

Abstract
Osteoporosis is an important clinical problem, affecting more than 50% of people over age 50 yr. Estrogen signaling is critical for maintaining proper bone density, and the identification of an endogenous selective estrogen receptor (ER) modulator, 27-hydroxycholesterol (27HC), suggests a mechanism by which nutritional/metabolic status can influence bone biology. With its levels directly correlated with cholesterol, a new possibility emerges wherein 27HC links estrogen and cholesterol signaling to bone homeostasis. In these studies, we found that increasing concentrations of 27HC, both by genetic and pharmacological means, led to decreased bone mineral density that was associated with decreased bone formation and increased bone resorption. Upon manipulation of endogenous estrogen levels, many of the responses to elevated 27HC were altered in such a way as to implicate ER as a likely mediator. In a model of postmenopausal bone loss, some pathologies associated with elevated 27HC were exacerbated by the absence of endogenous estrogens, suggesting that 27HC may act both in concert with and independently from classic ER signaling. These data provide evidence for interactions between estrogen signaling, cholesterol and metabolic disease, and osteoporosis. Patients with high cholesterol likely also have higher than average 27HC, perhaps putting them at a higher risk for bone loss and fracture. More studies are warranted to fully elucidate the mechanism of action of 27HC in bone and to identify ways to modulate this pathway therapeutically.
Elevation of the endogenous estrogen receptor modulator 27-hydroxycholesterol negatively impacts bone density.
There is increasing evidence that cholesterol metabolites, such as the oxysterols, are important signaling molecules that have both physiological and pathological activities in the cardiovascular system, liver, breast, and brain (1,2,3,4,5,6). Thus, in addition to evaluating the direct effects of cholesterol, there is now considerable interest in defining the biological activities of cholesterol catabolites on signaling pathways in different tissues. Although initially considered in the context of bile acid metabolism, it is now clear that the oxysterol 27-hydroxycholesterol (27HC) has signaling properties consistent with its having hormonal activity. In this regard, recent studies indicate that 27HC has activities in the cardiovascular system, the brain, and breast cancer (6,7,8), some of which are estrogen receptor (ER) dependent. In the cardiovascular system, 27HC competes with estrogen for binding to ER, and this along with data showing that 27HC is a partial ER agonist in cellular models of breast cancer led to the characterization of 27HC as an endogenous selective ER modulator (SERM) (6,9). The cytochrome p450 27A1 (CYP27A1)-dependent conversion of cholesterol to 27HC, which occurs predominately in macrophages, underlies the positive correlation between the cholesterol and 27HC levels found in blood.
The menopausal loss of estrogen is associated with increased bone turnover and, consequently, a 50% elevation in lifetime risk of developing osteoporosis (10). This increased bone turnover stems primarily from an increased number and activity of mature osteoclasts (OCs) (11,12,13). The signaling components that create the menopausal proresorptive environment have yet to be completely elucidated. Indeed, although decreased ER function is clearly the initiating event, the cellular targets and specific signaling pathways that enable estrogens to manifest regulatory actions over bone homeostasis remain to be determined. A dominant hypothesis is that estrogens have a greater influence on OC-dependent resorption than on osteoblastic bone formation. However, considerable evidence also indicates that ER regulates OC action through direct actions in osteoblasts (OBs) (14,15), thereby influencing the activity of both cell types. Of particular note is the observation that osteoprotegerin (OPG), a physiological inhibitor of the OC maturation process, is produced by OBs in an 17β-estradiol (E2)-dependent manner (16,17). These data illustrate the difficulties in ascribing specific cellular function in bone to estrogens and SERMs and highlight the potential complexities in defining the actions of endogenous SERMs such as 27HC in this target organ.
The observation that ER function can be modified in animal models of cardiovascular disease by altering blood levels of 27HC suggested that this oxysterol was likely to also impact physiological and pathologically important processes at additional ER target sites. Given that greater than 30% of the U.S. population are considered to have elevated cholesterol and, by inference, elevated 27HC, there is a need to define both the positive and negative actions of this oxysterol on ER biology. There is increasing evidence to suggest that individuals with elevated cholesterol exhibit reduced bone mineral density (BMD), with some studies suggesting that statins, which millions of people in the United States are currently taking, are associated with improved BMD (18,19). It is intriguing, given these links between cholesterol biology and BMD, that diagnosis of a cardiovascular event is a significant risk factor for subsequent hip fracture (20) and that low BMD is associated with an increased risk for cardiovascular events (21,22). Furthermore, genetic blockade of 27HC synthesis by mutation of the CYP27A1 gene, which increases cholesterol levels, leads to osteoporosis (23). However, a direct assessment of how 27HC levels correlate to bone pathology has not yet been performed. Clearly, although the etiology underlying these clinical observations remains to be identified, it seems prudent to probe the potential roles of 27HC and ER in these processes. Thus, a primary goal of this study was to evaluate the role of 27HC in bone and the impact that this has on estrogen action in this target tissue.
Materials and Methods
Breeding, care, and use of animals
All studies involving animals were approved by the Institutional Animal Care and Use Committee at Duke University. Animals were housed in a temperature-controlled room with a daily 12-h light, 12-h dark cycle and free access to water and chow (Laboratory Rodent Diet no. 5001). The Cyp27a1−/− and the Cyp7b1−/− strains, and their respective wild-type strains, were obtained with permission from Dr. E. Leitersdorf (Hadassah University Hospital, Jerusalem, Israel) and Dr. D. Russell (University of Texas Southwestern Medical Center, Dallas, TX), respectively. All studies began with adult (6 wk old) female mice. Mice received either no operation, sham operation, or ovariectomy at 6 wk of age and then were treated daily with placebo (corn oil or 2-hydroxypropyl-β-cyclodextrin), 40 mg/kg 27HC in 2-hydroxypropyl-β-cyclodextrin, or 10 μg/kg or 20 μg/kg E2. Animals were harvested at 10 wk of age for analysis.
Tissue harvest and analysis
Urine was collected over 10 h using metabolic cages within 3 d of the end of the study. Blood was collected by cardiac puncture after CO2 euthanasia, after which tissues were harvested. Serum osteocalcin was measured by enzyme immunoassay according to the manufacturer’s instructions (Biomedical Technologies Inc., Stoughton, MA). Urine deoxypyridinoline (DPD) cross-links were measured by enzyme immunoassay and normalized to creatinine according to the manufacturer’s instructions (Quidel, San Diego, CA). 27HC levels were measured by liquid chromatography-mass spectroscopy as described (7). Lumbar spine and femur were analyzed by dual-energy x-ray absorptiometry (DEXA) using a LUNARPIXIMUS bone densitometer (Lunar Corp., Madison, WI) as previously described (24). High-resolution quantitative computed tomography (qCT) (μCT40; Scanco Medical, Basserdorf, Switzerland), as previously described (24), was used to evaluate bone morphometry. Femur was analyzed by qCT at the midfemur (cortical) and diaphysis (trabecular) to obtain cortical thickness, percent bone fraction (bone volume/total volume), and trabecular number, thickness, and separation. For histology, tibias were fixed for 24 h in 10% buffered formalin and then transferred to 70% ethanol. After decalcification, embedding, and sectioning, the slides were stained with hematoxylin and eosin (H&E). Pictures were taken on an Axio Imager at ×5 magnification, and representative images are shown.
