

Abstract
Alternative splicing is considered a major mechanism for creating multicellular diversity from a limited repertoire of genes. Here, we performed the first study of genetic variation controlling alternative splicing patterns by comprehensively identifying quantitative trait loci affecting the differential expression of transcript isoforms in a large recombinant inbred population of Caenorhabditis elegans, using a new generation of whole-genome very-high-density oligonucleotide microarrays. Using 60 experimental lines, we were able to detect 435 genes with substantial heritable variation, of which 36% were regulated at a distance (in trans). Nonetheless, we find only a very small number of examples of heritable variation in alternative splicing (22 transcripts), and most of these genes colocalize with the associated genomic loci. Our findings suggest that the regulatory mechanism of alternative splicing in C. elegans is robust toward genetic variation at the genome-wide scale, which is in striking contrast to earlier observations in humans.
ALTERNATIVE splicing of pre-mRNAs is part of gene regulation and a major mechanism for increasing the protein repertoire and the resulting phenotypic diversity. Recently, in individual cases variations in number and ratio of splice variants have also been found in Caenorhabditis elegans in different developmental stages (Barberan-Soler and Zahler 2008b), tissues (Kuroyanagi et al. 2007), and genotypes (Fischer et al. 2008). However, the smaller number of alternative splicing patterns (Kim et al. 2007) and their strong evolutionary conservation in C. elegans (Barberan-Soler and Zahler 2008a) have been interpreted as signifying a fundamental difference in the way that worms and vertebrates generate diversity from their genetic information. The relative rarity of alternative splicing and the high degree of stabilizing selection are seen as having parallels in the limited cellular complexity and highly conserved, rigid developmental programs (Zhao et al. 2008) in worms compared to humans. If this is a general trend, and not restricted to just individual cases of splicing, the conservation of splicing patterns should be reflected at the whole-genome level.
In this article we explore this question by extending the genetical genomics strategy (Jansen and Nap 2001) to the characterization of the genetic factors contributing to variations in alternative splicing in 60 C. elegans recombinant inbred line (RIL) strains. This powerful new strategy, also known as expression genetics (Schadt et al. 2003), has emerged in recent years as a versatile tool to study the genetic basis of gene expression by integrating transcriptomics and classical quantitative genetics (Mackay et al. 2009). In this approach, molecular profiling on a large population of densely genotyped individuals is used to map genomic loci that modulate gene expression. This leads to the identification of expression quantitative trait loci (eQTL), i.e., polymorphic genetic loci that cause heritable differences in mRNA concentration. Using high-resolution tiling microarrays we were able to extend this concept to the detection of genetic determinants of alternative splicing (as)QTL and to the detailed quantification of the genetic robustness of the alternative splicing machinery in C. elegans on a genome-wide scale.
MATERIALS AND METHODS
Worm samples, genotyping, and Affymetrix GeneChips:
We used C. elegans recombinant inbred lines that were generated from a cross of N2 and CB4856 and were genotyped by Li et al. (2006). Age-synchronized C. elegans was cultured at 24° and the total RNA was isolated from the late L3 stage using the Trizol method. The RNA was cleaned using the QIAGEN (Valencia, CA) RNeasy Micro RNA cleanup kit. Double-stranded cDNA synthesis was done with the Affymetrix GeneChip WT double-stranded cDNA synthesis kit. We cleaned the cDNA using the GeneChip Sample Cleanup Module also from Affymetrix. For fragmentation and labeling, the GeneChip WT double-stranded DNA terminal labeling kit was used. The concentrations of RNA and cDNA were measured with a Nanodrop. After the fragmentation we determined the fragment size on a Nusieve 3:1 agarose gel. mRNA was hybridized to Affymetrix 1.0 C. elegans tiling arrays (2.9 million probes on each array) and the hybridization was done by ServiceXS (Leiden, The Netherlands). Since polymorphisms in the probe region can lead to spurious local eQTL (Alberts et al. 2007), 80,903 probes with known SNP (including predicted SNP; WS195 release) were removed for subsequent analysis. Each probe is annotated as exonic, intronic, or intergenic, when the entire probe of 25 bp falls in one of the three regions, respectively. Probes spanning exon–intron boundaries are labeled as boundary probes.
