzyme 2 (Me2; 7 voxels, enrichment score = 18.34) and calcium binding protein 39-like (Cab39l; 4 VMH voxels, enrichment score = 21.46) all express in few voxels yet appear to be truly enriched on the in situ images (data not shown). In other instances, a folded tissue edge (e.g. Gnal; enrichment score = 11.88; Supplementary Fig. 2c) or tissue debris lying in the ROI (e.g. Tctex1d1; enrichment score = 16.09; Supplementary Fig. 2d) is registered as expression.
Finally, a portion of the apparent false positives are likely true positives that are either differentially expressed across development, or were missed by the microarray experiment. It is not feasible to quantify the fraction of the ‘false positives’ that are true expressors, but many genes appear to be enriched in the ABA, although not identified by the microarray study. For example, Cylindromatosis (Cyld; score = 27.38; Supplementary Fig. 2e) and Patched-2 (Ptchd2, score = 23.65; Fig. 2d) are clearly enriched in the ABA, but neither is identified by the fetal microarray study.
2.6 Identification of genes with patterned or graded expression
To identify genes that are expressed non-uniformly or in a graded fashion across an ROI, for example, the mediolateral axis of the caudoputamen; (i) partitions are first defined along an axis (rostrocaudal, dorsoventral or mediolateral; roi_partition run mode); (ii) expression levels are quantified in each partition (roi_list mode); and (iii) these results are used to compute cross-axis gradient and regional patterning scores (calc_entropy mode). The gradient score describes how much of the total expression change from partition to partition occurs in the same numerical direction (Equation 3). A gene that expresses in ever-decreasing amounts across an axis receives a gradient score of −1, while ever-increasing amounts receives a gradient score of 1.
 |
(3) |
The regional patterning score (RPS) is a Shannon entropy-like score that sums the enrichment observed in each ROI partition, weighted by the occupancy of each partition (Equation 4). The RPS score decreases as a gene expresses more uniformly across the defined ROI partitions, and can become slightly negative if a gene is depleted in an ROI partition.
| (4) |
We performed regional patterning searches in the neocortex and caudoputamen to identify genes that were differentially expressed across these structures. Five rostrocaudal (R-C) bins were defined across the neocortex, as defined by the ABA cortex region after removal of voxels from the HPF and OLF. Similarly, five R-C and five mediolateral (M-L) bins were defined across the caudoputamen. The M-L bins were constructed to adjust to the width of the caudoputamen, which is greatest in the rostral and smallest in the caudal aspects. Gradient (Equation 3) and regional patterning scores (Equation 4) were computed for each gene to determine the gradient and non-uniformity of expression across the axis of interest, respectively.
The query results demonstrate that, although, most genes are uniformly expressed across the neocortex R-C and caudoputamen R-C and M-L axes, there exists a continuum of patterns, as quantified by the regional patterning and gradient scores (Figs 3 and 4; Supplementary Fig. 4). The distributions of gradient scores in all three structures exhibit a shared tri-modal shape that appears to arise for common reasons (Figs 3a and 4a; Supplementary Fig. 4a). The main peak over a gradient score of 0 is due to non-expressors as well as uniform expressors. In addition to genes that are actually expressed in perfect negative and positive gradients, the two smaller peaks at the extrema of −1 and +1 often correspond to expression calls that are from neighboring structures, but are registered on the edge of the first and last partitions, respectively. For example, the medial caudoputamen peak corresponds to expression in a neighboring ventricle, and the lateral peak to expression in the cortex, both of which are often registered on the edges of the caudoputamen (Supplementary Fig. 4a).
Fig. 3.

Patterned expression in the neocortex. (a) Gradient (Equation 3) and (b) patterning (Equation 4) scores were computed across five rostrocaudal neocortex bins (rainbow) for all ABA 3D expression profiles. In situ images and 3D neocortex-registered expression (inset) are shown for two genes with graded expression: (c) rostrally enriched multiple epidermal growth factor-like domains 11 (Megf11; gradient = −0.96; ABA image 71669548) and (d) caudally enriched S100 calcium-binding protein A3 (S100a3; gradient = 0.92; ABA image 73609669).