Histomorphometry
Lumbar vertebrae (L1–L4) were fixed in 70% ethanol for at least 72 h, followed by dehydration in 95% ethanol for 1 d and 100% ethanol for 6 d. The bones were embedded without demineralization in a mixture of methylmethacrylate-2-hydroxyethyl and methylacrylate (12.5:1) and subsequently sectioned at a thickness of 5 μm on a Reichert-Jung Supercut 2050 microtome using tungsten-carbide-tipped steel knives. Sections were taken from the dorsal spine passing through the middle of the lumbar spine. Analysis of the number of OBs per bone perimeter was carried out on Goldner Masson trichrome-stained sections in L2 and L3 using a light/epifluorescence microscope connected to a digitizing table and the OsteoMeasure histomorphometry system (OsteoMetrics Inc., Atlanta, GA). OC numbers per bone perimeter were identified by morphology (large multinucleated cells with cytoplasmic vesicles and intimate contact to bone) in the same Goldner Masson trichrome-stained sections. To assess dynamic histomorphometric indices, mice were given two injections of calcein (1 mg/ml) 7 d apart and killed 2 d after the last injection according to standard double-labeling protocol. Fluorochrome measurements to determine bone formation rate were made in two nonconsecutive, unstained sections per animal.
Alkaline phosphatase activity
Primary OBs were plated at 20,000 cells per well in phenol red-free medium containing 8% charcoal-stripped fetal bovine serum and penicillin/streptomycin in a 48-well plate. After overnight incubation, the cells were treated with the indicated ligand for 36 h, at which time the cells were lysed with 0.1% Triton X-100. Lysate was incubated with substrate solution [0.1 m glycine, 1 mm MgCl2, 1 mm ZnCl2, 1 mg/ml 4-nitrophenol phosphate (Sigma, St. Louis, MO)] for 30 min at 37 C. The reaction was stopped by addition of NaOH, and absorbance was read at 405 nm. Total protein was determined by the Bradford method. Absorbance values were normalized to total protein in each sample.
RNA isolation and quantitative RT-PCR
Primary OBs were seeded in phenol red-free α-MEM containing 8% charcoal-stripped fetal bovine serum and penicillin/streptomycin. RAW264.7 (mouse monocyte cell line) cells were seeded and grown in differentiating medium [α-MEM with 8% fetal bovine serum and supplemented with 30 ng/ml receptor activator of nuclear factor-κB ligand (RANKL) (R&D Systems) and 20 ng/ml macrophage colony-stimulating factor (R&D Systems)] for 8 d and then treated for 6 h with the indicated ligand. After the indicated time period, cells were harvested and total RNA was isolated using the Aurum Total RNA Mini Kit (Bio-Rad, Hercules, CA). One microgram of RNA was reverse transcribed using the iScript cDNA synthesis kit (Bio-Rad). The Bio-Rad iCycler Realtime PCR System was used to amplify and quantitate levels of target gene cDNA. Quantitative PCR was performed with 1 μl cDNA, 10 μm specific primers, and iQ SYBRGreen Supermix (Bio-Rad). Primer sequences (all mouse) are as follows: osteocalcin, forward AGACAAGTCCCACACAGCAG and reverse TTGGACATGAAGGCTTTGTC; RANKL, forward CAGCTATGATGGAAGGCTCA and reverse GACTTTATGGGAACCCGATG; OPG, forward TGCCGAGAGTGTAGAGAGGA and reverse CACAGAGGTCAATGTCTTGGA; alkaline phosphatase, forward GAGGGACGAATCTCAGGGTA and reverse TTTCAAGGTCTCTTGGGCTT; and cyclophilin, forward GAGCTGTTTGCAGACAAAGTTC and reverse CCCTGGCACATGAATCCTGG. Data are normalized to the Cyclophilin housekeeping gene and presented as fold induction over vehicle or normalized expression. Data are the mean ± sem for triplicate amplification reactions from one representative experiment.
Results
Elevations in 27HC negatively impact bone homeostasis
Because it has been established that 27HC is an endogenous SERM, it was of interest to evaluate the impact of manipulating the blood levels of this oxysterol on bone. To this end, we analyzed both trabecular and cortical bone using DEXA from mice that lack the enzyme responsible for the synthesis of 27HC (Cyp27a1−/− mice) and thus have no detectable 27HC. In female adult mice, there was no measurable difference in BMD in either the lumbar spine (trabecular bone) or the midfemur (cortical bone) region between Cyp27a1−/− mice and their wild-type (Cyp27a1+/+) counterparts (Fig. 1A). However, upon closer qCT analysis, significant changes were evident in the trabecular microarchitecture of female Cyp27a1−/− mice (Fig. 1B). Specifically, mice lacking 27HC had an increased number of trabeculae compared with wild-type animals, but these trabeculae were significantly thinner. Although a subtle phenotype, our data indicate that 27HC may modulate bone at either the homeostatic or the developmental level. There is evidence in humans that 27HC may play a role in bone, because patients who have inactivating mutations in the CYP27A1 gene, and thus cannot produce 27HC, tend to develop osteoporosis (23,25,26). Because we analyzed our model mice at a relatively young age (6–10 wk old), it is possible that aged mice develop decreased bone density stemming from the thinning trabeculae that are evident even at the young age. Overall, our data clearly indicate that the loss of 27HC alters bone composition and architecture.
Figure 1.

Altered trabecular bone in the absence of 27HC. The lumbar spine and femur from female mice at 10 wk of age were harvested for analysis. A, There was no significant difference in BMD between the Cyp27a1+/+ and Cyp27a1−/− mice. B, Trabecular (Trab.) morphometry was assessed by qCT on the femur. BV/TV, Bone volume/total volume. Data (n = 7–8) are shown as the mean ± sem. **, P < 0.01 by unpaired two-tailed t test on raw data.
