

Abstract
Since its discovery in 1997, phosphatase and tensin homologue deleted on chromosome 10 (PTEN) has become one of the most important molecules in tumor biology. Mutations, deletions or dysregulation of PTEN is found in many human tumors. Recent studies have extended the reach of PTEN to include diabetes and neurological diseases such as Parkinson’s and Autism. In this review, we summarize the traditionally characterized function of PTEN as the lipid phosphatase that dephosphorylates PI-3,4,5-P3, and several other newly discovered functions. The inhibition of the phosphatidylinositol-3-kinase (PI3K)/AKT signaling pathway may account for most of PTEN’s tumor suppressing function. However, other growth inhibiting functions of PTEN may not involve this pathway. PTEN can also inhibit growth through its protein phosphatase activity and in ways not related to its enzymatic activity at all. We survey the many functions and biochemical interactions of PTEN in cytoplasm, the nucleus and throughout the cell in this paper.
Keywords: PTEN, AKT, PI3K, Tumor, Nucleus
Signaling Network Facts.
PTEN possess both lipid and protein phosphatase activities
The lipid phosphatase activity of PTEN negatively regulate PI3K/AKT signaling
Nuclear PTEN may have effect independent of its enzymatic activities
PTEN is modified posttranslationally by phosphorylation and ubiquination. Phosphorylation and ubiquination affects its stability and cellular localization.
PTEN is the most deleted/mutated phosphatase found in human cancer.
Further insight into PTEN signaling can be found at http://ghr.nlm.nih.gov/gene=pten
Introduction
In 1997, three laboratories independently reported a tumor suppressor located on human chromosome 10q23. Sequence analysis demonstrated that this gene encodes a phosphatase with homology to tensin and auxilin. This gene was named PTEN for phosphatase and tensin homologue deleted on chromosome 10 (or MMAC1/TEP1) (B. Stiles, Groszer, Wang, Jiao, & Wu, 2004). Later studies demonstrated that PTEN is a negative regulator of a major cell growth and survival signaling pathway, namely the phosphatidylinositol-3-kinase (PI3K)/AKT signaling pathway (Downes et al., 2007; B. Stiles, Groszer et al., 2004). PTEN antagonizes the function of PI3K by dephosphorylating phosphatidylinositol-3,4.5-triphosphate (PI-3,4,5-P3) (Fig 1), a key product of PI3K responsible for the activation of downstream target molecules including AKT, an oncogenic protein (Downes et al., 2007). This function of PTEN has been the focus of many studies and is the foundation for the molecular mechanisms of PTEN as a tumor suppressor. Loss of PTEN has become an important marker for ensuring the sensitivity of chemotherapies targeted at PI3K/AKT signaling pathway (Neshat et al., 2001). Additionally, a role for PTEN that is independent of PI3K/AKT is currently being proposed and explored (Planchon, Waite, & Eng, 2008). Mouse models carrying deletion of Pten revealed that PTEN is indispensable for development (B. Stiles, Groszer et al., 2004; Suzuki, Nakano, Mak, & Sasaki, 2008). Studies performed with heterozygous Pten deletion mice and using the conditional deletion Cre-LoxP system have revealed much of the physiological function of PTEN (B. Stiles, Groszer et al., 2004; Suzuki et al., 2008). In this review, we will summarize the PTEN-controlled signaling pathways and the biological events involved in PTEN regulation.
Figure 1. Signaling network regulated by PTEN.