Data analysis:
Preprocessing of raw data:
The raw gene expression data from 60 microarrays (one RIL per array) were taken base-2 log transformed and then quantile normalized. Subsequently, the normalized intensity data were corrected for batch effects using the linear model
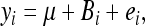 |
where yi is the gene's intensity on the ith microarray (i = 1, …, 60), μ is the mean, Bi is the batch effect defined as the date of hybridization and measurement and treated as a categorical variable, and ei is the residual error.
Differential expression between genotypes (eQTL):
We used a robust and powerful statistical approach to associate microarray probe intensity and genotype data in the face of widely different hybridization properties of individual probes. Instead of computing significance of a statistical test, we evaluated a nonparametric effect size [Cliff's Δ (Cliff 1996)] for all 3 million probes at each genomic marker. For each probe on the array we compute the eQTL effect size using Cliff's nonparametric Δ-statistic
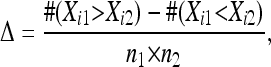 |
where n1 and n2 are the numbers of carriers of the N2 and the CB4856 allele, and #(Xi1 > Xi2) is the number of possible pairwise comparisons where the expression level of gene i in an N2 carrier is larger than in a CB4856 carrier. The genotype information of the 60 RILs was previously described (Li et al. 2006). For an individual probe, a value of Δ = 0.45 corresponds to a P-value = 0.001 in a Wilcoxon rank sum test (del Rosal et al. 2003).
As several positions in the genome show a strongly imbalanced genotype ratio (i.e., the number of RILs carrying the N2 allele is far larger than the number of RILs carrying the CB4856 allele at a particular locus), the corresponding threshold (Wilcoxon's U-value) for each marker at significance level P = 0.001 was obtained first, taking the locus-specific imbalance into account. Then, these values were converted into the corresponding threshold for the effect size (Cliff's D) on the basis of D = 2U/(n1n2) − 1 (del Rosal et al. 2003). The threshold of distorted genome regions is expected to be larger than that of balanced marker positions. These marker-dependent thresholds were applied in further analysis.
Summarizing the eQTL effect for exons:
To increase the robustness of the procedure, the median effect size of probes within each exon was taken as representing the expression QTL effect size of this exon for each genomic marker. Subsequently, the eQTL profile at the marker with maximal summarized eQTL effect was obtained. To achieve a reliable estimate of eQTL effect size, only exons covered by more than three probes were considered here. Transcripts with a summarized eQTL effect larger than the threshold for at least one exon were declared as having a significant eQTL and were used for further analysis.
Classification of eQTL:
There are 435 transcripts with a significant eQTL in total. They were examined in greater detail and manually classified as shown in Figure 1. By visualizing the intensity level and eQTL size of the entire transcript, we first classified transcripts as having a consistent eQTL if all annotated exons show the same eQTL pattern at a threshold of Δ = 0.45 and there is no additional eQTL signal in the presumed intron regions. In addition, there are eQTL patterns that indicate the need for revised gene definitions (but no evidence for difference in splicing), which can be subdivided into five subcategories: (1) new exons (at least two consecutive intron probes showing a similar expression level and eQTL size as the exon probes of the gene), (2) new introns (at least two consecutive exon probes showing a clear decrease of expression level and eQTL size compared to the other exon probes of the gene), (3) intron inclusions (all probes corresponding to an intron showing the same expression and eQTL size as the exon probes), (4) exon extensions (at least two intron probes next to an exon showing similar expression levels and eQTL size as the adjacent exon), and (5) intron extensions (at least the first or the last two exon probes showing a decrease of expression level and eQTL size compared to the other exon probes of the gene). Most interestingly, there are also eQTL patterns that indicate potential heritable differences in splicing, i.e., genes showing alternative splicing QTL (Kwan et al. 2008). These can be subdivided into three classes according to the position of the alternatively spliced exon: cassette exon, alternative initiation, or alternative termination, where the expression level of the exon of interest in all cases follows an allele-dependent pattern. Transcripts showing evidence for multiple types of variation, e.g., having various exons with different patterns of heritable difference, were classified as complex cases. Heterogeneous cases contain transcripts showing very diverse eQTL patterns across probes and exons and belonging to none of the above-mentioned categories.