To further define a role for 27HC in the bone, we determined the consequence of pathologically elevating 27HC in vivo by assessing parameters of bone quality in mice lacking the cytochrome P450 7B1 (CYP7B1) gene and therefore cannot efficiently metabolize 27HC and thus have elevated circulating levels of this oxysterol (0.75–1.24 μm) (7,27). Patients with mutations in the CYP7B1 gene have circulating 27HC levels of 2–3 μm, compared with normal concentrations ranging from 0.072–0.73 μm (7,28,29,30). In adult female mice, a significant decrease in lumbar spine BMD in Cyp7b1−/− mice compared with wild-type (Cyp7b1+/+) controls was observed (Fig. 2A). A more detailed examination by qCT imaging of the distal femur of the Cyp7b1−/− mice revealed a decrease in bone volume/total volume fraction, concurrent with an increase in trabecular separation, compared with wild-type controls (Fig. 2, B and C). These alterations likely stem from decreased trabecular number and thickness (Fig. 2, B and C). The loss in trabecular number was qualitatively confirmed using histological H&E staining of the proximal tibia (Fig. 2D). The decreased trabecular number in the Cyp7b1−/− mice directly contrasts with the Cyp27a1−/− mice, providing strong support for a role for 27HC in controlling this biological parameter. Cumulatively, our data suggest that elevated 27HC is associated with decreases in trabecular bone volume, an activity that would be expected to track with increased fracture risk.
Figure 2.

Pathological elevation of 27HC decreases trabecular bone density. The lumbar spine and femur from female mice at 10 wk of age were harvested for analysis. A, The BMD in the lumbar spine was significantly lower in Cyp7b1−/− compared with wild-type mice (unpaired two-tailed t test on raw data, P < 0.001). B, Trabecular (Trab.) morphometry was assessed by qCT on the femur. BV/TV, Bone volume/total volume. For A and B, data (n = 9–10) are shown as the mean ± sem. ***, P < 0.001; **, P < 0.01; *, P < 0.05 by unpaired two-tailed t test on raw data. C, qCT image of the spine from representative Cyp7b1−/− and Cyp7b1+/+ intact female mice. The 8-μm slice was decided at the same point via grossly determining the same bone epicondyle. D, H&E staining of the proximal tibia from representative Cyp7b1−/− and Cyp7b1+/+ intact female mice.
27HC has a significant impact on bone density and microarchitecture
Because other oxysterols besides 27HC, in particular 25-hydroxycholesterol, are also elevated in Cyp7b1−/− mice, we injected wild-type mice with 27HC to directly test the effects of 27HC on bone. Wild-type female mice were treated with exogenous 27HC to raise circulating levels without genetically altering the cholesterol/oxysterol metabolic pathways. Specifically, we elevated the circulating concentration of 27HC for 28 d in wild-type mice to about 4.5 μm (Supplemental Fig. 1A published on The Endocrine Society’s Journals Online web site at http://endo.endojournals.org) and found that this resulted in a significant decrease in BMD (Fig. 3A). This is similar to concentrations found in patients with mutations in the CYP7B1 gene, yet in atherosclerotic lesions, the concentration is much higher (in the millimolar range) (7). Our injection studies increased 27HC levels about 6-fold above the level found in the Cyp7b1−/− mice. Importantly, we observed similar modifications in the trabecular architecture in 27HC-treated mice as were seen in the Cyp7b1−/− mice (Fig. 3B), supporting the hypothesis that the bone phenotype in the Cyp7b1−/− mice is due to an elevation in 27HC as opposed to other potential substrates of the CYP7B1 enzyme. Gene expression analysis of the isolated calvaria from these animals demonstrated that 27HC treatment led to altered expression of bone markers, including the induction of OPG, a gene previously reported to be ER responsive (Fig. 3C) (31). Although the OB-associated gene OPG was up-regulated, the terminal OB differentiation marker osteocalcin was significantly decreased in calvaria of 27HC-treated mice. Interestingly, RANKL, a primary mediator of osteoclastogenesis, was strongly induced by 27HC. Together, these data provide direct evidence that 27HC is capable of altering bone integrity in part through altered gene expression and establishes a strong link between cholesterol and changes in BMD, a finding of tremendous potential clinical importance.
Figure 3.

27HC supplementation decreases bone parameters. Cyp7b1+/+ mice were treated with placebo or 27HC for 28 d before harvesting the lumbar spine, femur, and calvaria. A, Bones were subjected to DEXA analysis. Data (n = 6–8) are shown as the mean ± sem. *, P < 0.05 by unpaired two-tailed t test on raw data. B, Trabecular (Trab.) morphometry was assessed by qCT on the femur. BV/TV, Bone volume/total volume. Data (n = 5–8) are shown as the mean ± sem. *, P < 0.05; ^, P = 0.0506 by unpaired two-tailed t test on raw data. C, Gene expression was analyzed in the isolated calvaria. The expression for OPG, alkaline phosphatase, osteocalcin, and RANKL was quantified by qPCR and normalized to cyclophilin. Data (n = 3–5) are presented with respect to placebo (mean ± sem). *, P < 0.05 compared with placebo (unpaired t test).
Estrogen attenuates the negative impact of 27HC on the bone at some sites
Because estrogen signaling preserves BMD during adulthood, we were interested in determining whether the negative effects of 27HC in the bone were due to its ability to antagonize this particular hormone signaling pathway. As an initial step, we evaluated whether the consequences of elevated 27HC could be overcome by increasing circulating estrogen levels by supplementing ovary-intact female Cyp7b1−/− mice with daily injections of E2 for 28 d. It was anticipated that the higher-affinity ligand, E2, would outcompete 27HC for binding to ER. As a control for E2 delivery, we demonstrated that E2-treated mice exhibited a significant increase in uterine wet weight (Supplemental Fig. 1B). Interestingly, the E2-treated Cyp7b1−/− mice displayed increased BMD in both the lumbar spine and cortical bone compared with placebo-treated Cyp7b1−/− mice (Fig. 4A) (most significantly with the higher dose of E2), suggesting either that direct competition between E2 and 27HC for regulation of ER occurs in the bone as it does in the cardiovascular system or that E2 and 27HC activate alternate pathways that can compensate for one another.
Figure 4.

Estrogen attenuates the negative impact of 27HC on the bone. A, Cyp7b1−/− mice were treated with placebo or E2 (10 or 20 μg/kg) for 28 d. Data (n = 4–5) are shown as the mean ± sem. ***, P < 0.001; *, P < 0.05 compared with the placebo group (unpaired two-tailed t test on raw data). B, Trabecular (Trab.) morphometry was assessed by qCT on the femur. Mice were treated with placebo or 20 μg/kg E2 for 28 d. BV/TV, Bone volume/total volume. Data (n = 4–5) are presented as the mean ± sem. ***, P < 0.001; **, P < 0.01 compared with the placebo group (one-way ANOVA followed by Student-Newman-Keuls).