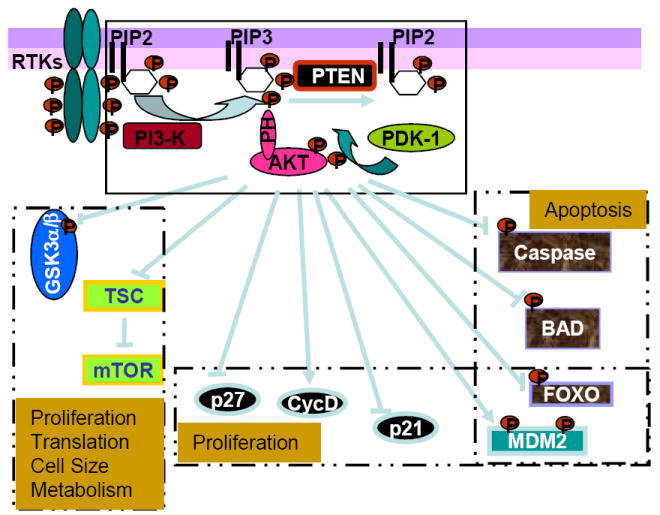
PTEN is the lipid phosphatase that negatively regulates the PI3K signaling pathway. PI3K is induced as a result of receptor binding and activation of receptor tyrosine kinase (RTK). PI3K phosphorylates the membrane lipid PIP2 to form PIP3. PTEN, the lipid phosphatase removes the phosphate and reduce PIP3 levels in the cell. By this action, PTEN blocks the receptor binding signal from propagating downstream. These downstream signals include a number of kinases that contain the pleckstrin homology (PH) domain such as PDK1 and AKT as well as others. Binding of PIP3 to the PH domain allows the exposure of residues on AKT that is critically needed for its activation. Binding of PIP3 to the PH domain also adds lipid moiety to the molecules and allows their association with membranes. The phosphorylation of AKT by PDK1 leads to its full activation. AKT, a proto-oncoprotein when activated acts as a central control for proliferation, cell survival, translation, cell size, and cell metabolism. AKT, being a serine/threonine kinase directly phosphorylates and regulates the molecules involved in these processes. Phosphorylation of GSK3α/β blocks their inhibitor activity on glycogen synthase and β-catenin. Phosphorylation of TSC2 blocks its ability to inhibit mTOR, thus allowing the downstream translational events to propagate. A prominent function of AKT on cell survival is mediate by its direct phosphorylation and negative regulation on caspases 3 and 9 as well as BAD, a proapoptosis factor. Phosphorylation of the forkhead transcription factor FOXO blocks the ligand dependent cell death as well as having an effect on cell proliferation. Phosphorylation of MDM2 by AKT leads to its cytoplasmic translocation and thus blocks its effects on p53 degradation. This is believed to be one of the mechanisms for PTEN and p53 to interact. AKT also has direct effect on cell cycle regulators like p21, p27 and cyclin D.
Structure and Functions of PTEN
The human PTEN gene encodes a 403 amino acid protein with two main domain structures: the catalytic phosphatase domain and the C2 domain (Das, Dixon, & Cho, 2003). The C2 domain and the short N-terminal sequence (also called PBD domain, phosphatidylinositol 2-phosphate binding domain) are crucial for the binding of PTEN to membrane lipids where its best characterized substrate PI-3,4,5-P3 is located. The c-terminal tail contains a PDZ binding motif and is thought to play a role in protein-protein interaction between PTEN and scaffolding proteins such as membrane associated guanylate kinase (MAGI) (Vazquez et al., 2001; Wu et al., 2000). The catalytic domain of PTEN is structurally similar to protein tyrosine phosphatases with few reported potential protein substrates including FAK and PTEN itself (Raftopoulou, Etienne-Manneville, Self, Nicholls, & Hall, 2004; Tamura et al., 1998). The discovery that PTEN dephosphorylates PI-3,4,5-P3, demonstrated for the first time that a lipid phosphatase may play an important role in cell growth and survival. Many studies using either primary tumors or cell lines have established PTEN to be the most deleted phosphatase and second most deleted gene next to P53 in human cancer.
The biological effects of PTEN are dominated by its ability to dephosphorylate the lipid substrate PI-3,4,5-P3 (Downes et al., 2007). PI-3,4,5-P3 is formed when PI3K is stimulated as a result of growth factors binding to their receptors coupled to PI3K. PI-3,4,5-P3 serves as a major signaling molecule for growth factor stimulation. The lipid phosphatase motif of PTEN leads to dephosphorylation of PI-3,4,5-P3 at the 3’ position (Downes et al., 2007). Through this action, PTEN inhibits the activation of downstream effector molecules including the serine/threonine kinase AKT and the protein kinase C (PKC). The activities of these effector molecules depend on the binding of PI-3,4,5-P3 to their pleckstrin homology domain for activation.