Figure 1.—

Classification of genes showing heritable expression variation (eQTL). The 435 transcripts were classified into different groups according to their eQTL pattern: consistent eQTLs (brown) showing the same expression differences between the two genotypes in all annotated exons of a gene, eQTL patterns indicating the need for revised gene definitions (green; 8.7%), and eQTL patterns showing potential heritable differences in splicing, i.e., alternative splicing QTL (Kwan et al. 2008). The latter (purple) are subdivided into three classes according to the position of the alternatively spliced exon; they comprise a total of 5% of all cases, compared to 55% of a total of 324 transcripts with significant eQTL that showed heritable isoform changes in humans (Kwan et al. 2008). Complex cases (black) contain indications for multiple event types, e.g., various exons with different patterns of heritable difference. Some cases (10.6%) show very heterogeneous eQTL patterns across probes and exons.
To validate the classification procedure, all classifications were performed independently by two researchers, and special cases were checked in more detail. A complete list of classifications is available in supporting information, Table S1 and the corresponding plots for all genes are available at www.wormplot.org.
Permutation:
A permutation approach was used to estimate the empirical false discovery rates for the detection of genetically regulated alternative splicing. We permute sample labels in the genotype matrix and keep the correlation structure between traits and the correlation structure between markers; this makes this empirical procedure perfectly suited to a nonbiased estimation of the significance under the multiple-dependence properties of the data (Breitling et al. 2008). The permuted data were reanalyzed for all genes at chromosome IV to keep the computational burden within reasonable limits: we repeated the QTL detection and classification as we did for the real data. On the basis of a total of 67,000 permuted instances of genes, we estimated the false discovery rate for the genetically regulated alternative splicing case being <1%.
Deleted genes:
We validated our ability to detect heritable expression differences by examining published gene deletions in CB4856 worms (Maydan et al. 2007). These genes should show consistently variable expression according to the local genotype. Of 531 CB4856-deleted genes, ∼10% (53 genes) are detected as differentially expressed in our experiment. All of these genes show consistent eQTL across all probes with larger expression in the N2 allele, well above our threshold. This confirms the sensitivity of our approach.
Comparison with a previous experiment:
As a further validation step, we compared the detected eQTL to those observed in an earlier study using cDNA microarrays (Li et al. 2006). Nearly half of the top 500 highly expressed genes (231 genes) are shared in the two experiments. The eQTL effect size also shows strong correlation (locally regulated QTL, r = 0.72; distantly regulated, r = 0.48). Several strong distant eQTL were found in both experiments including ZK488.6, F10D2.9 (fat-7), F56H6.5 (gmd-2), C38D9.2, T21E8.1 (pgp-6), C05A9.1 (pgp-5), and F15D4.5.
Quantitative changes in alternative splicing:
Generally, the genetic effect on the abundance of transcript isoforms can be quantitative rather than qualitative (shifts in isoform ratios, rather than on–off effects). We calculated the expected effect size for all possible shifts of isoform ratio, assuming that two isoforms differ only by the presence or the absence of one exon and that there is no overall expression difference (Figure 2). It turns out that the difference in abundance of transcript isoforms should be at least ∼1.86-fold to be picked up in our study. This means that our method has sufficient power to identify quantitative changes in isoform ratio like 90:10 (allele 1) → 20:80 (allele 2) or 60:40 (allele 1) → 12:88 (allele 2).