Using qCT analysis, we demonstrated that E2 treatment of ovary-intact Cyp7b1−/− mice increased trabecular thickness and bone volume/total volume fraction, two parameters known to be sensitive to ER signaling (Fig. 4B). Furthermore, cortical thickness was increased in the E2-treated Cyp7b1−/− mice, mimicking what was observed in a patient with a loss-of-function mutation in ER (32). These data suggest that 27HC directly affects the ability of ER to maintain normal cortical thickness as well as other parameters of bone architecture. Importantly, however, it was observed that trabecular number, and thereby trabecular separation, were not significantly impacted by E2 treatment, implicating targets in addition to ER as being involved in 27HC action in bone.
The consequence of 27HC in a model of postmenopausal bone loss
Because the highest prevalence of osteoporosis occurs in postmenopausal women, we wanted to determine the effect of 27HC under conditions of chronic estrogen deprivation. In bone, a partial ER agonist would be expected to manifest proresorptive activities in the presence of normal estrogen levels yet protective/sparing activity under estrogen-depleted conditions. As an example, the SERM tamoxifen decreases BMD in premenopausal women but partially blocks postmenopausal loss of BMD and perhaps improves BMD in elderly men as well (33,34). Therefore, we hypothesized that if 27HC was functioning solely as a SERM, it should exhibit bone-sparing activities in ovariectomized (OVX) mice. To confirm the involvement of E2 and ER in changes brought on by OVX, we supplemented OVX mice with daily injections of either placebo or 10 μg/kg E2. Uterine wet weight measurements confirmed that endogenous production of E2 decreased in OVX mice, and supplementation of OVX mice with E2 blocked the associated decrease in uterine wet weight (Supplemental Fig. 1C). Because we did not control for reproductive cycle in these experiments, the uterine wet weight measured here cannot be directly compared with Supplemental Fig. 1B. As expected, mice that underwent OVX gained more body weight over the 28-d study period, most significantly in the wild-type OVX placebo-treated group (Supplemental Fig. 1C).
In the lumbar spine region, the OVX-induced bone loss observed in wild-type mice could be inhibited by exogenous administration of E2 (Fig. 5A). Notably, however, the already low BMD at this site in Cyp7b1−/− mice was not further reduced upon OVX. Furthermore, the low baseline BMD in these knockout mice was not increased to any significant degree upon treatment with E2. This finding may imply that a critically low bone mass had already been achieved in these animals before OVX, thereby masking the effects of estrogen deprivation. Alternatively, as proposed above, these data are also consistent with there being a target other than ER that is required for 27HC action in bone. Regardless, it is clear from our data that 27HC does not directly mimic the action of therapeutic SERMs that exhibit partial agonist activity in the bone (i.e. tamoxifen and raloxifene), because 27HC does not protect against OVX-induced bone loss in this model system.
Figure 5.

A model of postmenopausal osteoporosis is exacerbated by high levels of 27HC. The lumbar spine and femur were harvested for analysis. A, Mice (6 wk old) were assigned to sham surgery or OVX with placebo (OVX P) or E2 (OVX E) treatment for 28 d. Data (n = 9–10) are shown as the mean ± sem. B, Trabecular (Trab.) morphometry was assessed by qCT on the femur. BV/TV, Bone volume/total volume. Data (n = 9–10) are shown as the mean ± sem. Mice were assigned to sham surgery or OVX with placebo (OVX P) or E2 (OVX E) treatment for 28 d. Different letters denote statistical significance (one-way ANOVA followed by Student-Newman-Keuls, P < 0.05).
In wild-type mice, the BMD in the cortical bone (midfemur) was unaffected by OVX. However, in Cyp7b1−/− mice, the cortical BMD was significantly reduced by OVX (Fig. 5A), suggesting that elevated 27HC increased the sensitivity of the cortical bone microenvironment to E2 deprivation. This finding has significant implications for the management of osteoporosis risk in postmenopausal women with high cholesterol, remembering that levels of 27HC are positively associated with those of cholesterol.
Wild-type and Cyp7b1−/− mice exhibited a loss of trabeculae after OVX, which increased trabecular separation (Fig. 5B). The change in trabecular number induced by OVX was not significantly affected by supplementation with E2 in either wild-type or Cyp7b1−/− mice. It is therefore likely that 27HC impacts trabecular formation or destruction in an ER-independent manner. Trabecular thickness was maintained by E2 treatment in OVX wild-type mice. Intriguingly, although trabecular thickness was not significantly altered by OVX in Cyp7b1−/− mice, treatment of these mice with E2 increased trabecular thickness to wild-type ovary-intact levels; we also observed increased trabecular thickness in ovary-intact Cyp7b1−/− mice treated with supraphysiological levels of E2 (Fig. 4B). These activities are likely due to anabolic actions of E2 at these sites. After OVX, both trabecular number and thickness decreased, resulting in an increase in trabecular separation in both wild-type and Cyp7b1−/− mice; however, the E2-mediated increase in trabecular thickness was not enough to significantly decrease trabecular separation. These data could implicate two (or more) mechanisms for 27HC action in the bone: direct competition for ER binding and/or the regulation of parallel pathways that influence trabecular number and thickness and thus bone microarchitecture. Furthermore, it suggests that 27HC can result in irreparable damage to bone under conditions of E2 deprivation.
Elevated osteoclastic bone resorption and reduced bone formation underlie the low BMD in Cyp7b1−/− mice
The actions of OBs and OCs coordinately regulate homeostatic bone formation and resorption. The osteoporotic phenotype of the female Cyp7b1−/− mice is indicative of a disruption in this homeostasis, either resulting from decreased bone formation and/or increased bone resorption. During the process of bone resorption, DPD cross-links are excreted unmetabolized in urine and thus are a surrogate for resorptive activity. Interestingly, sham-operated Cyp7b1−/− mice have increased urine DPD compared with wild-type animals, and this was not significantly altered by either OVX or E2 treatment, suggestive of an ER-independent effect of 27HC on bone resorption (Fig. 6A).
Figure 6.

Deregulated OB and OC function upon increasing 27HC. A, Urine was collected and analyzed for DPD cross-links. *, P < 0.05 compared with the sham-operated Cyp7b1+/+ group (one-way ANOVA followed by Student-Newman-Keuls); ***, P < 0.001. B, Serum was collected and analyzed for osteocalcin by ELISA. ^, P < 0.05 compared with the sham-operated Cyp7b1−/− group (one-way ANOVA followed by Student-Newman-Keuls). C, Lumbar spine was analyzed by histomorphometry. **, P < 0.01 by unpaired two-tailed t test on raw data.