Because of its biological function as a lipid phosphatase, PTEN’s function has been traditionally characterized in the cytoplasm and at the plasma membrane. The best characterized biological function of PTEN is through its ability to negatively regulate the proto-oncogenic protein AKT (Fig 1). Activation of AKT leads to a number of cellular functions including cell growth, survival, cell migration and differentiation, cell and organ size control, metabolism and others (for detailed review, see (Manning & Cantley, 2007)). Direct phosphorylation of downstream targets by AKT mediates these cellular processes regulated by PTEN-PI3K-AKT signaling cascade. Proapoptotic factor BAD, caspases 3 & 9 are among the direct substrates of AKT that play significant roles in cell survival. Phosphorylation of these molecules by AKT renders them inactive and thus leads to cell survival phenotypes. Through phosphorylating forkhead transcriptional factors and inducing their binding to 14-3-3 proteins, AKT blocks their translocation to the nucleus where they can induce gene transcription regulating cell proliferation, survival and metabolic changes. Some cell cycle modulators such as p21, p27 and MDM2 are also directly regulated by AKT.
The PTEN regulated signalings cross talk with several other signaling pathways (Fig 2). Two substrates of AKT play key roles in this cross talk. GSK3α/β is phosphorylated by AKT on Serine 21/9 which inhibits their activity (Manning & Cantley, 2007). GSK3β, a key regulator in Wnt signaling, phosphorylates and targets β-catenin for ubiquitin-mediated degradation. The crosstalk between PTEN and Wnt signaling may underscore some of the effects of PTEN in the regulation of stem cell maintenance and G0-G1 cell cycle regulation (Backman et al., 2001; Groszer et al., 2001; Kwon et al., 2001; Yilmaz et al., 2006; Zhang et al., 2006). Another substrate of AKT, tuberous sclerosis complex 2 (TSC2) plays a key role in incorporating controls of metabolism, cell size with growth and proliferation (Leung & Robson, 2007). TSC 2, encoding tuberin is responsible for a rare multi-symptom genetic disease, tuberous sclerosis. The heterodimer of TSC1/2 is critical for suppressing mTOR function (Fig 1). TSC is inhibited by PI3K/AKT and upregulated by LKB-AMPK, a signaling cascade responding to energy levels (Y. Li, Corradetti, Inoki, & Guan, 2004). The placement of TSC2 in the PI3K signaling pathway explained some of the clinical similarities between tuberous sclerosis and Pten mutation predisposition syndromes such as Cowden’s disease. It also highlighted a possible involvement of metabolism in cell proliferation and survival.
Figure 2. Crosstalks between PTEN and other signaling networks.
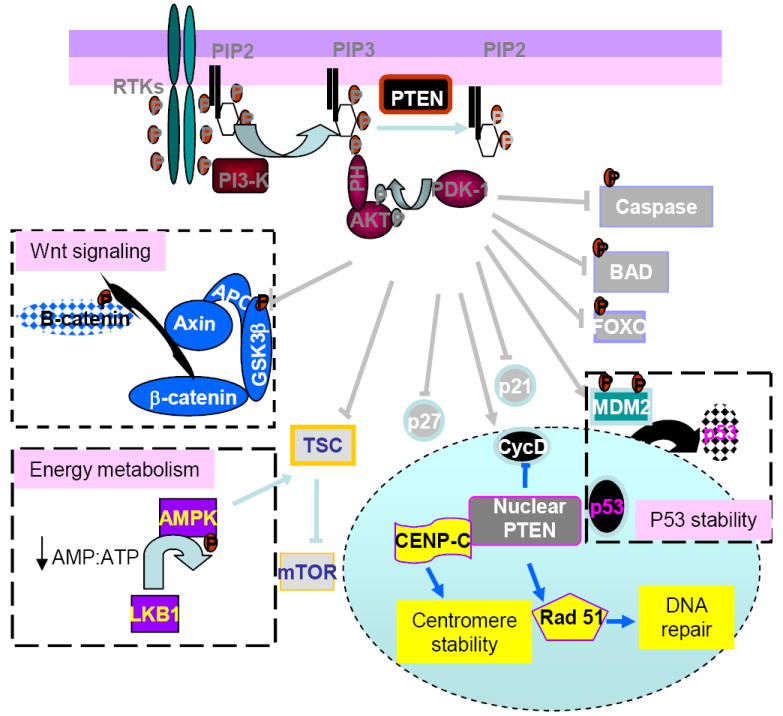
The PTEN regulated PI3K/AKT signaling cross talks with several other signaling pathways. These pathways include the Wnt signaling pathway, the energy signaling pathway, as well as p53 signaling. Through inhibiting GSK3β, activation of AKT leads to the stabilization of β-catenin, a critical factor in Wnt signaling. PI3K/AKT signal also cross talks with energy signaling pathway at TSC. Whereas AKT phophorylates and inhibit TSC, AMPK, a kinase activated in response to an increase in AMP:ATP ratio phosporylates and activates TSC. In addition to its function primarily through AKT in the cytoplasm, nuclear PTEN may have its unique functions. These include direct association with p53, CENP and regulation of rad 51 to control the later stages of the cell cycle and DNA repair mechanisms.