Fig. 4.

Patterned expression in the caudoputamen. (a) Gradient (Equation 3) and (b) patterning (Equation 4) scores were computed across five rostrocaudal caudoputamen bins (rainbow) for all ABA 3D expression profiles. In situ images and caudoputamen voxel calls (inset) are shown for two genes with graded expression: (c) rostrally enriched sodium–calcium exchanger NCX3 isoform (Slc8a3; gradient = −0.84; ABA image 72577486) and (d) caudally enriched Ras-specific guanine nucleotide-releasing factor 2 (Rasgrf2; gradient = 0.94; ABA image 69791544).
The patterning scores also suggest that most genes express uniformly across the queried structures. These distributions exhibit a similar shape for all three queries, with a peak at a value of 0, corresponding to uniform expression and long tails towards higher values, corresponding to patterned expression (Figs 3b and 4b; Supplementary Fig. 4b). The distribution is smoother for the neocortex than for the caudoputamen. There are two likely explanations for this observation. First, the edge effect observed in the gradient distributions of the caudoputamen queries, especially across the M-L axis, are larger in magnitude than that of the neocortex R-C query. These outliers correspond to the small peaks of high-patterning scores observed in both the R-C and M-L caudoputamen queries (Fig. 4b; Supplementary Fig. 4b). Second, because the caudoputamen is smaller than the neocortex, it is sampled less thoroughly by the in situ slices. This reduced sampling leads to a more significant discrete binning effect across the ROI partitions, resulting in more fluctuation in the patterning score distribution. Although it is not possible to systematically quantify the prevalence of edge and sampling artifacts, or their exact effects on the patterning scores, we found these kinds of errors in ∼5% (n = 2 of 37) of the ABA datasets for the genes identified in the VMH microarray study (Supplementary Table 1).
The gradient distributions may reflect the underlying cellular architecture of the structures. For example, the neocortical R-C gradient distribution is biased towards rostral enrichment (Fig. 3a), while the caudoputamen R-C gradient is more symmetric and slightly biased towards caudal enrichment (Fig. 4a). This may correspond to the known variation of cell density and morphology along the neocortical R-C axis (Benavides-Piccione et al., 2006; Elston, 2003; Schuz and Palm, 1989). In contrast, although there are regional neurochemical differences in the caudoputamen, it is often considered a structurally and cytoarchitectonically homogeneous tissue (Glynn and Ahmad, 2002; Nieuwenhuys, 1998), and variability in cellular composition does not exist along its R-C axis.
2.7 Genome-wide distribution of expression patterns and the diversity of neuronal cell types
Beyond queries for particular regions and patterns of interest, the comprehensive nature of the ABA also allows unique insight into the diversity of expression patterns and, potentially, of neuronal cell types that exist in the mouse brain. The specificity of gene expression in the brain was quantified for each gene by computing the RPS across all 210 atlas regions (Fig. 5a). The initial ABA report used the number of expressing voxels as a measure of specificity, suggesting that most genes expressed in specific patterns (Lein et al., 2007). Here, we find that although most genes express relatively sparsely (Fig. 5b), they do so in patterns that are not specific with respect to anatomical regions (Fig. 5a). Comparing the number of expressing voxels to the RPS demonstrates a range of patterning for genes with comparable voxel occupancy, suggesting that the RPS score is a useful additional dimension to quantify gene expression specificity (Fig. 5b). For example, Rgs5 (Fig. 5c and e) and Gabra4 (Fig. 5d and f) have detectable expression in a similar number of voxels, however the former is expressed in a dispersed fashion across several brain regions, while the latter is enriched in the thalamus, caudoputamen and cortex.
Fig. 5.
