

Abstract
We studied the physiometabolic effects of a mitochondrial DNA (mtDNA) heteroplasmic point mutation, the A-->G3260 transition associated with maternally inherited myopathy and cardiomyopathy. To eliminate the possible influence of the autochthonous nuclear gene set, we fused myoblast-derived cytoplasts of a patient with a human tumoral cell line deprived of mtDNA (Rho degrees). The presence and amount of the mutant G3260 vs the wild-type A3260 were measured by solid phase minisequencing. We observed a marked reduction of the percentage of mutant mtDNA in the culture system compared with that measured in the donor's muscle biopsy, suggesting the presence of negative selection against the mutation. Furthermore, stable mitotic segregation of the two mtDNA populations was observed in 18 of 19 transformant clones, suggesting the presence of intraorganelle and possibly intracellular homoplasmy in the precursor cells of the donor. Several indexes of mtDNA-related respiratory capacity, including oxygen consumption, complex I- and complex IV-specific activities, and lactate production, were markedly abnormal in the clones containing a high proportion of mutant mtDNA, as compared with those containing homoplasmic wild-type mtDNA, possibly because of impaired mitochondrial protein synthesis. We conclude that (a) the A-->G3260 transition is indeed responsible for the mitochondrial disorder identified in the donor patient, and (b) transformant cybrid system gives direct evidence of the mitochondrial origin of a genetic disorder and should be adopted for the evaluation of the pathogenic potential of the mtDNA mutations.
Full text
PDF