The majority of PTEN studies focus on the enzymatic function of PTEN and its role in antagonizing the PI3K/AKT signaling pathway. Recent evidence indicates that it may have effects beyond its ability to dephosphorylate PIP3. The ability of PTEN to directly dephosphorylate protein substrates is still being explored even though the identities of the substrates remain illusive. In earlier studies, PTEN was reported to be a protein exclusively located in the cytoplasm while more recent evidence shows that PTEN can be both cytoplasmic and nuclear (Planchon et al., 2008; B. Stiles, Wang et al., 2004; B. L. Stiles et al., 2006). Analysis of tumor cells vs. control cells also indicated that the nuclear PTEN plays an important role in inhibiting tumorigenesis (Planchon et al., 2008). A lysine mutation K289E in Cowden’s syndrome later led to the discovery of ubiquination as a mechanism controlling the shuttling between the cytoplasm and the nucleus (Chi et al., 1998). Upon mono ubiquination by its E3 ligase, PTEN is imported to the nucleus (Trotman et al., 2007). Deubiquination in the nucleus keeps it from reentering the cytoplasm where it is degraded. The nuclear PTEN is thought to play a role independent of its lipid phosphatase activity although existence of PI-3,4,5-P3 in the nucleus has been documented. In the nucleus, PTEN may promote p53 stability and transcriptional activity by directly associating with p53 (Tang & Eng, 2006). This is independent of the effect of PTEN on MDM2 through AKT activity (Freeman et al., 2003). On the other hand, recent studies also indicated that p53 may be induced to compensate for the loss of Pten in some situations(Chen et al., 2005). Nuclear PTEN is also capable of dephosphorylating MAPK and directly regulating cyclin D1 in addition to its effect through AKT (Y. H. Shen et al., 2006). Further evidence shows that PTEN physically interacts with the centromere specific binding protein C (CENP-C) to increase centromere stability as well as regulating DNA break repair through upregulation of Rad 51 transcription (W. H. Shen et al., 2007). Nuclear PTEN also regulates histone acetylation through interaction with acetylase PCAF and p300/CBP and may impose another level of regulation on p53 (A. G. Li et al., 2006; Okumura et al., 2006).
Regulation of PTEN
PTEN is regulated on several levels. PTEN RNA expression is regulated by several transcriptional factors (For detailed description and references, see(Tamguney & Stokoe, 2007). PTEN was first found to be a TGFβ-regulated gene (D. M. Li & Sun, 1997). In TGFβ sensitive cells, PTEN is readily down-regulated by TGFβ treatment. Other negative regulators of PTEN transcription include NFkB and c-JUN. The tumor suppressor p53 interacts with PTEN on several levels including transcription. p53 directly binds to the promoter of PTEN to regulate its transcription. In response to UV irradiation, PTEN is also induced to promote cell death under the transcriptional regulation of an early growth response gene EGR-1. When these mechanisms are lost, either p53, PTEN or EGR-1, cells fail to induce cell death in response to UV damage leading to tumor development. Another notable regulator of PTEN transcription is PPARγ. PPARγ is the orphan nuclear receptor responsible for the effect of the antidiabetic drugs of the thiazolidinedione class. This class of drugs binds to PPARγ as its ligand and induces its activation. The activated PPARγ controls the transcription of a number of genes involved in metabolism, rendering the drugs effective for diabetes. The fact that PPARγ activation also induces PTEN expression and inhibition of AKT leads to the hypothesis that these drugs may be used for cancer therapy as well. Other more recent discoveries includes Sprouty homologue (SPRY2), active transcriptional factor 2 (ATF2) and microRNAs such as miR 19 and miR 21(Meng et al., 2007).