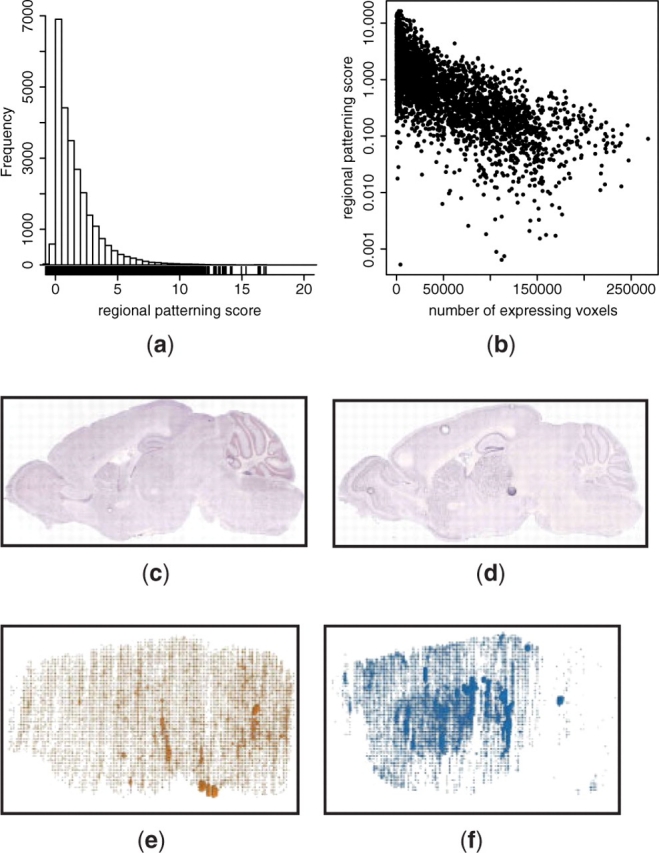
Distribution of gene expression specificity in the mouse brain. (a) Distribution of RPS (Equation 4) of all ABA 3D expression profiles (sagittal and coronal). The maximum observed RPS is 39 (data not shown). (b) Comparison of the RPS to the number of voxels with detectable expression in the coronal subset of ABA 3D expression profiles. (c and e) Regulator of G-protein signaling 5 (Rgs5; 38,501 voxels; RPS=0.14; ABA image 71548305) is distributed across the brain. (d and f) gamma-aminobutryic acid (GABA-A) receptor, subunit alpha 4 (Gabra4; 26,983 voxels; RPS=2.8; ABA image 75251433) is more specifically expressed, with enrichment in the thalamus, caudoputamen and cortex. Although both genes express in a similar number of voxels, they exhibit significant differences in expression specificity.
This analysis provides a molecular basis for discussions of neuronal cell type diversity. The spatial expression data in the ABA are most obviously useful for identifying region-specific genes. However, cell types in the brain are not necessarily, or even predominantly, region-specific. If we assume a parsimonious genomic definition of cell types, it is likely that cell types are defined by a single or small number of genes more often than by several genes. Previous microarray analysis of 12 neuronal sub-populations suggest that this is a reasonable assumption (Sugino et al., 2006). Sugino et al. found that although functional cell types often exhibit differential expression of a large battery of genes, as few as five genes are necessary to uniquely identify a particular cell type (Sugino et al., 2006). Following from this assumption and the observed distributions of voxel occupancy and regional patterning (Fig. 5a and b), it is likely that most cell types are dispersed throughout the brain (e.g. GABA-ergic neurons) rather than restricted to a brain region (e.g. cerebellar Purkinje cells). Although the definition of a cell type is still debated and its catalog in the brain far from complete (Masland, 2004; Yuste, 2005), it is likely that there is a spectrum of regional specificity and a hierarchical resolution to its definition that spans these two extreme scenarios.
3 DISCUSSION
We presented a tool for 3D searches of expression data in the ABA (Fig. 1), demonstrated its accuracy and utility in identifying genes with specific (Fig. 2, Table 1; Supplementary Fig. 1–3 and Table 1) and patterned expression (Figs 3 and 4; Supplementary Fig. 4). We will now discuss potential improvements to ALLENMINER performance and the utility of ALLENMINER for designing genetic strategies to access restricted neuronal sub-types.
3.1 Future improvements
The comparison of VMH-enriched genes identified by ALLENMINER to those identified by microarray analysis suggested properties of the registered ABA data, such as in situ artifacts, that affect ALLENMINER queries (Supplementary Figs 2 and 3). Although these artifacts are best handled at the image analysis stage of the ABA processing and registration pipeline, some of them, such as the particulate and bubble artifacts, can likely be identified in the 3D registrations and the corresponding voxels flagged for ALLENMINER analysis.