Figure 2.—

Schematic illustration (A) and power of detection (B) for quantitative changes in alternative splicing. (A) We consider a transcript with two alternative splicing forms: the second exon is included in isoform 1 but excluded in isoform 2 (cassette exon). Under allele A, x% of the entire transcript amount is of isoform 1, while isoform 2 is expressed at (1 − x)%. Similarly, under allele B, the isoform 1 is expressed at y% and isoform 2 at (1 − y)%. Without loss of generality, we assume that the total transcript amount is 1, and thus the detected signal for the second exon is x and y under alleles 1 and 2, respectively. The difference between these signals (x − y) is detected as our asQTL effect. (B) The asQTL effect size changes for different combinations of x and y. The white dashed line corresponds to our QTL threshold; changes in transcript isoform ratios outside the dashed line are reliably detectable for the population size used.
RESULTS AND DISCUSSION
Here, we performed the first genome-wide analysis of genetic variation of alternative splicing in C. elegans using a comprehensive tiling microarray. We used 60 recombinant inbred lines of a cross between two very diverse strains, Bristol (N2) and Hawaii (CB4856), which have been genotyped using 121 markers (Li et al. 2006). By using tiling array data, with multiple probes targeting every exon of each gene, we obtained a more comprehensive and sensitive picture of heritable variation of gene expression than possible with previous technologies. It also allows us to dissect the genetic component for differences in isoform-specific gene expression. Thus we can detect asQTL, the genome regions controlling variation in isoform-specific expression. Two categories of asQTL can be distinguished, i.e., those that map in close vicinity to the gene itself (local) and those that map elsewhere in the genome (distant). Local activity can be explained, for example, by altered functional motifs in exonic splicing enhancers that will affect the splicing activity. The mechanism of distant regulation is often more complicated and can possibly be explained by a polymorphism in an auxiliary splicing factor (e.g., SR protein) that modulates the activity of the spliceosome. In this case we would expect to see a genetic master regulator at the locus of the splicing factor controlling isoform ratios for large groups of transcripts.
Using nonparametric effect size estimates, corrected for genotype imbalance (materials and methods) and corresponding to a P-value of 0.001 (Wilcoxon's test), we detected 435 genes with substantial heritable variation for at least one exon. The comparison of gene position and associated polymorphisms shows that most eQTL map in close proximity to the affected gene (local eQTL: 277 genes or 64%; Figure 3). There are 158 eQTL mapping to another chromosome (distant eQTL). Two hundred sixty-seven genes show higher expression in carriers of the N2 allele than in CB4856 carriers, including 53 cases of known gene deletions in the CB4856 strain (Maydan et al. 2007).
Figure 3.—

Mapping location (A) and type (B) of heritable variation in gene expression. (A) Each dot represents a single transcript. The physical position of each transcript is indicated on the y-axis, and the position of the locus that is most strongly associated with variation of the corresponding transcript level is shown on the x-axis. Transcripts on and off the diagonal are locally and distantly regulated, respectively. The different symbols/colors discriminate the parental allele of the eQTL that caused a higher expression (N2 is indicated by a red cross and CB4856 by a blue dot). Transcripts that physically overlap with another gene on the genome according to the WormBase genome annotation are shown in pink (N2>CB4856) and light blue (N2<CB4856). (B) The same eQTL with a different coloring scheme: colors discriminate consistent eQTL (brown) and asQTL (purple). The relative rarity of the latter category is clearly visible. The few cases that are observed are mostly restricted to the cis diagonal; i.e., they are caused by local variation of the gene sequence close to the affected splice site. Differences in regulation associated with variation in trans-acting splicing factors seem to be extremely rare. Transcripts with revised gene definition are indicated in green. Genes with complex and heterogeneous eQTL patterns are shown in black.