PTEN protein has a relatively long half life and is speculated to be regulated by posttranslational modifications. Indeed, PTEN is modified by phosphorylation, acetylation, ubiquitination and oxidation (For detailed description and references, see (Gericke, Munson, & Ross, 2006)). These modifications contribute to PTEN’s stability, activity, subcellular localization and protein interactions. Two kinase candidates are reported to phosphorylate PTEN, casein kinase 2 (CK2) and GSK3β, both on the C-terminal tail. Phosphorylation of PTEN by CK2 on S370 and S385 leads to the stabilization of PTEN. The resultant effect on its catalytic activity is unclear. GSK3β phosphorylation on S362 and T366 may represent a negative feedback loop for PTEN/PI3K signaling pathway. The C2 domain of PTEN has multiple potential phosphorylation sites for modification. One of them, S380 is a candidate site associated with Gliomas in which PTEN mutation is high. RhoA associated kinase and PI3K p110δ subunits are thought to play roles in regulating PTEN through various phosphorylation sites including those residing at the C2 domain. Acetylation of PTEN occurs on two lysine residues 125 and 128 by the action of PCAF. These acetylations affect the substrate specificity of PTEN towards PI-3,4,5-P3. In the presence of hydrogen peroxide, PTEN inactivation is thought to be a result of the formation of a disulphide bond between Cys124 and Cys71. This inactivation is reversible and may account for the regulation of PTEN by environmental changes.
Poly-ubiquination is a common mechanism to tag proteins for targeted proteosome degradation. PTEN is also subjected to such proteosome mediated degradation. An E3 ubiquitin ligase for PTEN has been identified to be NEDD4-1 (neuron precursor cell expressed, developmentally downregulated-4-1) though disagreement is still being debated (Fouladkou et al., 2008; W. H. Shen et al., 2007; Trotman et al., 2007). Interestingly, NEDD4-1 appears to both enhance PTEN function and lead to its degradation. NEDD4-1 can both monoubiquitinate and polyubiquitinate PTEN through regulations that are yet to be determined. Monoubiquination of PTEN on lysine residues K13 and K289 is essential for its shuttling into the nucleus where additional growth inhibitory functions of PTEN independent of PI3K/AKT may occur. In the cytoplasm, polyubiquination of PTEN targets it to proteosome degradation also using NEDD4-1 as the E3 ubiquitin ligase. Thus, it appears, through regulating PTEN stability and nuclear shuttling, NEDD4-1 can have both tumor suppressive and oncogenic functions. How NEDD4-1 balances these two tasks and the mechanisms regulating the switch remains to be determined.
PTEN and human disease
PTEN was first discovered by analyzing a segment of chromosome 10 found to be associated with prostate cancer and gliomas(Eng, 2003). Since then, PTEN has been deemed one of the most important genes in human cancer. Approximately 30% of human cancer carry some kind of mutations/deletions or dysregulated PTEN expression, most notably gliomas, prostate and endometrium cancers. This astonishing figure marked PTEN as the second most deleted/mutated gene in human cancer next to p53 which is found to be mutated in 50% of human cancer. Germline deletion/mutation of PTEN is associated with several autosomal dominant tumor predisposition syndromes including Cowden’s disease (CS), Bannayan-Zonana syndrome (BZS) and Lhermitte-Duclos disease (LDD) (Eng, 2003). These patients often suffer from hamartomas in multiple organs with a tendency toward malignant transformation. In addition to hamartomas syndrome, a subset of the patients with CS and BZS also display macrocephaly (Buxbaum et al., 2007; Herman et al., 2007). This discovery led to further identification of PTEN as a gene associated with Autism where a high percentage of patients present macrocephaly as part of the symptoms (Butler et al., 2005). These analyses greatly expanded the role of PTEN to include non-neoplastic diseases. Neurological diseases such as Parkinson’s are associated with PTEN function through PINK 1 (PTEN induced kinase-1) and DJ-1 and the commonly speculated interaction between PTEN and oxidative damage (Gasser, 2007). Due to its role in regulating PI3K/AKT signaling, PTEN is thought to be a regulator in insulin signaling and thus may play a role in metabolic syndromes such as diabetes. Experimental evidence have demonstrated that when Pten is deleted, animals demonstrate insulin sensitivity phenotype (B. Stiles, Wang et al., 2004; B. L. Stiles et al., 2006). Cowden’s disease patients who are carriers of Pten mutation also presented with improved insulin sensitivity (Iida et al., 2000). Genetic evidence indicates that lack of one of the AKT isoforms, AKT2 may contribute to familial type II diabetes (George et al., 2004).