The distributions of expression gradient in the neocortex and caudoputamen highlighted the edge effect that results from expression in neighboring tissues being registered within the ROI (Fig. 3a and 4a; Supplementary Fig. 4a). The precise registration of regional boundaries is a difficult problem that requires single voxel accuracy. However, the impact of these errors can likely be minimized by peeling away the outer layer of voxels from the ROI definition. This would reduce the contribution of voxels that have been mis-registered from neighboring tissues, such as the cortical expression registered as caudoputamen, to the gradient and patterning scores.
The biological and technical non-uniformities in the expression data confound precise quantitative analysis of the spatial expression patterns. For example, the rostral bias in the gradient distribution of the neocortex (Fig. 3a) may reflect rostrocaudal variation in cell morphology, size and density (Benavides-Piccione et al., 2006; Elston, 2003; Schuz and Palm, 1989). Similarly, the in situ sampling of tissue slices across the brain series often varies between genes. ALLENMINER currently does not correct for these non-uniformities. One possible way to correct for cell density is to normalize the observed expression levels or gradient scores using a ubiquitous or pan-neuronal marker, for example, that captured by the ABA fractional area metric (Lein et al., 2007). However, this strategy would not correct for the tissue sampling differences across partitions, as the sampling often varies from in situ series to series. Similarly, sparse tissue sampling is not currently distinguished from very patterned expression. Non-uniform sampling might be corrected for by computing a normalization factor proportional to the number of tissue slices that correspond to each ROI partition. Although, it is not possible to count the exact number of slices in an ROI using only the 3D XPR data, ALLENMINER provides an estimate of this number based on the reported ABA sectioning intervals of 100 μm in the coronal and 200 μm in the sagittal series (run mode estimate_roi_sampling). Expression levels quantified in ROI with a low-estimated sampling are likely to be less reliable than in more frequently sampled ROI.
An application programming interface (API) to the ABA has recently been released that provides standardized access to the ABA data (http://mouse.brain-map.org/api/index.html), facilitating the development of custom software. ALLENMINER development began before the API was released, and so we developed parsers for the ABA data to which access was required, such as the 3D expression profiles. However, the search algorithm we describe here is equally applicable to implementation with the API.
Currently, ALLENMINER only uses the 3D registered expression data in the ABA. However, the original 2D in situ images may contain additional information, such as the shape of the hybridization signal in each soma, nucleus or dendrite. This high-resolution information about the shape and distribution of the in situ signal may be useful in the classification of neuronal sub-types and can possibly be extracted by applying image analysis techniques to the raw in situ images.
3.2 Designing neurogenetic strategies
ALLENMINER may also be useful in identifying genes or combinations of genes that express in a specific region or cell type of the mouse brain. Although, the resolution of the registered in situ data precludes precise definition of cell types, the regionally patterned genes that are identified by ALLENMINER include ion channels, solute carriers and neurotransmitter machinery that can provide a link between genomic definitions and traditional electrophysiological and neurochemical definitions of cell type. Further integration of ABA expression data with other publicly available databases, such as the list of currently available transgenic mouse lines (MGD; Bult et al., 2008) may highlight brain regions that can be specifically addressed by intersectional strategies (Luo et al., 2008).
The ABA is a rich collection of spatially resolved 3D gene expression patterns in the mouse brain. ALLENMINER enables an unlimited repertoire of ‘virtual’ experiments using the ABA, and we expect this to facilitate the generation and validation of neurobiological hypotheses.
4 MATERIALS AND METHODS
4.1 Obtaining ABA data
The list of genes available from the ABA was downloaded (http://mouse.brain-map.org/pdf/allGenes.csv). For each gene, an XML file was downloaded that describes the available in situ series and the riboprobes used to generate them. The image series identifiers were extracted from the XML files and the corresponding 3D expression (.XPR) files were downloaded (25 636 XPR files corresponding to 21 201 genes). The 3D reference atlas that describes the brain structures to which each voxel belongs, as well as the neuroanatomical hierarchy of these structures, was retrieved from the Annotation100.sva and BrainStructures.csv files, respectively, of the publicly available API package.