As a marker of bone formation, we analyzed serum osteocalcin levels and found no difference between ovary-intact wild-type and Cyp7b1−/− mice (Fig. 6B). Cyp7b1−/− mice subjected to OVX had lower osteocalcin levels, regardless of treatment with placebo or E2, than sham-operated Cyp7b1−/− mice. Further studies are necessary to determine whether ER expressed in OBs mediates the effects of 27HC on bone formation and whether this signaling leads to impaired bone mineralization or response to bone injury. Gene expression analysis of the calvaria of the wild-type and Cyp7b1−/− mice subjected to OVX and treatment with E2 did not yield further clues as to the mechanism of 27HC in bone physiology (Supplemental Fig. 2). However, in 27HC-treated wild-type mice, osteocalcin mRNA was significantly lower than placebo-treated mice, thereby indicating a lack of mature OBs (Fig. 3C).
To further delineate what cell type(s) might be responsible for the disruptive effects of 27HC on bone homeostasis, we performed histomorphometric analysis of the lumbar spine. To our surprise, Cyp7b1−/− mice have significantly more OBs than wild-type mice (Fig. 6C). Although more numerous, perhaps as a result of a compensatory response to elevated bone resorption, the OBs present in the bones of Cyp7b1−/− mice are likely to be either immature or dysfunctional, as evidenced by the decreased bone formation rate, a readout of in vivo OB activity (Fig. 6C). This also supports our gene expression findings that elevated 27HC led to increased expression of the OB marker OPG but a decrease in osteocalcin, the marker of mature OBs. OC numbers were not different among experimental groups, indicating that the OCs present may have increased activity in Cyp7b1−/− mice.
27HC acts as a partial ER agonist in primary pre-OB cells
In an effort to implicate ER in the observed bone pathology associated with elevated 27HC, we tested the effects of 27HC on alkaline phosphatase activity in primary calvarial pre-OB cells. Our findings clearly indicate that 27HC increased alkaline phosphatase activity in a dose-dependent manner (Fig. 7A). Furthermore, the activity of 27HC in this context is ER dependent, as shown by the ability of ICI 182,780, the pure ER antagonist, to block the increase in alkaline phosphatase activity (Fig. 7B). However, 27HC at 1 μm did not increase alkaline phosphatase activity to the same extent as 10 nm E2. In fact, 27HC attenuated the E2-mediated increase in activity, reducing it to the level seen with 27HC alone. Therefore, in these pre-OB cells, 27HC behaves as a classical ER partial agonist by decreasing the beneficial effects of E2.
Figure 7.
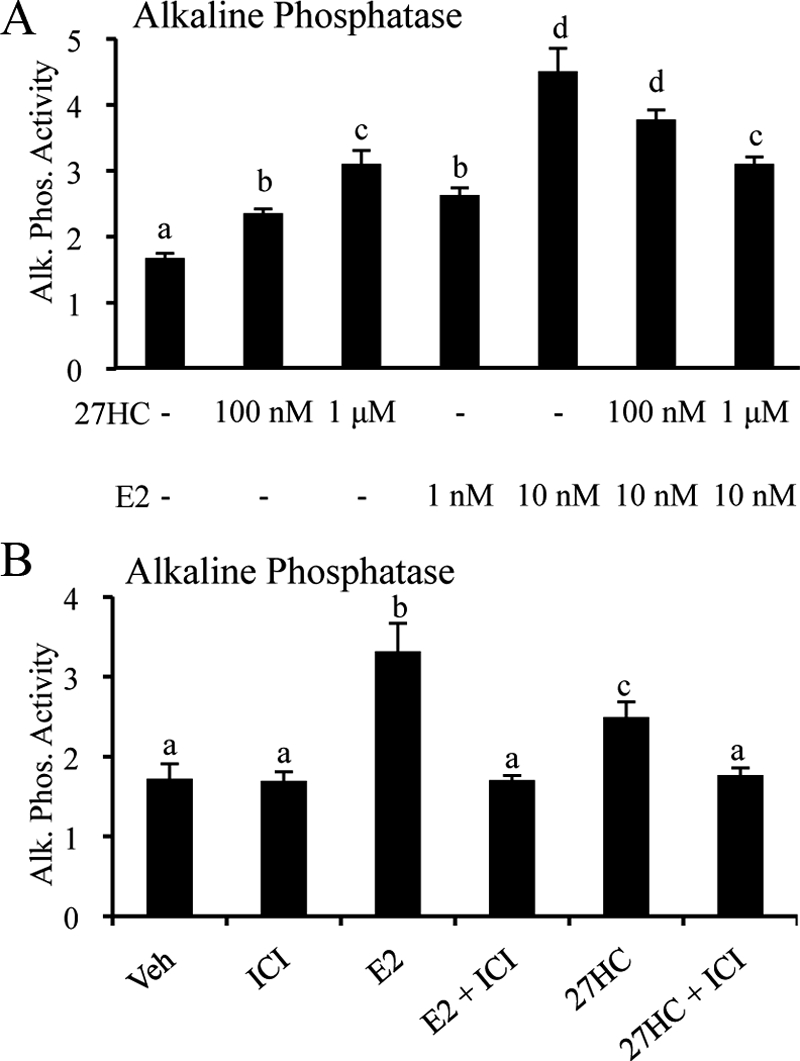
27HC acts as a partial ER agonist in primary calvarial pre-OBs. A, The effect of 27HC alone or in combination with E2 on alkaline phosphatase (Alk. Phos.) activity was assessed at the indicated dose. B, The effect of E2 (10 nm) and 27HC (1 μm) on alkaline phosphatase (Alk. Phos.) activity is blocked by the pure antiestrogen (1 μm ICI 182,780). Calvarial pre-OBs were treated for 36 h with vehicle or the indicated ligand, after which time alkaline phosphatase activity was determined. Data (n = 6) are expressed with respect to total protein (mean ± sem). Different letters denote statistical significance (one-way ANOVA followed by Student-Newman-Keuls, P < 0.05).
Because 27HC has been shown to bind to and compete with E2 for both ERα and ERβ (7), it is possible that the observed effects of 27HC on bone are mediated in part by ERβ. However, ERα mRNA was found to be 4-fold higher than ERβ mRNA in primary calvarial pre-OBs (Supplemental Fig. 3A). Furthermore, when these cells were treated with the selective ERβ agonists ERB-041 or DPN [2,3-bis(4-hydroxyphenyl)-propionitrile], we observed only minimal activation of alkaline phosphatase (Supplemental Fig. 3B). Cotreatment with either selective ERβ agonist did not significantly change the activity of 27HC or E2. During osteoclastogenesis in RAW264.7 cells, ERβ expression was consistently lower than that of ERα, despite the fact that ERβ transcript levels increased over the duration of the differentiation process (Supplemental Fig. 3C). OC differentiation was confirmed by the significant time-dependent increase in the OC marker cathepsin K (Supplemental Fig. 3C). Although we cannot rule out a contribution from ERβ, when taken together, these data suggest that ERα is the predominant mediator of 27HC, at least in cellular models of bone. Further work is required to fully delineate the roles of ERα vs. ERβ in mediating the effects of 27HC in bone.