Taken as a whole, this evidence suggests that in addition to regulating cell growth and survival, PTEN is also a multifunction protein that has a wide spectrum of functions in many cellular processes. It is not clear how these processes may interact with each other and how one may modulate the balance to treat each of the pathological conditions resulted from malfunction of PTEN. Recent studies have reestablished the theory that metabolic changes observed with tumor cells are not side effects but major contributors to the progression of the malignancy. How tumor suppressors like PTEN may regulate this process to control the progression of the tumor growth is an area that needs further study. The mechanisms behind the regulation of oxidative damage and mitochondrial function by PTEN may provide clues to such an understanding.
Acknowledgments
We acknowledge Dr. Bryan Stiles for his help in editing this manuscript. Funding source: DK075928-01A1 and USC liver Center pilot grant.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Backman SA, Stambolic V, Suzuki A, Haight J, Elia A, Pretorius J, et al. Deletion of Pten in mouse brain causes seizures, ataxia and defects in soma size resembling Lhermitte-Duclos disease. Nat Genet. 2001;29(4):396–403. doi: 10.1038/ng782. [DOI] [PubMed] [Google Scholar]
- Butler MG, Dasouki MJ, Zhou XP, Talebizadeh Z, Brown M, Takahashi TN, et al. Subset of individuals with autism spectrum disorders and extreme macrocephaly associated with germline PTEN tumour suppressor gene mutations. J Med Genet. 2005;42(4):318–321. doi: 10.1136/jmg.2004.024646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buxbaum JD, Cai G, Chaste P, Nygren G, Goldsmith J, Reichert J, et al. Mutation screening of the PTEN gene in patients with autism spectrum disorders and macrocephaly. Am J Med Genet B Neuropsychiatr Genet. 2007;144B(4):484–491. doi: 10.1002/ajmg.b.30493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Trotman LC, Shaffer D, Lin HK, Dotan ZA, Niki M, et al. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature. 2005;436(7051):725–730. doi: 10.1038/nature03918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi SG, Kim HJ, Park BJ, Min HJ, Park JH, Kim YW, et al. Mutational abrogation of the PTEN/MMAC1 gene in gastrointestinal polyps in patients with Cowden disease. Gastroenterology. 1998;115(5):1084–1089. doi: 10.1016/s0016-5085(98)70078-2. [DOI] [PubMed] [Google Scholar]
- Das S, Dixon JE, Cho W. Membrane-binding and activation mechanism of PTEN. Proc Natl Acad Sci U S A. 2003;100(13):7491–7496. doi: 10.1073/pnas.0932835100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Downes CP, Ross S, Maccario H, Perera N, Davidson L, Leslie NR. Stimulation of PI 3-kinase signaling via inhibition of the tumor suppressor phosphatase, PTEN. Adv Enzyme Regul. 2007;47:184–194. doi: 10.1016/j.advenzreg.2006.12.018. [DOI] [PubMed] [Google Scholar]
- Eng C. PTEN: one gene, many syndromes. Hum Mutat. 2003;22(3):183–198. doi: 10.1002/humu.10257. [DOI] [PubMed] [Google Scholar]
- Fouladkou F, Landry T, Kawabe H, Neeb A, Lu C, Brose N, et al. The ubiquitin ligase Nedd4-1 is dispensable for the regulation of PTEN stability and localization. Proc Natl Acad Sci U S A. 2008;105(25):8585–8590. doi: 10.1073/pnas.0803233105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman DJ, Li AG, Wei G, Li HH, Kertesz N, Lesche R, et al. PTEN tumor suppressor regulates p53 protein levels and activity through phosphatase-dependent and -independent mechanisms. Cancer Cell. 2003;3(2):117–130. doi: 10.1016/s1535-6108(03)00021-7. [DOI] [PubMed] [Google Scholar]