The XPR files were linked to the knownGenes in the mouse mm9 UCSC genome database through riboprobe identifiers and the knownToAllenBrain table [Karolchik et al. (2008); http://genome.ucsc.edu]. In addition, knownGene, knownToLocusLink, knownToRefSeq, all_est, estOrientInfo, gbCdnaInfo, all_mrna, kgXref, tables were obtained from the UCSC mm9 mouse genome database. The in situ images presented here can be accessed using their ABA image identifiers. For example, image 73429295 is available at http://mouse.brain-map.org/viewImage.do?imageId=73429295.
4.2 Mapping microarray probes to ABA XPR files
The microarray study of amygdala, cerebellum, hippocampus, olfactory bulb and periaqueductal gray in 3-week-old female mice identified 452 probes enriched at least 3.5-fold in one of the five regions (Zirlinger et al., 2001). Enriched Affymetrix probe set IDs (Mu11Ka, Mu11Kb, Mu19Ka, Mu19Kb, Mu19Kc platforms) were mapped to mm9 UCSC knownGenes using the Entrez Gene ID from the Affymetrix annotation file (http://www.affymetrix.com) and UCSC genome browser KnownToLocusLink table. In cases where the Entrez GeneID was not available in the annotation file or the knownToLocusLink table, the aligned chromosome location (available for Mu11k, but not Mu19k) was used to search the knownGene table for the gene that contained the probe in the correct orientation. In total, 410 probes were mapped to 377 knownGenes, 299 of which mapped to the ABA (via knownToAllenBrain).
The microarray study of the fetal VMH identified 50 genes that are the most enriched compared to the rest of the HY (Kurrasch et al., 2007). Of these genes, 46 mapped to UCSC knownGenes table and 37 also mapped to the ABA through the UCSC knownToAllenBrain table.
4.3 Availability
ALLENMINER is implemented in Perl except for a single routine written in Python. It is freely available under the GPL v3 license at http://research.janelia.org/davis/allenminer. The results of enrichment analysis for all ABA reference brain regions, as well as all the results presented in this article, are also available for download.
Supplementary Material
ACKNOWLEDGEMENTS
We are grateful to the Allen Brain Institute, in particular, Susan Sunkin and Mike Hawrylycz, for making the Allen Brain Atlas expression data and API publicly available. We thank Lee Henry, Alla Karpova and Albert Lee (HHMI) for useful discussion and Goran Ceric for managing Janelia's high-performance computing resources.
Conflict of Interest: none declared.
REFERENCES
- Benavides-Piccione R, et al. Dendritic size of pyramidal neurons differs among mouse cortical regions. Cereb. Cortex. 2006;16:990–1001. doi: 10.1093/cercor/bhj041. [DOI] [PubMed] [Google Scholar]
- Bult CJ, et al. The mouse genome database (MGD): mouse biology and model systems. Nucleic Acids Res. 2008;36:D724–D728. doi: 10.1093/nar/gkm961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahoy JD, et al. A transcriptome database for astrocytes, neurons, and oligodendrocytes: a new resource for understanding brain development and function. J. Neurosci. 2008;28:264–278. doi: 10.1523/JNEUROSCI.4178-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbit LH, Janak PH. Inactivation of the lateral but not medial dorsal striatum eliminates the excitatory impact of pavlovian stimuli on instrumental responding. J. Neurosci. 2007;27:13977–13981. doi: 10.1523/JNEUROSCI.4097-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elston GN. Cortex, cognition and the cell: new insights into the pyramidal neuron and prefrontal function. Cereb. Cortex. 2003;13:1124–1138. doi: 10.1093/cercor/bhg093. [DOI] [PubMed] [Google Scholar]
- Glynn G, Ahmad SO. Three-dimensional electrophysiological topography of the rat corticostriatal system. J. Comp. Physiol. A Neuroethol. Sens. Neural Behav. Physiol. 2002;188:695–703. doi: 10.1007/s00359-002-0341-7. [DOI] [PubMed] [Google Scholar]