Discussion
27HC modulates bone properties with a complex relationship to estrogen signaling
Numerous links have already been established between cholesterol levels and BMD, and although the mechanisms underlying this association are largely unknown, our data implicate 27HC as a key component of this process. Specifically, we note that female mice with a pathological elevation of 27HC, secondary to a disruption of the catabolic enzyme CYP7B1, exhibit decreased BMD in the lumbar spine accompanied by significant defects in the architecture of trabecular bone in both the spine and femur. The elevated 27HC in these mice (∼0.75 μm) reflects the upper levels found in healthy humans (0.15–0.73 μm), although local production in cases such as atherosclerotic plaques can reach the millimolar range (5,7). When Cyp7b1−/− mice undergo OVX to induce a postmenopausal-like state, they experience dramatic loss of cortical bone in the midfemur region. This implies that 27HC, although a partial agonist in cellular models of breast cancer, antagonizes ER signaling in the bone as it does in the cardiovascular system.
As with all genetic models, assigning a function to a particular signaling molecule must be done with caution. Although 27HC is overproduced in the Cyp7b1−/− mice, other changes are also evident, such as increased levels of 25-hydroxycholesterol and changes in bile acid metabolism in the liver, processes that could impact steroidogenesis. However, the cholesterol and E2 levels in these mice are not significantly altered (7). Of significance, we demonstrated that treatment of wild-type mice with exogenous 27HC mimicked the phenotype in Cyp7b1−/− mice, thus implicating 27HC as the primary determinant of the observed bone phenotype. Given that a diet high in cholesterol (the Western diet) has also been shown to increase circulating levels of 27HC (7), the public health consequences of our findings are likely to be significant.
Our study of the Cyp27a1−/− mice, which completely lack 27HC, provided further evidence of a role for 27HC in the bone. These female mice, at adulthood, did not have significantly different BMD from wild-type mice but did exhibit significant changes in trabecular bone architecture. The increased trabecular number could indicate a developmental impact of 27HC, an impact on normal age-related trabecular loss, or an effect on OB function; whether these depend on signaling through ER remains to be determined. On the contrary, both the absence and elevation of 27HC decreased trabecular thickness, a factor known to be under ER control, suggesting that a critical balance of ER activity is necessary to maintain proper trabecular thickness.
The relationship between estrogen and 27HC in the bone
Evidence in support of 27HC impacting ER signaling in the bone, either through direct competition for receptor binding or through parallel pathways, is provided by studies in which the effect of exogenous E2 supplementation on the Cyp7b1−/− phenotypes was assessed. Some parameters that were significantly altered by elevated 27HC were normalized when Cyp7b1−/− mice were treated with E2; however, others were unchanged. Perhaps the inability of E2, or in the clinical setting hormone replacement therapy, to be completely effective at blocking menopausal bone loss is due to the confounding presence of 27HC (35). This may suggest that hormone replacement therapy in combination with a low-cholesterol diet may have increased efficacy. Interestingly, trabecular number was decreased in OVX mice, but this may not be solely due to loss of endogenous E2 production because E2 supplementation does not block this decrease. This may indicate that 27HC, acting through a target other than ER, is causing irreparable harm to bone. It is important to remember that the effects of OVX are multifactorial, with changes also occurring in FSH and inhibins A and B, all of which complicate the ability to define the specific roles of ER and E2 in isolation in OVX-induced bone loss (36). Additionally, there are reported effects of E2 on trabecular bone in the ERα/ERβ double-knockout mouse, suggesting perhaps that E2 signals in an ER-independent manner under certain circumstances (37). Another hypothesis is that different compartments in the bone use ERα vs. ERβ and classical (estrogen response element) vs. nonclassical (activator protein 1/Fos/Jun, nuclear factor-κB, Sp1) signaling pathways to differing extents (37,38), and these pathways may very well be differentially regulated by 27HC, although there is not currently evidence to support this hypothesis.
Under conditions of high 27HC, a block exists at the level of differentiation, maturation, or function of OBs (as indicated by decreased osteocalcin levels in 27HC-treated mice). Without mature fully functional OBs, an important regulator of OC activity is lost, leading to increased OC activity (indicated by increased mRNA expression of RANKL and increased levels of DPD) and therefore perhaps increased bone resorption. 27HC may be a pivotal regulator of the balance between OBs and OCs, and under pathological conditions of high 27HC, the combination of decreased bone formation and increased bone resorption could lead to an osteoporotic-like phenotype of low BMD and diminished trabecular bone architecture.
Potential 27HC-mediated regulation of other signaling pathways in the bone
Although we clearly show that 27HC induces the ER-responsive gene OPG in vivo and behaves as a partial ER agonist in vitro by inducing alkaline phosphatase, our data suggest that 27HC likely influences multiple signaling pathways in coordination with ER. However, as of yet, the identity of these pathways is unknown. Recent data suggest that both isoforms of liver X receptor (LXRα and LXRβ) play an important role in homeostatic bone maintenance, although the data are limited (39). Interestingly, the cognate ligand for LXR is 22(R)HC, an oxysterol known to influence differentiation of bone cell precursors. Although an initial study suggested that 27HC did not significantly regulate LXR (7), studies by our group and others indicate that 27HC increases the transcriptional activity of LXR and thus may be a second endogenous ligand for these receptors (our unpublished data) (40,41). Therefore, it is possible that some ER-independent activities of 27HC in the context of the bone can be attributed to regulation of LXR. Further studies are necessary to ascertain the contribution of signaling through ER vs. LXR, and perhaps other pathways as well, in mediating the biological effects of 27HC.