- Gasser T. Update on the genetics of Parkinson’s disease. Mov Disord. 2007;22(Suppl 17):S343–350. doi: 10.1002/mds.21676. [DOI] [PubMed] [Google Scholar]
- George S, Rochford JJ, Wolfrum C, Gray SL, Schinner S, Wilson JC, et al. A family with severe insulin resistance and diabetes due to a mutation in AKT2. Science. 2004;304(5675):1325–1328. doi: 10.1126/science.1096706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gericke A, Munson M, Ross AH. Regulation of the PTEN phosphatase. Gene. 2006;374:1–9. doi: 10.1016/j.gene.2006.02.024. [DOI] [PubMed] [Google Scholar]
- Groszer M, Erickson R, Scripture-Adams DD, Lesche R, Trumpp A, Zack JA, et al. Negative regulation of neural stem/progenitor cell proliferation by the Pten tumor suppressor gene in vivo. Science. 2001;294(5549):2186–2189. doi: 10.1126/science.1065518. [DOI] [PubMed] [Google Scholar]
- Herman GE, Butter E, Enrile B, Pastore M, Prior TW, Sommer A. Increasing knowledge of PTEN germline mutations: Two additional patients with autism and macrocephaly. Am J Med Genet A. 2007;143(6):589–593. doi: 10.1002/ajmg.a.31619. [DOI] [PubMed] [Google Scholar]
- Iida S, Ono A, Sayama K, Hamaguchi T, Fujii H, Nakajima H, et al. Accelerated decline of blood glucose after intravenous glucose injection in a patient with Cowden disease having a heterozygous germline mutation of the PTEN/MMAC1 gene. Anticancer Res. 2000;20(3B):1901–1904. [PubMed] [Google Scholar]
- Kwon CH, Zhu X, Zhang J, Knoop LL, Tharp R, Smeyne RJ, et al. Pten regulates neuronal soma size: a mouse model of Lhermitte-Duclos disease. Nat Genet. 2001;29(4):404–411. doi: 10.1038/ng781. [DOI] [PubMed] [Google Scholar]
- Leung AK, Robson WL. Tuberous sclerosis complex: a review. J Pediatr Health Care. 2007;21(2):108–114. doi: 10.1016/j.pedhc.2006.05.004. [DOI] [PubMed] [Google Scholar]
- Li AG, Piluso LG, Cai X, Wei G, Sellers WR, Liu X. Mechanistic insights into maintenance of high p53 acetylation by PTEN. Mol Cell. 2006;23(4):575–587. doi: 10.1016/j.molcel.2006.06.028. [DOI] [PubMed] [Google Scholar]
- Li DM, Sun H. TEP1, encoded by a candidate tumor suppressor locus, is a novel protein tyrosine phosphatase regulated by transforming growth factor beta. Cancer Res. 1997;57(11):2124–2129. [PubMed] [Google Scholar]
- Li Y, Corradetti MN, Inoki K, Guan KL. TSC2: filling the GAP in the mTOR signaling pathway. Trends Biochem Sci. 2004;29(1):32–38. doi: 10.1016/j.tibs.2003.11.007. [DOI] [PubMed] [Google Scholar]
- Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129(7):1261–1274. doi: 10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng F, Henson R, Wehbe-Janek H, Ghoshal K, Jacob ST, Patel T. MicroRNA-21 regulates expression of the PTEN tumor suppressor gene in human hepatocellular cancer. Gastroenterology. 2007;133(2):647–658. doi: 10.1053/j.gastro.2007.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neshat MS, Mellinghoff IK, Tran C, Stiles B, Thomas G, Petersen R, et al. Enhanced sensitivity of PTEN-deficient tumors to inhibition of FRAP/mTOR. Proc Natl Acad Sci U S A. 2001;98(18):10314–10319. doi: 10.1073/pnas.171076798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okumura K, Mendoza M, Bachoo RM, DePinho RA, Cavenee WK, Furnari FB. PCAF modulates PTEN activity. J Biol Chem. 2006;281(36):26562–26568. doi: 10.1074/jbc.M605391200. [DOI] [PubMed] [Google Scholar]
- Planchon SM, Waite KA, Eng C. The nuclear affairs of PTEN. J Cell Sci. 2008;121(Pt 3):249–253. doi: 10.1242/jcs.022459. [DOI] [PubMed] [Google Scholar]
- Raftopoulou M, Etienne-Manneville S, Self A, Nicholls S, Hall A. Regulation of cell migration by the C2 domain of the tumor suppressor PTEN. Science. 2004;303(5661):1179–1181. doi: 10.1126/science.1092089. [DOI] [PubMed] [Google Scholar]