- Gong S, et al. A gene expression atlas of the central nervous system based on bacterial artificial chromosomes. Nature. 2003;425:917–925. doi: 10.1038/nature02033. [DOI] [PubMed] [Google Scholar]
- Grahn JA, et al. The cognitive functions of the caudate nucleus. Prog. Neurobiol. 2008;86:141–155. doi: 10.1016/j.pneurobio.2008.09.004. [DOI] [PubMed] [Google Scholar]
- Karolchik D, et al. The UCSC Genome Browser Database: 2008 update. Nucleic Acids Res. 2008;36:D773–D779. doi: 10.1093/nar/gkm966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khattra J, et al. Large-scale production of SAGE libraries from microdissected tissues, flow-sorted cells, and cell lines. Genome Res. 2007;17:108–116. doi: 10.1101/gr.5488207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurrasch DM, et al. The neonatal ventromedial hypothalamus transcriptome reveals novel markers with spatially distinct patterning. J. Neurosci. 2007;27:13624–13634. doi: 10.1523/JNEUROSCI.2858-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau C, et al. Exploration and visualization of gene expression with neuroanatomy in the adult mouse brain. BMC Bioinformatics. 2008;9:153. doi: 10.1186/1471-2105-9-153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CK, et al. Quantitative methods for genome-scale analysis of in situ hybridization and correlation with microarray data. Genome Biol. 2008;9:R23. doi: 10.1186/gb-2008-9-1-r23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lein ES, et al. Genome-wide atlas of gene expression in the adult mouse brain. Nature. 2007;445:168–176. doi: 10.1038/nature05453. [DOI] [PubMed] [Google Scholar]
- Luo L, et al. Genetic dissection of neural circuits. Neuron. 2008;57:634–660. doi: 10.1016/j.neuron.2008.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masland RH. Neuronal cell types. Curr. Biol. 2004;14:R497–R500. doi: 10.1016/j.cub.2004.06.035. [DOI] [PubMed] [Google Scholar]
- McClellan KM, et al. Development of the ventromedial nucleus of the hypothalamus. Front. Neuroendocrinol. 2006;27:193–209. doi: 10.1016/j.yfrne.2006.02.002. [DOI] [PubMed] [Google Scholar]
- Ng L, et al. Neuroinformatics for genome-wide 3D gene expression mapping in the mouse brain. IEEE/ACM Trans. Comput. Biol. Bioinform. 2007;4:382–393. doi: 10.1109/tcbb.2007.1035. [DOI] [PubMed] [Google Scholar]
- Nieuwenhuys R. The Central Nervous System of Vertebrates. Berlin: Springer; 1998. [Google Scholar]
- Oldham MC, et al. Functional organization of the transcriptome in human brain. Nat. Neurosci. 2008;11:1271–1282. doi: 10.1038/nn.2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuz A, Palm G. Density of neurons and synapses in the cerebral cortex of the mouse. J. Comp. Neurol. 1989;286:442–455. doi: 10.1002/cne.902860404. [DOI] [PubMed] [Google Scholar]
- Su AI, et al. A gene atlas of the mouse and human protein-encoding transcriptomes. Proc. Natl Acad. Sci. USA. 2004;101:6062–6067. doi: 10.1073/pnas.0400782101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugino K, et al. Molecular taxonomy of major neuronal classes in the adult mouse forebrain. Nat. Neurosci. 2006;9:99–107. doi: 10.1038/nn1618. [DOI] [PubMed] [Google Scholar]
- Yuste R. Origin and classification of neocortical interneurons. Neuron. 2005;48:524–527. doi: 10.1016/j.neuron.2005.11.012. [DOI] [PubMed] [Google Scholar]
- Zapala MA, et al. Adult mouse brain gene expression patterns bear an embryologic imprint. Proc. Natl Acad. Sci. USA. 2005;102:10357–10362. doi: 10.1073/pnas.0503357102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zirlinger M, et al. Amygdala-enriched genes identified by microarray technology are restricted to specific amygdaloid subnuclei. Proc. Natl Acad. Sci. USA. 2001;98:5270–5275. doi: 10.1073/pnas.091094698. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.