Conclusion
Given the increasing prevalence of obesity and high cholesterol, a better understanding of the numerous ways that cholesterol impacts signaling and ultimately organ function is clearly important. The positive association between cholesterol and 27HC levels suggests that a significant portion of the population is also experiencing a potentially pathological elevation in 27HC. Furthermore, obesity and cholesterol increase with age and thus inversely correlate with estrogen levels in females. Our data suggest that a certain range of 27HC levels are required to maintain a normal bone phenotype and that 27HC impacts estrogen/ER signaling in the bone in such a way that removal of endogenous estrogens exacerbates the effects of 27HC. We provide compelling data to support future epidemiological studies directed toward an understanding of the link between cholesterol, 27HC, and BMD and further studies that evaluate the impact of lowering cholesterol on bone homeostasis in postmenopausal women. A complete mechanistic understanding of the ways that 27HC alters signaling pathways through ER, LXR, and other pathways will allow a better assessment of risk for osteoporosis in obese/hypercholesterolemic individuals and will aid in appropriate selection or development of therapeutics.
Supplementary Material
Acknowledgments
We thank the members of the McDonnell laboratory for their support on this project.
Footnotes
This work was supported by Department of Defense Breast Cancer Research Program Predoctoral Traineeship Award BC050609 (to C.D.D.); National Institutes of Health Grants 5R37DK048807 (to D.P.M), NIH P01AG004875 (to S.K. and R.M.), and R01 DK064353 (to D.G.P.); Arthritic Foundation Investigator Award (to D.G.P.), and National Institute of Diabetes and Digestive and Kidney Diseases Grant 1P30DK079328 (to M.U.).
Disclosure summary: The authors have nothing to disclose.
First Published Online May 25, 2010
Abbreviations: BMD, Bone mineral density; CYP27A1, cytochrome p450 27A1; DEXA, dual-energy x-ray absorptiometry; DPD, deoxypyridinoline; E2, 17β-estradiol; ER, estrogen receptor; 27HC, 27-hydroxycholesterol; H&E, hematoxylin and eosin; LXR, liver X receptor; OB, osteoblast; OC, osteoclast; OPG, osteoprotegerin; OVX, ovariectomized; qCT, quantitative computed tomography; RANKL, receptor activator of nuclear factor-κB ligand; SERM, selective ER modulator.
References
- Björkhem I, Cedazo-Minguez A, Leoni V, Meaney S 2009 Oxysterols and neurodegenerative diseases. Mol Aspects Med 30:171–179 [DOI] [PubMed] [Google Scholar]
- Björkhem I, Andersson O, Diczfalusy U, Sevastik B, Xiu RJ, Duan C, Lund E 1994 Atherosclerosis and sterol 27-hydroxylase: evidence for a role of this enzyme in elimination of cholesterol from human macrophages. Proc Natl Acad Sci USA 91:8592–8596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olkkonen VM, Lehto M 2004 Oxysterols and oxysterol binding proteins: role in lipid metabolism and atherosclerosis. Ann Med 36:562–572 [DOI] [PubMed] [Google Scholar]
- Javitt NB 2002 25R,26-Hydroxycholesterol revisited: synthesis, metabolism, and biologic roles. J Lipid Res 43:665–670 [PubMed] [Google Scholar]
- Brown AJ, Jessup W 1999 Oxysterols and atherosclerosis. Atherosclerosis 142:1–28 [DOI] [PubMed] [Google Scholar]
- DuSell CD, Umetani M, Shaul PW, Mangelsdorf DJ, McDonnell DP 2008 27-Hydroxycholesterol is an endogenous selective estrogen receptor modulator. Mol Endocrinol 22:65–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umetani M, Domoto H, Gormley AK, Yuhanna IS, Cummins CL, Javitt NB, Korach KS, Shaul PW, Mangelsdorf DJ 2007 27-Hydroxycholesterol is an endogenous SERM that inhibits the cardiovascular effects of estrogen. Nat Med 13:1185–1192 [DOI] [PubMed] [Google Scholar]
- Kim WS, Chan SL, Hill AF, Guillemin GJ, Garner B 2009 Impact of 27-hydroxycholesterol on amyloid-β peptide production and ATP-binding cassette transporter expression in primary human neurons. J Alzheimers Dis 16:121–131 [DOI] [PubMed] [Google Scholar]
- DuSell CD, McDonnell DP 2008 27-Hydroxycholesterol: a potential endogenous regulator of estrogen receptor signaling. Trends Pharmacol Sci 29:510–514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riggs BL, Khosla S, Melton 3rd LJ 1998 A unitary model for involutional osteoporosis: estrogen deficiency causes both type I and type II osteoporosis in postmenopausal women and contributes to bone loss in aging men. J Bone Miner Res 13:763–773 [DOI] [PubMed] [Google Scholar]
- Manolagas SC 2000 Birth and death of bone cells: basic regulatory mechanisms and implications for the pathogenesis and treatment of osteoporosis. Endocr Rev 21:115–137 [DOI] [PubMed] [Google Scholar]
- Kalu DN, Salerno E, Liu CC, Ferarro F, Arjmandi BN, Salih MA 1993 Ovariectomy-induced bone loss and the hematopoietic system. Bone Miner 23:145–161 [DOI] [PubMed] [Google Scholar]
- García Palacios V, Robinson LJ, Borysenko CW, Lehmann T, Kalla SE, Blair HC 2005 Negative regulation of RANKL-induced osteoclastic differentiation in RAW264.7 cells by estrogen and phytoestrogens. J Biol Chem 280:13720–13727 [DOI] [PubMed] [Google Scholar]
- Krum SA, Miranda-Carboni GA, Hauschka PV, Carroll JS, Lane TF, Freedman LP, Brown M 2008 Estrogen protects bone by inducing Fas ligand in osteoblasts to regulate osteoclast survival. EMBO J 27:535–545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearns AE, Khosla S, Kostenuik PJ 2008 Receptor activator of nuclear factor κB ligand and osteoprotegerin regulation of bone remodeling in health and disease. Endocr Rev 29:155–192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofbauer LC, Khosla S, Dunstan CR, Lacey DL, Spelsberg TC, Riggs BL 1999 Estrogen stimulates gene expression and protein production of osteoprotegerin in human osteoblastic cells. Endocrinology 140:4367–4370 [DOI] [PubMed] [Google Scholar]