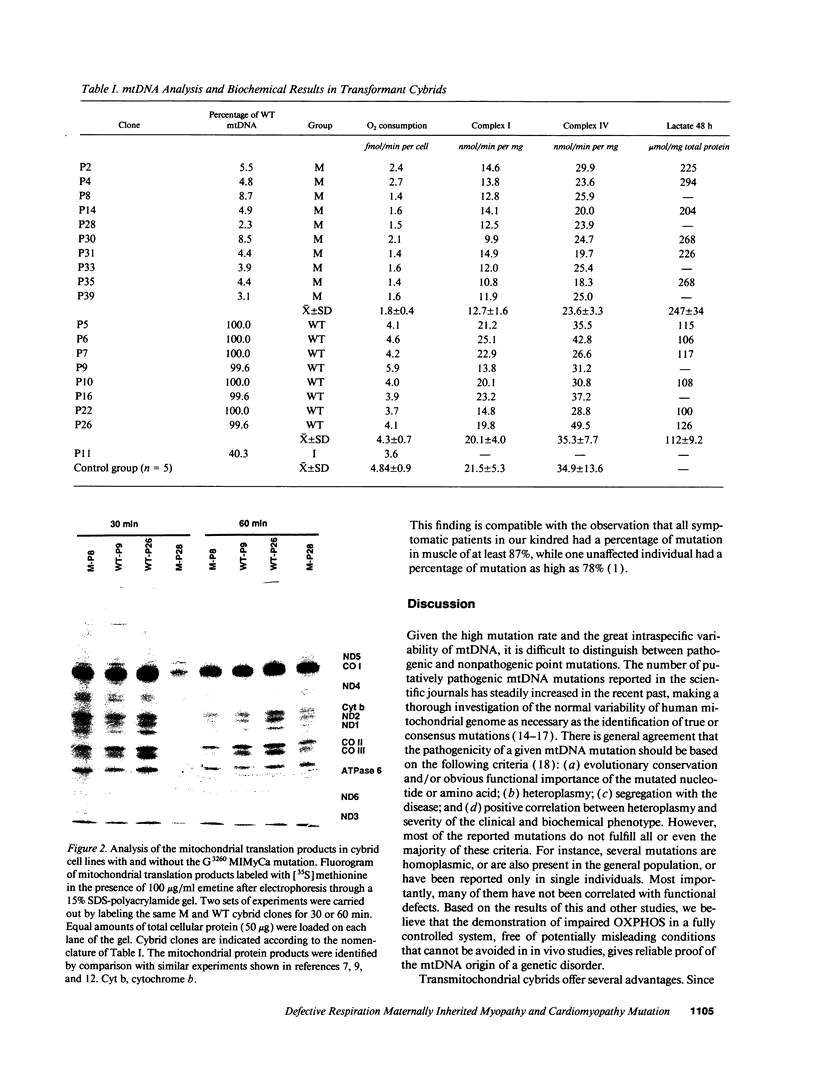


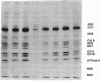
Images in this article
Selected References
These references are in PubMed. This may not be the complete list of references from this article.
- Anderson S., Bankier A. T., Barrell B. G., de Bruijn M. H., Coulson A. R., Drouin J., Eperon I. C., Nierlich D. P., Roe B. A., Sanger F. Sequence and organization of the human mitochondrial genome. Nature. 1981 Apr 9;290(5806):457–465. doi: 10.1038/290457a0. [DOI] [PubMed] [Google Scholar]
- Askanas V., Engel W. K. A new program for investigating adult human skeletal muscle grown aneurally in tissue culture. Neurology. 1975 Jan;25(1):58–67. doi: 10.1212/wnl.25.1.58. [DOI] [PubMed] [Google Scholar]
- Boulet L., Karpati G., Shoubridge E. A. Distribution and threshold expression of the tRNA(Lys) mutation in skeletal muscle of patients with myoclonic epilepsy and ragged-red fibers (MERRF). Am J Hum Genet. 1992 Dec;51(6):1187–1200. [PMC free article] [PubMed] [Google Scholar]
- Chomyn A., Meola G., Bresolin N., Lai S. T., Scarlato G., Attardi G. In vitro genetic transfer of protein synthesis and respiration defects to mitochondrial DNA-less cells with myopathy-patient mitochondria. Mol Cell Biol. 1991 Apr;11(4):2236–2244. doi: 10.1128/mcb.11.4.2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eubanks J. H., Selleri L., Hart R., Rosette C., Evans G. A. Isolation, localization, and physical mapping of a highly polymorphic locus on human chromosome 11q13. Genomics. 1991 Nov;11(3):720–729. doi: 10.1016/0888-7543(91)90080-x. [DOI] [PubMed] [Google Scholar]
- Howell N., McCullough D. A., Kubacka I., Halvorson S., Mackey D. The sequence of human mtDNA: the question of errors versus polymorphisms. Am J Hum Genet. 1992 Jun;50(6):1333–1340. [PMC free article] [PubMed] [Google Scholar]
- King M. P., Attardi G. Human cells lacking mtDNA: repopulation with exogenous mitochondria by complementation. Science. 1989 Oct 27;246(4929):500–503. doi: 10.1126/science.2814477. [DOI] [PubMed] [Google Scholar]
- King M. P., Koga Y., Davidson M., Schon E. A. Defects in mitochondrial protein synthesis and respiratory chain activity segregate with the tRNA(Leu(UUR)) mutation associated with mitochondrial myopathy, encephalopathy, lactic acidosis, and strokelike episodes. Mol Cell Biol. 1992 Feb;12(2):480–490. doi: 10.1128/mcb.12.2.480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LOWRY O. H., ROSEBROUGH N. J., FARR A. L., RANDALL R. J. Protein measurement with the Folin phenol reagent. J Biol Chem. 1951 Nov;193(1):265–275. [PubMed] [Google Scholar]
- Laemmli U. K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970 Aug 15;227(5259):680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- Marzuki S., Noer A. S., Lertrit P., Thyagarajan D., Kapsa R., Utthanaphol P., Byrne E. Normal variants of human mitochondrial DNA and translation products: the building of a reference data base. Hum Genet. 1991 Dec;88(2):139–145. doi: 10.1007/BF00206061. [DOI] [PubMed] [Google Scholar]
- Shoffner J. M., Lott M. T., Lezza A. M., Seibel P., Ballinger S. W., Wallace D. C. Myoclonic epilepsy and ragged-red fiber disease (MERRF) is associated with a mitochondrial DNA tRNA(Lys) mutation. Cell. 1990 Jun 15;61(6):931–937. doi: 10.1016/0092-8674(90)90059-n. [DOI] [PubMed] [Google Scholar]
- Suomalainen A., Kollmann P., Octave J. N., Söderlund H., Syvänen A. C. Quantification of mitochondrial DNA carrying the tRNA(8344Lys) point mutation in myoclonus epilepsy and ragged-red-fiber disease. Eur J Hum Genet. 1993;1(1):88–95. doi: 10.1159/000472391. [DOI] [PubMed] [Google Scholar]
- Yoneda M., Chomyn A., Martinuzzi A., Hurko O., Attardi G. Marked replicative advantage of human mtDNA carrying a point mutation that causes the MELAS encephalomyopathy. Proc Natl Acad Sci U S A. 1992 Dec 1;89(23):11164–11168. doi: 10.1073/pnas.89.23.11164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeviani M., Gellera C., Antozzi C., Rimoldi M., Morandi L., Villani F., Tiranti V., DiDonato S. Maternally inherited myopathy and cardiomyopathy: association with mutation in mitochondrial DNA tRNA(Leu)(UUR). Lancet. 1991 Jul 20;338(8760):143–147. doi: 10.1016/0140-6736(91)90136-d. [DOI] [PubMed] [Google Scholar]
- Zeviani M., Moraes C. T., DiMauro S., Nakase H., Bonilla E., Schon E. A., Rowland L. P. Deletions of mitochondrial DNA in Kearns-Sayre syndrome. Neurology. 1988 Sep;38(9):1339–1346. doi: 10.1212/wnl.38.9.1339. [DOI] [PubMed] [Google Scholar]