A large majority of eQTL (319 or 70.4%) lead to a consistent differential expression across all exons of the affected gene. Interestingly, the genetic effects (eQTL size) of these consistent eQTL show a strong correlation (Spearman's ρ = 0.78) with a previous experiment using cDNA micorarrays (Li et al. 2006). As shown in Figure 1, 8.7% of cases show evidence for a necessary refinement of existing gene definitions, predominantly by expanding known exons (plotted results for all genes are available at www.wormplot.org for a detailed examination). In contrast to the large number of consistent eQTL, we find only 22 genes that show evidence for genetic variation of alternative splicing, i.e., an exon-specific asQTL (Figure 4). This genome-wide evidence for the genetic robustness of the alternative splicing machinery is consistent with the earlier indication that individual alternative splicing events in C. elegans are highly conserved and hardly tolerate genetic variation (Barberan-Soler and Zahler 2008a). Note, however, that variation in alternative splicing events restricted to a specific cell or tissue type can be diluted in measurements on whole-worm mRNA. In addition, 77% of asQTL were found to be locally regulated. This agrees with recent findings that alternative splicing can be regulated without involvement of an auxiliary splicing factor, by cis-acting RNA sequences that can function as a splicing silencer (Yu et al. 2008).
Figure 4.—

Expression intensity and eQTL effect per probe along the genome for selected genes. (A) Detecting consistent heritable differences in gene expression with high resolution. Nearly 300 probes cover the area of this gene, Y87G2A.5. Exon probes show consistently high expression (median intensity = 9.64), compared to intron probes. However, there is huge variation between probes, which makes the clear delimitation of exon boundaries challenging. In contrast, there is a clear and highly consistent differential expression between carriers of different alleles (N2 and CB4856). This so-called eQTL effect, indicated by red bars, is highly reproducible across all exon probes within the gene. In this example, the average expression difference between the two alleles is ∼2.4-fold. In total, there are 306 genes with similar consistent expression differences. It should be noted that a majority of genes show consistently lower intensity (and thus lower eQTL effect) in the 3′-untranslated region (UTR) indicating the decaying end of transcript (Kolmogorov–Smirnov test, P-value <2.2 × 10−16). (B) Refining existing gene definition. The exon probes within gene T21E8.1 show consistently higher expression for individuals carrying the N2 allele than for those carrying the CB4856 allele. Additionally, several adjacent probes within the sixth intron show the same differential expression pattern, suggesting that this intron contains a previously unannotated additional exon. This would not have been detectable based on the absolute expression levels, due to the high interprobe variability. We find a total of 41 genes that require refined definition according to the eQTL pattern, mostly extensions of known exons and redefinitions of the transcript start and end sites. (C and D) Detecting heritable variation in alternative splicing. These genes do not show heritable expression differences in general, but individual exons show consistently lower signal for carriers of the CB4856 allele. This suggests that these exons are specifically removed by alternative splicing in one of the two alleles. In both cases, this alternative splicing variation is determined by a local sequence variation (QTL mapping in cis). The first example (Y69H2.3) was confirmed experimentally by Barberan-Soler and Zahler (2008a). We find 22 comparable instances of heritable differences in splicing patterns.
Most of the reported asQTL detected in our study have strong genetic effects (qualitative on–off patterns) and we found only a few cases of subtle quantitative effects on alternative splicing. However, this does not mean that this on–off behavior is a general property of alternative splicing patterns, but rather that despite the large population used in this study, technical noise and biological variation might limit our ability to detect subtle shifts in isoform proportions. To detect more quantitative effects (Figure 2), more precise technology such as deep-sequencing would be required. Even then, reliable detection of changes in isoform proportions will depend on extremely large read numbers.
Our genome-wide study provides the first genome-wide evidence supporting earlier hypotheses that in C. elegans the alternative splicing machinery exhibits a general genetic robustness, and only a minor fraction of genes show heritable variation in splicing forms and relative abundance. This observation points to a profound difference in the regulation of the alternative splicing machinery compared to that in humans (Kwan et al. 2008), which parallels the differences in cellular diversity and developmental flexibility in the two species and has important consequences for interpreting future studies using C. elegans as a model organism for metazoan splicing.