- Simonet WS, Lacey DL, Dunstan CR, Kelley M, Chang MS, Lüthy R, Nguyen HQ, Wooden S, Bennett L, Boone T, Shimamoto G, DeRose M, Elliott R, Colombero A, Tan HL, Trail G, Sullivan J, Davy E, Bucay N, Renshaw-Gegg L, Hughes TM, Hill D, Pattison W, Campbell P, Sander S, Van G, Tarpley J, Derby P, Lee R, Boyle WJ 1997 Osteoprotegerin: a novel secreted protein involved in the regulation of bone density. Cell 89:309–319 [DOI] [PubMed] [Google Scholar]
- Lupattelli G, Scarponi AM, Vaudo G, Siepi D, Roscini AR, Gemelli F, Pirro M, Latini RA, Sinzinger H, Marchesi S, Mannarino E 2004 Simvastatin increases bone mineral density in hypercholesterolemic postmenopausal women. Metabolism 53:744–748 [DOI] [PubMed] [Google Scholar]
- Rosenson RS, Tangney CC, Langman CB, Parker TS, Levine DM, Gordon BR 2005 Short-term reduction in bone markers with high-dose simvastatin. Osteoporos Int 16:1272–1276 [DOI] [PubMed] [Google Scholar]
- Sennerby U, Melhus H, Gedeborg R, Byberg L, Garmo H, Ahlbom A, Pedersen NL, Michaëlsson K 2009 Cardiovascular diseases and risk of hip fracture. JAMA 302:1666–1673 [DOI] [PubMed] [Google Scholar]
- Browner WS, Seeley DG, Vogt TM, Cummings SR 1991 Non-trauma mortality in elderly women with low bone mineral density. Study of Osteoporotic Fractures Research Group. Lancet 338:355–358 [DOI] [PubMed] [Google Scholar]
- von der Recke P, Hansen MA, Hassager C 1999 The association between low bone mass at the menopause and cardiovascular mortality. Am J Med 106:273–278 [DOI] [PubMed] [Google Scholar]
- Keren Z, Falik-Zaccai TC 2009 Cerebrotendinous xanthomatosis (CTX): a treatable lipid storage disease. Pediatr Endocrinol Rev 7:6–11 [PubMed] [Google Scholar]
- Gesty-Palmer D, Flannery P, Yuan L, Corsino L, Spurney R, Lefkowitz RJ, Luttrell LM 2009 A β-arrestin-biased agonist of the parathyroid hormone receptor (PTH1R) promotes bone formation independent of G protein activation. Sci Transl Med 1:1ra1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berginer VM, Shany S, Alkalay D, Berginer J, Dekel S, Salen G, Tint GS, Gazit D 1993 Osteoporosis and increased bone fractures in cerebrotendinous xanthomatosis. Metabolism 42:69–74 [DOI] [PubMed] [Google Scholar]
- Federico A, Dotti MT, Loré F, Nuti R 1993 Cerebrotendinous xanthomatosis: pathophysiological study on bone metabolism. J Neurol Sci 115:67–70 [DOI] [PubMed] [Google Scholar]
- Rosen H, Reshef A, Maeda N, Lippoldt A, Shpizen S, Triger L, Eggertsen G, Björkhem I, Leitersdorf E 1998 Markedly reduced bile acid synthesis but maintained levels of cholesterol and vitamin D metabolites in mice with disrupted sterol 27-hydroxylase gene. J Biol Chem 273:14805–14812 [DOI] [PubMed] [Google Scholar]
- Schüle R, Siddique T, Deng HX, Yang Y, Donkervoort S, Hansson M, Madrid RE, Siddique N, Schöls L, Björkhem I 2010 Marked accumulation of 27-hydroxycholesterol in SPG5 patients with hereditary spastic paresis. J Lipid Res 51:819–823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirayama T, Mizokami Y, Honda A, Homma Y, Ikegami T, Saito Y, Miyazaki T, Matsuzaki Y 2009 Serum concentration of 27-hydroxycholesterol predicts the effects of high-cholesterol diet on plasma LDL cholesterol level. Hepatol Res 39:149–156 [DOI] [PubMed] [Google Scholar]
- Lee CY, Huang SH, Jenner AM, Halliwell B 2008 Measurement of F2-isoprostanes, hydroxyeicosatetraenoic products, and oxysterols from a single plasma sample. Free Radic Biol Med 44:1314–1322 [DOI] [PubMed] [Google Scholar]
- Zallone A 2006 Direct and indirect estrogen actions on osteoblasts and osteoclasts. Ann NY Acad Sci 1068:173–179 [DOI] [PubMed] [Google Scholar]
- Smith EP, Specker B, Bachrach BE, Kimbro KS, Li XJ, Young MF, Fedarko NS, Abuzzahab MJ, Frank GR, Cohen RM, Lubahn DB, Korach KS 2008 Impact on bone of an estrogen receptor-α gene loss of function mutation. J Clin Endocrinol Metab 93:3088–3096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powles TJ, Hickish T, Kanis JA, Tidy A, Ashley S 1996 Effect of tamoxifen on bone mineral density measured by dual-energy x-ray absorptiometry in healthy premenopausal and postmenopausal women. J Clin Oncol 14:78–84 [DOI] [PubMed] [Google Scholar]
- Doran PM, Riggs BL, Atkinson EJ, Khosla S 2001 Effects of raloxifene, a selective estrogen receptor modulator, on bone turnover markers and serum sex steroid and lipid levels in elderly men. J Bone Miner Res 16:2118–2125 [DOI] [PubMed] [Google Scholar]
- Practice Committee of American Society for Reproductive Medicine 2008 Estrogen and progestogen therapy in postmenopausal women. Fertil Steril 90(5 Suppl):S88–S102 [DOI] [PubMed] [Google Scholar]
- Martin TJ, Gaddy D 2006 Bone loss goes beyond estrogen. Nat Med 12:612–613 [DOI] [PubMed] [Google Scholar]
- Lindberg MK, Weihua Z, Andersson N, Movérare S, Gao H, Vidal O, Erlandsson M, Windahl S, Andersson G, Lubahn DB, Carlsten H, Dahlman-Wright K, Gustafsson JA, Ohlsson C 2002 Estrogen receptor specificity for the effects of estrogen in ovariectomized mice. J Endocrinol 174:167–178 [DOI] [PubMed] [Google Scholar]
- Syed FA, Mödder UI, Fraser DG, Spelsberg TC, Rosen CJ, Krust A, Chambon P, Jameson JL, Khosla S 2005 Skeletal effects of estrogen are mediated by opposing actions of classical and nonclassical estrogen receptor pathways. J Bone Miner Res 20:1992–2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson KM, Norgård M, Windahl SH, Hultenby K, Ohlsson C, Andersson G, Gustafsson JA 2006 Cholesterol-sensing receptors, liver X receptor α and β, have novel and distinct roles in osteoclast differentiation and activation. J Bone Miner Res 21:1276–1287 [DOI] [PubMed] [Google Scholar]
- Fu X, Menke JG, Chen Y, Zhou G, MacNaul KL, Wright SD, Sparrow CP, Lund EG 2001 27-Hydroxycholesterol is an endogenous ligand for liver X receptor in cholesterol-loaded cells. J Biol Chem 276:38378–38387 [DOI] [PubMed] [Google Scholar]
- Song C, Liao S 2000 Cholestenoic acid is a naturally occurring ligand for liver X receptor α. Endocrinology 141:4180–4184 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.