- Shen WH, Balajee AS, Wang J, Wu H, Eng C, Pandolfi PP, et al. Essential role for nuclear PTEN in maintaining chromosomal integrity. Cell. 2007;128(1):157–170. doi: 10.1016/j.cell.2006.11.042. [DOI] [PubMed] [Google Scholar]
- Shen YH, Zhang L, Gan Y, Wang X, Wang J, LeMaire SA, et al. Up-regulation of PTEN (phosphatase and tensin homolog deleted on chromosome ten) mediates p38 MAPK stress signal-induced inhibition of insulin signaling. A cross-talk between stress signaling and insulin signaling in resistin-treated human endothelial cells. J Biol Chem. 2006;281(12):7727–7736. doi: 10.1074/jbc.M511105200. [DOI] [PubMed] [Google Scholar]
- Stiles B, Groszer M, Wang S, Jiao J, Wu H. PTENless means more. Dev Biol. 2004;273(2):175–184. doi: 10.1016/j.ydbio.2004.06.008. [DOI] [PubMed] [Google Scholar]
- Stiles B, Wang Y, Stahl A, Bassilian S, Lee WP, Kim YJ, et al. Liver-specific deletion of negative regulator Pten results in fatty liver and insulin hypersensitivity [corrected] Proc Natl Acad Sci U S A. 2004;101(7):2082–2087. doi: 10.1073/pnas.0308617100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stiles BL, Kuralwalla-Martinez C, Guo W, Gregorian C, Wang Y, Tian J, et al. Selective deletion of Pten in pancreatic beta cells leads to increased islet mass and resistance to STZ-induced diabetes. Mol Cell Biol. 2006;26(7):2772–2781. doi: 10.1128/MCB.26.7.2772-2781.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki A, Nakano T, Mak TW, Sasaki T. Portrait of PTEN: messages from mutant mice. Cancer Sci. 2008;99(2):209–213. doi: 10.1111/j.1349-7006.2007.00670.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamguney T, Stokoe D. New insights into PTEN. J Cell Sci. 2007;120(Pt 23):4071–4079. doi: 10.1242/jcs.015230. [DOI] [PubMed] [Google Scholar]
- Tamura M, Gu J, Matsumoto K, Aota S, Parsons R, Yamada KM. Inhibition of cell migration, spreading, and focal adhesions by tumor suppressor PTEN. Science. 1998;280(5369):1614–1617. doi: 10.1126/science.280.5369.1614. [DOI] [PubMed] [Google Scholar]
- Tang Y, Eng C. PTEN autoregulates its expression by stabilization of p53 in a phosphatase-independent manner. Cancer Res. 2006;66(2):736–742. doi: 10.1158/0008-5472.CAN-05-1557. [DOI] [PubMed] [Google Scholar]
- Trotman LC, Wang X, Alimonti A, Chen Z, Teruya-Feldstein J, Yang H, et al. Ubiquitination regulates PTEN nuclear import and tumor suppression. Cell. 2007;128(1):141–156. doi: 10.1016/j.cell.2006.11.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vazquez F, Grossman SR, Takahashi Y, Rokas MV, Nakamura N, Sellers WR. Phosphorylation of the PTEN tail acts as an inhibitory switch by preventing its recruitment into a protein complex. J Biol Chem. 2001;276(52):48627–48630. doi: 10.1074/jbc.C100556200. [DOI] [PubMed] [Google Scholar]
- Wu Y, Dowbenko D, Spencer S, Laura R, Lee J, Gu Q, et al. Interaction of the tumor suppressor PTEN/MMAC with a PDZ domain of MAGI3, a novel membrane-associated guanylate kinase. J Biol Chem. 2000;275(28):21477–21485. doi: 10.1074/jbc.M909741199. [DOI] [PubMed] [Google Scholar]
- Yilmaz OH, Valdez R, Theisen BK, Guo W, Ferguson DO, Wu H, et al. Pten dependence distinguishes haematopoietic stem cells from leukaemia-initiating cells. Nature. 2006;441(7092):475–482. doi: 10.1038/nature04703. [DOI] [PubMed] [Google Scholar]
- Zhang J, Grindley JC, Yin T, Jayasinghe S, He XC, Ross JT, et al. PTEN maintains haematopoietic stem cells and acts in lineage choice and leukaemia prevention. Nature. 2006;441(7092):518–522. doi: 10.1038/nature04747. [DOI] [PubMed] [Google Scholar]