Acknowledgments
This work was supported by European Union grant FP7 PANACEA 222936 and The Netherlands Organization for Scientific Research, grant NWO-86504001.
Supporting information is available online at http://www.genetics.org/cgi/content/full/genetics.110.119677/DC1.
References
- Alberts, R., P. Terpstra, Y. Li, R. Breitling, J. P. Nap et al., 2007. Sequence polymorphisms cause many false cis eQTLs. PLoS ONE 2 e622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barberan-Soler, S., and A. M. Zahler, 2008a. Alternative splicing and the steady-state ratios of mRNA isoforms generated by it are under strong stabilizing selection in Caenorhabditis elegans. Mol. Biol. Evol. 25 2431–2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barberan-Soler, S., and A. M. Zahler, 2008b. Alternative splicing regulation during C. elegans development: splicing factors as regulated targets. PLoS Genet. 4 e1000001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breitling, R., Y. Li, B. M. Tesson, J. Fu, C. Wu et al., 2008. Genetical genomics: spotlight on QTL hotspots. PLoS Genet. 4 e1000232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cliff, D., 1996. Answering ordinal questions with ordinal data using ordinal statistics. Multivariate Behav. Res. 31 331–350. [DOI] [PubMed] [Google Scholar]
- del Rosal, A. B., C. San Luis and A. Sanchez-Bruno, 2003. Dominance statistics: a simulation study on the d statistic. Qual. Quant. 37 303–316. [Google Scholar]
- Fischer, S. E., M. D. Butler, Q. Pan and G. Ruvkun, 2008. Trans-splicing in C. elegans generates the negative RNAi regulator ERI-6/7. Nature 455 491–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen, R. C., and J. P. Nap, 2001. Genetical genomics: the added value from segregation. Trends Genet. 17 388–391. [DOI] [PubMed] [Google Scholar]
- Kim, E., A. Magen and G. Ast, 2007. Different levels of alternative splicing among eukaryotes. Nucleic Acids Res. 35 125–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuroyanagi, H., G. Ohno, S. Mitani and M. Hagiwara, 2007. The Fox-1 family and SUP-12 coordinately regulate tissue-specific alternative splicing in vivo. Mol. Cell. Biol. 27 8612–8621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwan, T., D. Benovoy, C. Dias, S. Gurd, C. Provencher et al., 2008. Genome-wide analysis of transcript isoform variation in humans. Nat. Genet. 40 225–231. [DOI] [PubMed] [Google Scholar]
- Li, Y., O. A. Alvarez, E. W. Gutteling, M. Tijsterman, J. Fu et al., 2006. Mapping determinants of gene expression plasticity by genetical genomics in C. elegans. PLoS Genet. 2 e222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackay, T. F., E. A. Stone and J. F. Ayroles, 2009. The genetics of quantitative traits: challenges and prospects. Nat. Rev. Genet. 10 565–577. [DOI] [PubMed] [Google Scholar]
- Maydan, J. S., S. Flibotte, M. L. Edgley, J. Lau, R. R. Selzer et al., 2007. Efficient high-resolution deletion discovery in Caenorhabditis elegans by array comparative genomic hybridization. Genome Res. 17 337–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schadt, E. E., S. A. Monks, T. A. Drake, A. J. Lusis, N. Che et al., 2003. Genetics of gene expression surveyed in maize, mouse and man. Nature 422 297–302. [DOI] [PubMed] [Google Scholar]
- Yu, Y., P. A. Maroney, J. A. Denker, X. H. Zhang, O. Dybkov et al., 2008. Dynamic regulation of alternative splicing by silencers that modulate 5′ splice site competition. Cell 135 1224–1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, Z., T. J. Boyle, Z. Bao, J. I. Murray, B. Mericle et al., 2008. Comparative analysis of embryonic cell lineage between Caenorhabditis briggsae and Caenorhabditis elegans. Dev. Biol. 314 93–99. [DOI] [PMC free article] [PubMed] [Google Scholar]